
Alfa-talasemie u 45 českých rodin a 37 rodin cizinců žijících v České republice: přehled literatury a molekulárně-genetická diagnostika
Alpha-thalassemia in 45 Czech and 37 immigrant families: review of the literature and molecular diagnosis
Alpha-thalassemia is the most common monogenic gene disorder in the world. It is an inherited autosomal recessive disorder characterized by reduced production of the α-globin chains of hemoglobin. It belongs, together with other types of thalassemias and hemoglobinopathies, to common globin gene disorders widely distributed due to their natural selection through protection of carriers against severe malaria. Alpha-thalassemia most frequently results from deletions removing segments of DNA from the α-globin gene cluster on chromosome 16 and is characterized by a microcytic hypochromic anemia, with a clinical phenotype varying from almost asymptomatic to a lethal hemolytic anemia. Alpha-thalassemia is especially frequent in populations originating from the South East Asia, Mediterranean region, Africa and Indian subcontinent, with rare occurrence in Central Europe. However, also in our region the presence of α-thalassemia should be considered as a possible cause of hereditary microcytic anemia. Specialized diagnostic approaches that are needed for correct diagnosis of α-thalassemia involve molecular analysis required to confirm the hematological and biochemical observations. The aim of this study is to report the variable phenotype and genotype of these disease states in the Czech families and in immigrants living in the Czech Republic.
KEY WORDS:
hemoglobinopathy – α-thalassemia – HbH disease – molecular diagnostics
Autoři:
M. Divoká 1; M. Partschová 2; D. Pospíšilová 3; M. Orviská 1; A. Lapčíková 1; K. Indrák 1; J. Čermák 4; V. Divoký 1,2
Působiště autorů:
Hemato-onkologická klinika, Lékařská fakulta Univerzity Palackého a Fakultní nemocnice Olomouc
1; Ústav biologie, Lékařská fakulta Univerzity Palackého v Olomouci
2; Dětská klinika, Lékařská fakulta Univerzity Palackého v Olomouci a Fakultní nemocnice Olomouc
3; Ústav hematologie a krevní transfuze, Praha
4
Vyšlo v časopise:
Transfuze Hematol. dnes,22, 2016, No. 3, p. 201-209.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
Alfa-talasemie jsou nejrozšířenější monogenně dědičná onemocnění na světě. Jedná se o vrozené anémie s autozomálně recesivní dědičností charakterizované kvantitativní redukcí syntézy α-globinových řetězců molekuly hemoglobinu. Společně s dalšími typy talasemií a se strukturními hemoglobinovými variantami jsou řazeny do skupiny chorob s poruchou tvorby nebo struktury globinových řetězců – hemoglobinopatií. Výskyt většiny hemoglobinopatií je soustředěn především do malarických oblastí. Globinové mutace poskytují heterozygotním nosičům ochranu před malárií, pravděpodobně proto došlo v průběhu evoluce k selektivnímu zvýhodnění mutantních alel globinových genů v oblastech s vysokým výskytem malárie.
Alfa-talasemie jsou nejčastěji způsobeny delecemi úseků DNA v α-globinovém lokusu na 16. chromozomu a vyznačují se mikrocytární hypochromní anémií s různými fenotypovými stupni závažnosti – od téměř asymptomatických forem až po život ohrožující hemolytické anémie. Jsou rozšířeny zvláště v jihovýchodní Asii, ve Středomoří, v Africe a Indii, ve střední Evropě jsou vzácné. Přesto i v našich zeměpisných šířkách je na přítomnost α-talasemie nutné myslet jako na možnou příčinu vrozené mikrocytární anémie. Pro správnou diagnostiku α-talasemií je nutný soubor hematologických, biochemických a molekulárně-genetických vyšetření. Cílem tohoto sdělení je upozornit na variabilní fenotyp a genotyp těchto onemocnění v České republice u českých rodin i rodin cizinců žijících v ČR.
KLÍČOVÁ SLOVA:
hemoglobinopatie – α-talasemie – onemocnění HbH – molekulárně-genetická diagnostika
ÚVOD
Alfa-talasemie je nejrozšířenější monogenně dědičné onemocnění na světě, asi 5 % světové populace nese některou z α-talasemických alel [1]. Alfa-talasemie, spolu s dalšími typy talasemií a s endemickými hemoglobinovými variantami byly selektovány v průběhu evoluce v malarických oblastech, neboť heterozygotům přinášejí výhodu v protekci proti malarickému parazitu Plasmodium falciparum (2; světovou distribuci α-talasemií znázorňuje obr. 1). Talasemie i hemoglobinové varianty se mohou ovšem vyskytnout v každé etnické skupině a vzhledem ke vzrůstající migraci obyvatel se stále častěji rozšiřují do oblastí střední a severní Evropy, do severní Ameriky a také Austrálie. Právě ve střední Evropě bylo dříve toto onemocnění lékaři opomíjeno.

Díky historické i současné imigraci se talasemie a hemoglobinové varianty objevují stále častěji i v naší populaci. V České republice a na Slovensku jsou talasemie nejčastější příčinou vrozené mikrocytární anémie [3], ale pozornost byla dosud věnována téměř výhradně β-talasemiím [3–6]. Alfa-talasemie byly dosud v ČR charakterizovány na molekulární úrovni jen výjimečně [3, 4]; v jednom případě se jednalo o nový unikátní molekulární mechanismus vedoucí k α-talasemii [7, 8]. V tomto sdělení přinášíme přehled literatury a vlastní zkušenosti s diagnostikou α-talasemických alel. Uvádíme variabilní fenotyp a molekulárně-genetickou analýzu těchto onemocnění u 45 rodin českého původu a 37 rodin cizinců žijících v České republice.
GENOTYPOVÁ A FENOTYPOVÁ VARIABILITA α-TALASEMIÍ
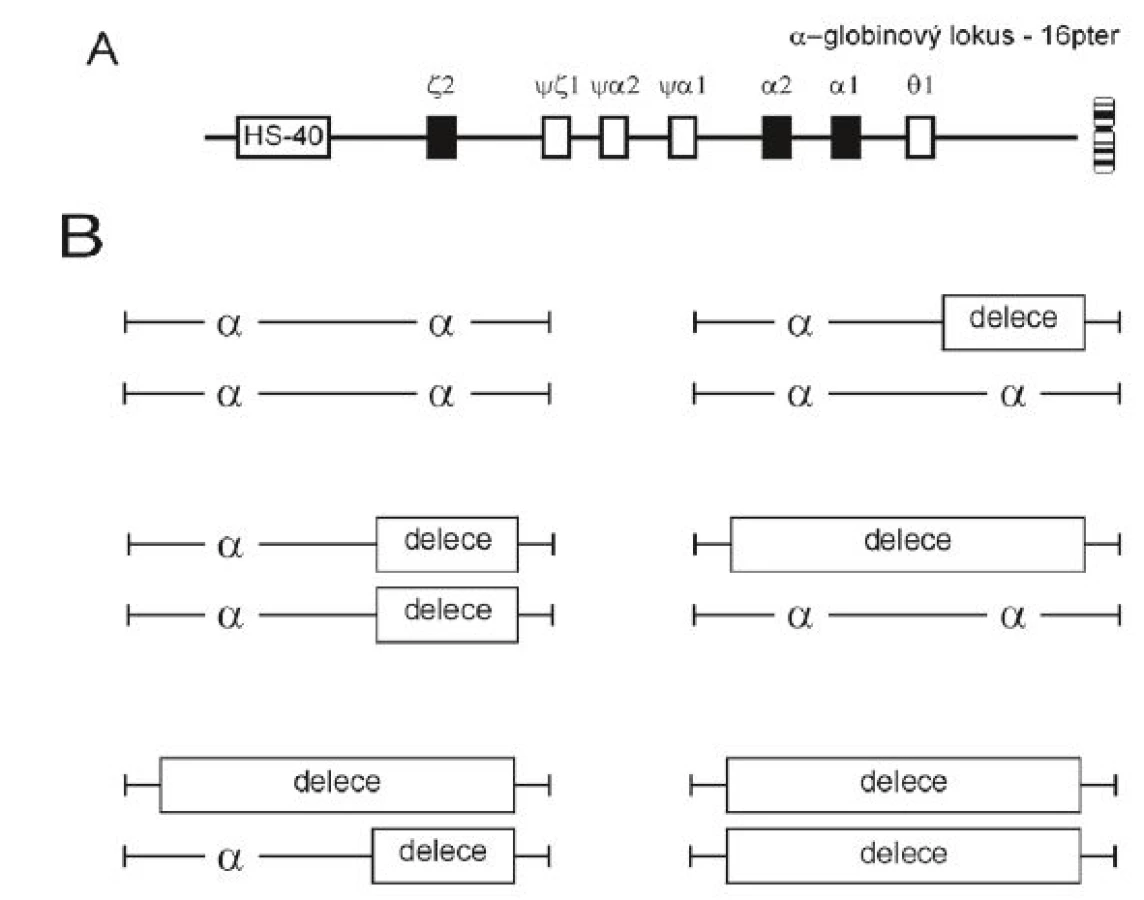
Poměr α a β-globinových řetězců v molekule hemoglobinu (Hb) je fyziologicky jedna ku jedné. Nerovnováha vzájemného poměru globinových řetězců je charakteristická pro talasemie; α-talasemie se vyznačují redukovanou produkcí α-globinových řetězců. Alfa-talasemie jsou většinou způsobeny delecí funkčních α-globinových genů, méně často jsou α-globinové geny inaktivovány bodovou mutací. Haploidní lidský genom má dva α-globinové geny (obr. 2A), to znamená, že normální diploidní genotyp je αα/αα. Normální sada čtyř funkčních α-globinových genů může být snížena o 1, 2, 3 nebo o všechny 4 kopie genu a z toho následně vyplývají rozdílné klinické projevy a zvyšující se závažnost onemocnění (obr. 2B). Míru snížené produkce α-globinových řetězců z lokusu vyjadřuje označení α+-talasemie a α0-talasemie. U α+--talasemie je jen jeden ze dvou α-globinových genů na chromozomu deletován (–α/αα) nebo inaktivován bodovou mutací (αND/αα, kde ND značí „nondeletional“; dříve αT/αα). U α0-talasemie jsou oba α-globinové geny na chromozomu inaktivovány delecí nebo bodovou mutací. Stupeň mikrocytózy, hypochromázie a anémie závisí na počtu mutovaných α-globinových genů a dobře koreluje s mírou redukce syntézy α-globinových řetězců [1].
KLINICKÉ FORMY α-TALASEMIÍ
Název α-talasemie obecně zahrnuje všechna onemocnění s deficitem tvorby α-globinových řetězců, které jsou součástí tetrametru Hb molekuly. Všechny lidské Hb molekuly jsou tetramery, sestávající ze dvou různých párů globinových řetězců. Jeden pár globinových řetězců je produkován geny α-globinového lokusu (ve fetálním období i v dospělosti jsou to α2 neboli HBA2 a α1 neboli HBA1 geny, jejichž kódující sekvence jsou identické). Druhý pár řetězců je produkován geny β-globinového lokusu (ve fetálním období jsou to γG neboli HBG2 a γA neboli HBG1 geny, v dospělosti β neboli HBB gen). Snížená produkce α-globinových řetězců vede ve fetálním období k nadbytku γ-globinových řetězců, které se formují do γ4 tetramerů, a vzniká tak Hb Bart’s, v období dospělosti k nadbytku β-globinových řetězců, které se formují do β4 tetramerů za vzniku nestabilního HbH.
Na základě fenotypu rozlišujeme u nosičství pro α-talasemii dva typy: tiché nosičství s inaktivací (většinou delecí) jednoho α-globinového genu (–α/αα), které se v krevním obraze téměř neprojevuje a prokázat jej lze jen pomocí molekulárně genetického vyšetření. Výskyt tohoto genotypu je v České republice častý. U druhého typu nazývaného nosičství α-talasemie se jedná nejčastěji o deleci dvou α-globinových genů. Řadíme sem tedy buď homozygoty pro α+-talasemii (–α/–α), nebo heterozygoty pro α0-talasemii (– –/αα) (viz obr. 2B). Nosičství α-talasemie se projevuje mírnou mikrocytární anémií se zvýšeným počtem erytrocytů. Výskyt těchto jedinců je v České republice relativně vzácný.
HbH onemocněním (někdy označovaným jako choroba HbH) trpí jedinci se ztrátou tří α-globinových genů, v případě deleční formy α-talasemie se jedná o jedince s (– –/–α) genotypem, jde tedy o složené heterozygoty pro α+-talasemii a α0-talasemii (obr. 2B). Tito nemocní většinou produkují méně než 30 % normální hladiny α-globinu, trpí středně těžkou anémií (Hb mezi 80–100 g/l) s různým zastoupením HbH (0,8–40 %), mají splenomegalii (která může být závažná). Samotný HbH má vysokou afinitu ke kyslíku a je nestabilní [10]. Choroba HbH výrazně neovlivňuje kvalitu života; v České republice se vyskytuje ojediněle u imigrantů, respektive jejich potomků (viz dále). Hb Bart’s (γ4) syndrom nebo hydrops fetalis je stav neslučitelný se životem, jedná se o homozygotní stav pro α0-talasemii, tedy o chybění všech čtyř α-globinových genů (– –/– –). Většinou se rodí novorozenci s hydropsem a ascitem, velkými játry, erytroblastózou a retikulocytózou. Nejvíce hemoglobinu v erytrocytech u novorozenců s Hb Bart’s hydrops fetalis syndromem je tvořeno fyziologicky nefunkčními homotetramery γ4 a β4. Mají také proměnlivé množství embryonálního Hb Portland, což je u těchto dětí jediný funkční hemoglobin a jediný přenašeč kyslíku, který je drží při životě do narození [11].
Vzácně jsou známé případy dlouhých delecí, které postihují kromě α-globinových genů také další geny v jejich okolí. Toto onemocnění je asociované s poruchami vývoje (zahrnující také duševní poruchy) a je označováno jako α-talasemie/syndrom mentální retardace asociovaný s chromozomem 16 (ATR-16 syndrom; 12). Byly popsány také případy vzácné formy mentální retardace spojené s α-talasemií, způsobené abnormalitami ATRX genu ležícím na chromozomu X (ATR-X syndrom). U těchto nemocných je duševní porucha více závažná a dysmorfické změny jsou nápadně podobné mezi jednotlivými pacienty. Gen ATRX kóduje protein ze skupiny helikáz/ATPáz, který ovlivňuje chromatinovou strukturu a transkripci různých genů, mezi nimi i α-globinových genů. Mutace ATRX vedou ke globálnímu potlačení exprese těchto genů, mezi nimi i genů důležitých pro normální rozvoj CNS. U nás (v ČR nebo na Slovensku) jsme dosud ATR syndrom nediagnostikovali. Vzácné jsou získané formy α-talasemie, asociované s myelodysplastickým syndromem (syndrom ATMDS). Ve většině případů je ATMDS syndrom asociován se získanými mutacemi v ATRX genu [12].
MOLEKULÁRNÍ PODSTATA α-TALASEMIÍ
Deleční α+-talasemie
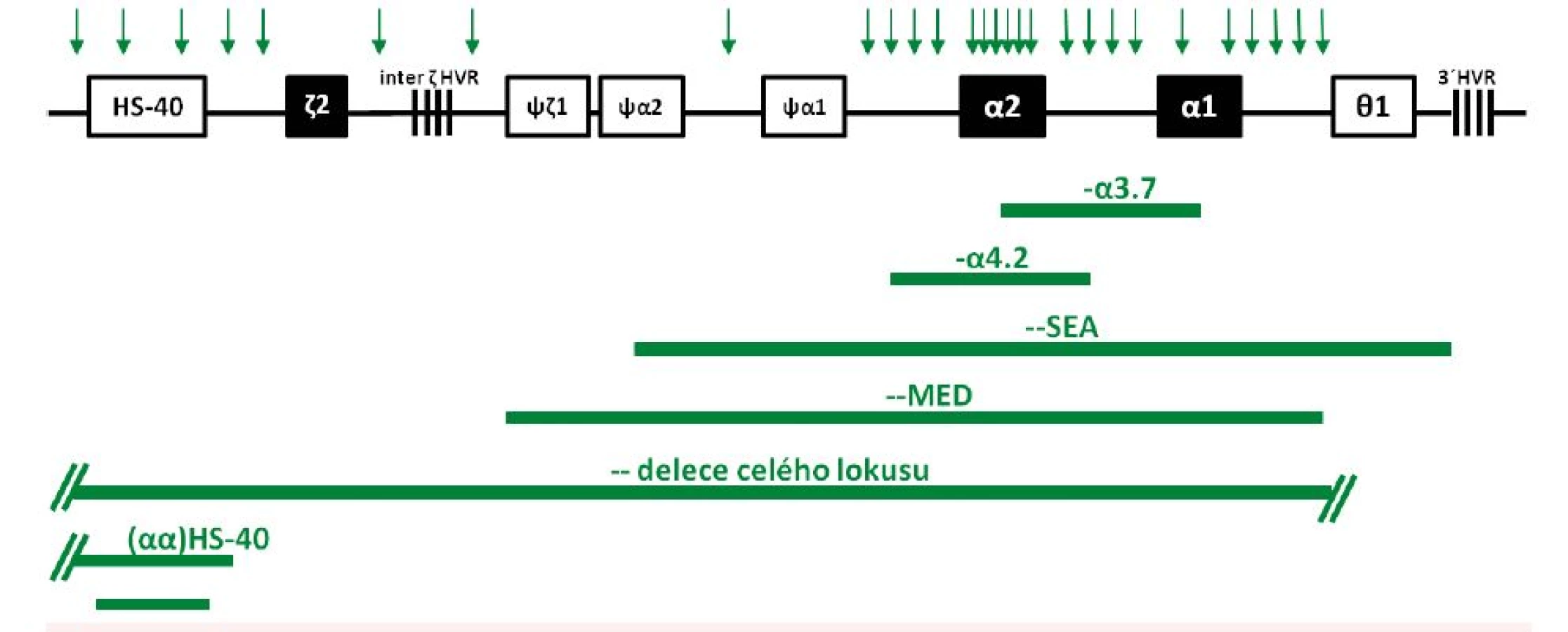
Vzhledem k tomu, že dva α-globinové geny v lokusu vznikly v evoluci duplikací, vyznačuje se tato oblast DNA několika homologiemi, které mohou vést k nerovnoměrným reciprokým rekombinacím (interchromozomálně, méně často intrachromozomálně). Výsledkem je vznik alel s jedním funkčním α-globinovým genem a alel s triplikacemi α-globinových genů [13]. Vzniká tak velmi častá 3.7 kb dlouhá α-talasemická delece (s variantami označovanými jako I, II a III lišícími se místem rekombinace), produkující chromozom pouze s jedním funkčním α-genem (–α3.7) (viz obr. 1, obr. 3) a α-triplikovaná alela (αααanti3.7) bez talasemického efektu. Stejně tak reciproká rekombinace v průběhu meiózy mezi homologními segmenty DNA způsobuje běžnou 4.2 kb dlouhou deleci (–α4.2) (viz obr. 1 a 3) a αααanti4.2 chromozom (13). Další případy rekombinací mezi takto vzniklými chromozomy (α, αα a ααα) pak mohou způsobit kvadruplikaci α genů (αααα) nebo kvintuplikaci (ααααα) a další přeskupení [14]. Na rozdíl od delecí vedoucích k talasemickému fenotypu se samotné zmnožení α-globinových genů u jinak zdravých jedinců klinicky většinou neprojevuje. Problém ovšem nastává u dvojitých heterozygotů pro alelu se zmnoženými α-globinovými geny a β-talasemickou alelu – dochází tak ke zvýšení nerovnováhy α a β řetězců a ke zvýraznění chudokrevnosti, což bylo vzácně potvrzeno i u nemocných v ČR [6, 15].
Deleční α0-talasemie
V současné době je známo asi 50 typů delecí HBA lokusu, které buď kompletně, nebo částečně deletují oba α-globinové geny s následným zastavením syntézy α-globinových řetězců in vivo [13]. Nejčastější jsou delece – –MED rozšířená mezi populacemi ve Středomoří a – –SEA vyskytující se převážně v jihovýchodní Asii [1] (viz obr. 1). Homozygoti pro tento typ delecí trpí Hb Bart’s hydrops fetalis syndromem. Složení heterozygoti pro tento typ delece a delece pro α+-talasemii, která způsobí chybění jednoho α-globinového genu, mají HbH onemocnění. Tyto zatím zmíněné typy delecí postihují pouze HBA lokus a vyskytují se i v ČR (viz níže). Jiné delece postihují i jiné přiléhající geny, což nemusí mít výrazný vliv na zhoršení fenotypu. Některé známé delece odstraňují i ζ-gen současně s α-geny, a i když heterozygoti vykazují normální vývoj, u homozygotů je nepravděpodobné přežití období časného stadia gravidity vzhledem k tomu, že ani embryonální (ζ2γ2) a ani fetální (α2γ2) hemoglobiny nemohou být syntetizovány [13]. Tyto typy delecí byly zjištěny i v české populaci (viz níže).
Vzácné delece vedoucí k α0-talasemii odebírají regulační oblast HS-40, která se nachází 40 kb 5’ od ζ-globinového genu (viz obr. 2A), přičemž zanechávají α-globinové geny intaktní. HS-40 je esenciální pro α-globinovou expresi, delece této oblasti vede k úplné ztrátě exprese α-globinových genů v poloze cis. Fenotyp je tedy shodný jako u vzácných delecí celého HBA lokusu. Tyto delece se sporadicky vyskytují v různých populacích [10], vzácně i u českých rodin (viz níže, obr. 3, tab. 1).
Vznik α-talasemie způsobené zastavením transkripce neaktivním chromatinem u polské rodiny žijící v České republice byl popsán jako nový mechanismus vzniku tohoto onemocnění [16]. Delece dlouhá 18 kb zahrnuje α1 gen a přilehlou 3’oblast a způsobí utlumení exprese α2 globinového genu v poloze cis. Díky deleci se do lokusu přemístí represivní chromatin, který zastaví α2 transkripci [7, 8]. Heterozygoti pro tuto mutaci trpí mírnou hypochromní mikrocytární anémií, mají snížený poměr syntézy α/β globinových řetězců a HbH inkluze v erytrocytech.
Nedeleční formy α-talasemie (αND)
Alfa-talasemie jsou častěji způsobeny genovými delecemi, bodové mutace (jednonukleotidové záměny) vedoucí k α-talasemii jsou vzácnější [10, 13]. Alfa-globinové geny mutované v kanonických sekvencích HBA1 a HBA2 ovlivňujících α-globinovou genovou expresi se označují jako (αND nebo dříve αT); bodová talasemická mutace může postihnout α2 (αNDα) nebo α1 (ααND) globinový gen. Většina těchto mutací ovlivňuje úpravu (přepis a post-transkripční zpracování) mRNA, mRNA translaci nebo α-globinovou stabilitu. Bodové mutace mohou vést k částečnému (α+) nebo úplnému (α0) utlumení genové exprese, respektive produkce funkčních α-globinových řetězců. Je známo několik desítek bodových mutací způsobujících α-talasemii; mnohé z nich jsou běžné i ve Středomoří a na Blízkém východě (obr. 1) a lze je tedy očekávat i v české a slovenské populaci. Aktualizovaný seznam těchto mutací je dostupný v databázi HbVar, http://globin.cse.psu.edu/hbvar. Některé bodové mutace v terminačním kodonu vedou k prodlouženým Hb variantám s talasemickým fenotypem, jako např. Hb Constant Spring, nejběžnější nedeleční α-talasemická mutace častá zvláště v některých oblastech jihovýchodní Asie. Jiné mutace způsobují vysoce nestabilní α-globinové varianty, např. Hb Adana (10). Nedávno byla popsána odchylka jednoho nukleotidu v negenové oblasti globinového klastru, která vede k novému molekulárnímu mechanismu vzniku α-talasemie (17). Jedná se o záměnu v sekvenci regulačního jednonukleotidového polymorfismu (SNP, “Single Nucleotide Polymorphism”), kterou se vytváří nový transkripčně aktivní promotor v normálně transkripčně neaktivní oblasti ψζ1. Tato abnormální transkripční aktivace interferuje a zeslabuje expresi α-globinových genů.
Diagnóza a diagnostické metody
Iniciální laboratorní testování by mělo zahrnovat kompletní krevní obraz s popisem červených krvinek, bioanalytické testy zahrnující měření hladiny HbA2, HbF a elektroforézu Hb. Dříve bylo pro potvrzení α-talasemie nezbytné měření poměru syntézy α/β globinových řetězců radioaktivně značeným leucinem; redukovaný poměr na méně než ∼0,8 svědčil pro α-talasemii [18]. V současné době převažují molekulární analýzy, které potvrdí podezření na α-talasemii. Elektroforéza Hb slouží k separaci abnormálních hemoglobinových frakcí, je důležitá mimo jiné pro detekci Hb variant s α-talasemickým fenotypem. Precipitovaný HbH je v erytrocytech prokazován inkubací s brilliant-kresylovou modří (obr. 4A).

MOLEKULÁRNÍ ANALÝZA
Molekulární diagnostice napomáhá etnický původ vyšetřovaných jedinců; rozdílné spektrum α-talasemických mutací bývá nalézáno v rozdílných populacích. Dříve byla většina přeskupení HBA genů v rámci HBA lokusu charakterizována pomocí Southern blotu (obr. 4B). V současné době existují rychlé screeningové metody pro detekci delecí a jiných změn v HBA lokusu.

Pro detekci sedmi nejčastějších α-talasemických delecí bylo vytvořeno multiplexní PCR [19] (obr. 4C). Tato metoda se používá pro určení dvou nejčastějších α+-talasemických delecí (–α3.7; –α4.2) a dalších 5 α0-talasemických delecí, mezi nimi – –MED a – –SEA. Novější MLPA (Multiplex Ligation-dependent Probe Amplification) je založena na ligaci více párů prób (obvykle dlouhých), které hybridizují napříč k analyzované oblasti. Následuje semikvantitativní amplifikace s použitím univerzálních tag PCR primerů a poté fragmentační analýza (obr. 4D). Tato metoda je hodnotnou alternativou k Southern blotu a doplňkovou k multiplexní PCR při zjišťování jak známých tak neznámých delecí způsobujících α-talasemii [20].


DIFERENCIÁLNÍ DIAGNOSTIKA
V některých případech mají nosiči α+-talasemie normální hematologické výsledky, speciálně nosiči –α3.7 delece a nosiči nedelečních mutací v α1 genu. Někteří jedinci mohou mít normocytózu nebo hraniční hypochromazii bez anémie. Tato skutečnost může být zjištěna pouze náhodně při rutinní molekulární analýze pro záchyt hemoglobinopatií.
Příležitostně, speciálně v zemích, kde je talasemie vzácná, mohou být nosiči α-talasemie nesprávně označení za pacienty s anémií z nedostatku železa, zvláště pokud není status železa u pacienta správně zhodnocen. Vždy je nutná molekulární analýza, zvláště u tichého nosičství nebo nosičství α-talasemie k potvrzení diagnózy, protože i nosiči α-talasemické alely můžou vykazovat vyšší riziko přetížení organismu železem [21].
VÝSKYT α-TALASEMIÍ V ČESKÉ REPUBLICE – PŘEHLED LITERÁRNÍCH ÚDAJŮ
Alfa-talasemie byly považovány v České republice za velmi vzácné; jen několik případů bylo charakterizováno na molekulární úrovni tradiční metodou Southernova přenosu a hybridizace [3, 4]. Kromě −α3.7 delece zjištěné u několika jedinců byl u jedné rodiny polského původu žijící v ČR popsán již zmíněný nový typ unikátní delece 18 kb DNA postihující α1 gen a přilehlou 3‘-oblast, která vedla překvapivě k utlumení exprese α2 globinového genu v poloze cis, tj. k fenotypu, který je asociován s α-tal-1 genotypem [16]. Bylo zjištěno, že 18 kb delece přemístila do klastru represivní chromatin, který spolu s protismyslnými transkripty přilehlého genu (gen, jenž je přepisován proti směru exprese α-globinových genů) zastaví α2 transkripci [7, 8]. Vzácně byly také detekovány triplikace α-globinových genů [3, 4], někdy vedoucí ke zhoršení klinického projevu β-talasemie [22].
DIAGNOSTIKA α-TALASEMIÍ V ČESKÉ REPUBLICE – NOVÉ VÝSLEDKY
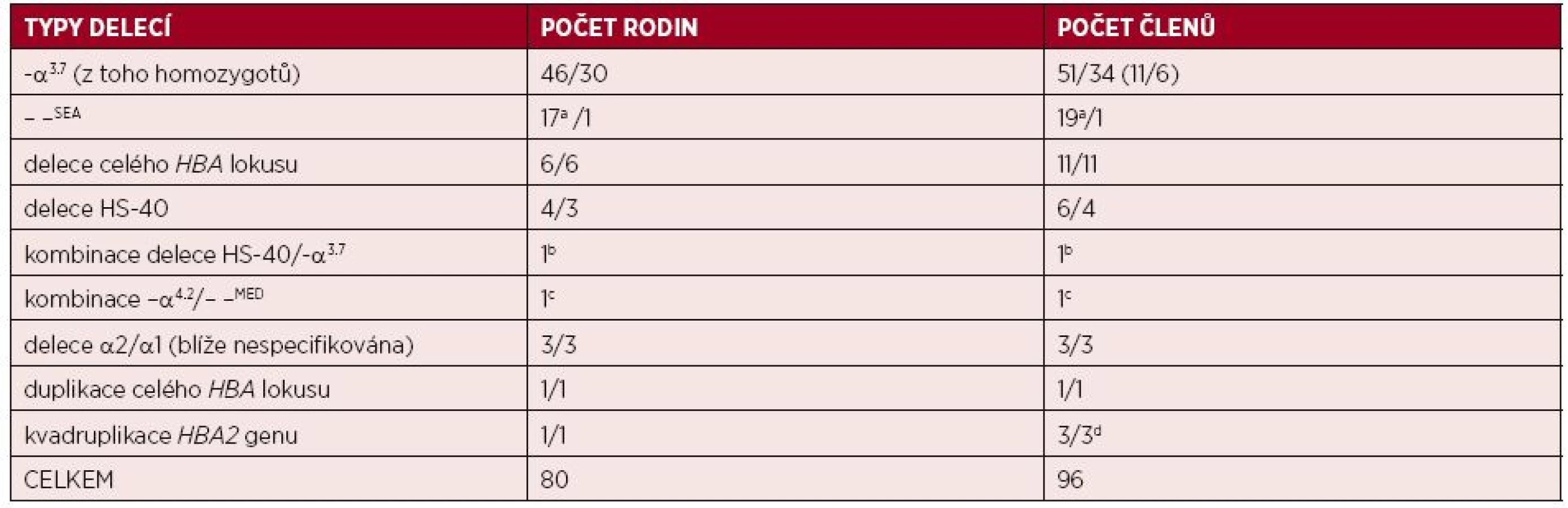
V průběhu posledních 10 let došlo k významnému rozšíření spektra α-talasemických mutací i počtu molekulárně-geneticky diagnostikovaných jedinců v ČR. Ke zpřesnění diagnostiky přispělo využití nových molekulárně-genetických přístupů – multiplexní PCR a MLPA pro diagnostiku delečních forem α-talasemií a sekvenační analýzy HBA genů pro nedeleční formy α-talasemií. V souboru 82 rodin (98 členů) s podezřením na α-talasemii jsme odhalili u 78 rodin (92 jedinců) různé delece v rámci HBA lokusu; 45 rodin (53 jedinců) bylo českých, bez předků z malarických oblastí nejméně ve třech generacích rodu, ostatní byli cizinci žijící v ČR (viz tab. 1). U dvou jiných českých rodin (4 jedinců) jsme odhalili multiplikaci α-globinových genů (viz tab. 1) a u dalších dvou imigrantů jsme odhalili nedeleční α-talasemickou mutaci. Většina diagnostikovaných jedinců měla mírnou mikrocytární hypochromní anémii, s normálními hladinami HbA2 a HbF, se standardní elektroforézou Hb a mírně zvýšeným počtem erytrocytů. Pro možnou přítomnost α-talasemické alely svědčil rodinný výskyt fenotypu, v případě rodin cizinců i etnický původ. Ve většině případů se jednalo o tiché nosičství nebo nosičství α-talasemie, s běžnými typy delecí jednoho nebo dvou α-globinových genů, které jsou známé ze Středozemí nebo z jihovýchodní Asie (viz obr. 1 a 3). Rozsáhlé delece celého HBA lokusu, včetně regulační oblasti HS-40, byly detekovány u 6 českých rodin (11 členů) s klinickými projevy odpovídajícími nosičství α-talasemie (jednalo se vesměs o heterozygoty, viz tab. 1). U 4 rodin (z nich 3 českého původu) jsme detekovali různě dlouhé delece regulační oblasti HS-40, ale s intaktními α-globinovými geny (viz obr. 3), s fenotypem odpovídajícím ztrátě dvou α-globinových genů (nosičství α-talasemie) u 5 jedinců, a u 1 pacientky jedné rodiny s klinickými projevy odpovídajícími ztrátě tří α-globinových genů (choroba HbH). Molekulární analýzou této pacientky s italskými předky jsme zjistili, že fenotyp HbH onemocnění je u ní způsoben inaktivací dvou α-globinových genů zmíněnou delecí HS-40 na jednom chromozomu v kombinaci s delecí jednoho α-globinového genu (−α3.7) na druhém chromozomu. Stejně závažný fenotyp, tj. chorobu HbH, jsme detekovali u imigranta ze severní Afriky, u kterého byl příčinný genotyp kombinací dvou častých delecí ze Středozemí (–α4.2/– –MED) (viz tab. 1).
Nedeleční formy α-talasemie jsme zatím diagnostikovali jen u dvou imigrantů žijících v ČR, u obou se jednalo o mutaci HBA2:c.95+2_95+6delTGAGG, označovanou také jako α-thal-2 (-5nt) (23), která se běžně vyskytuje ve Středomoří.
ZÁVĚR
První systematická molekulárně-genetická analýza α-talasemií v ČR provedená na selektovaném souboru 82 rodin (98 jedinců) odhalila novými diagnostickými přístupy 92 jedinců ze 78 rodin s delecemi v rámci HBA lokusu; 45 rodin (53 jedinců) bylo prokazatelně českého původu. Další 2 české rodiny (4 členové) měli v genotypu multiplikaci α-globinových genů a u dalších dvou imigrantů byla odhalena nedeleční α-talasemická mutace. U většiny diagnostikovaných jedinců s α-talasemií minor (37 cizinců/35 Čechů), byly nalezeny delece a u 2 cizinců bodové mutace α-globinových genů běžné v malarických oblastech. Vzácně jsme odhalili rozsáhlé delece celého HBA lokusu nebo delece regulační oblasti lokusu HS-40, ale s intaktními α-globinovými geny; tyto typy delecí byly častěji zjištěny u českých rodin než u imigrantů. Podobné typy delecí jsou velmi sporadicky detekovány v různých populacích, včetně původních obyvatel západní a severní Evropy. U 3 českých rodin nebylo možné deleci HBA klastru blíže specifikovat. Prioritně byl v ČR charakterizován genetický podklad dvou pacientů s onemocněním HbH, první měl italské předky, druhý byl imigrant z Afriky. Heterogenita α-talasemií je v ČR zřejmě daleko větší než se dosud soudilo. Navíc, α-talasemické alely budou se stupňující se migrací v ČR postupně přibývat, a s tím bude stoupat i nutnost jejich přesné diagnostiky založené na molekulárně-genetických analýzách.
Seznam použitých zkratek
Hb – hemoglobin
ND – „nondeletional“, nedeleční
kb – kilobáze
ATR – „α-thalassemia with mental retardation“
SNP – „Single Nucleotide Polymorphism“, jednonukleotidový polymorfismus
PCR – polymerázová řetězová reakce
MLPA – „Multiplex Ligation-dependent Probe Amplification“
Podíl autorů na rukopisu
DM – napsání rukopisu, provádění analýz, interpretace výsledků, příprava finální verze rukopisu
PM – provádění analýz, revize rukopisu
PD – léčba pacientů, připomínkování rukopisu
OM – analýzy pacientských vzorků
LA – analýzy pacientských vzorků
IK – léčba pacientů, připomínkování rukopisu
ČJ – léčba pacientů, připomínkování rukopisu
DV – příprava finální verze rukopisu, interpretace výsledků
Poděkování
Lékařům hematologických pracovišť v České republice a na Slovensku za poskytnutí pacientských vzorků a za spolupráci.
Tato práce byla podpořena granty IGA_LF_2016_001 a IGA_LF_2016_014.
Čestné prohlášení autora
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Doručeno do redakce dne 18. 5. 2016.
Přijato po recenzi dne 22. 6. 2016.
RNDr. Martina Divoká
Hemato-onkologická klinika, Lékařská fakulta Univerzity Palackého a FN Olomouc
Hněvotínská 3
775 15 Olomouc
email: martina.divoka@fnol.cz
Zdroje
1. Piel FB, Weatherall DJ. The α-thalassemias. N Engl J Med 2014; 371(20): 1908–1916.
2. Weatherall DJ, Williams TN, Allen SJ, O’Donnell A. The population genetics and dynamics of the thalassemias. Hematol Oncol Clin North Am 2010; 24(6): 1021–1031.
3. Indrák K, Divoký V, Brabec V, et al. Molekulárně genetická charakte-ristika α, α a αα thalassémií u 139 heterozygotů z 56 nepříbuzných rodin českého a slovenského původu. Vnitř Lék 1993; 39(10): 969–978.
4. Indrak K, Brabec V, Indrakova J, et al. Molecular characterization of α-thalassemia in Czechoslovakia. Hum Genet 1992; 88(4): 399–404.
5. Divoký V., Walczysková S, Pospíšilová D, et al. Některé vzácnější formy hereditárních anémií vyskytující se v dospělé populaci v ČR a SR – α-talasemie a nestabilní hemoglobinové varianty. Vnitř Lék 2005; 51: 741–748.
6. Divoka M, Partschova M, Kucerova J, et al. Molecular Characterization of b-Thalassemia in the Czech and Slovak Populations: Mediterranean, Asian and Unique Mutations. Hemoglobin 2016: 1–7. DOI: 10.3109/03630269.2016.1152581.
7. Barbour VM, Tufarelli C, Sharpe JA, et al. α-thalassemia resulting from a negative chromosomal position effect. Blood, 2000; 96(3): 800–807.
8. Tufarelli C, Stanley JA, Garrick D, et al. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet 2003; 34(2): 157–165.
9. Divoký V, Indrák K, Mojzíková R. Hemoglobinopatie: talasémie a strukturální Hb varianty. In: Molekulární hematologie. Praha: Galén, 2013, s. 270– 283. ISBN 978-80-7262-942-8.
10. Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis 2010; 5: 13. DOI: 10.1186/1750-1172-5-13.
11. Chui DH. Alpha-thalassemia: Hb H disease and Hb Barts hydrops fetalis. Ann N Y Acad Sci 2005; 1054: 25–32.
12. Higgs DR, Buckle VJ, Gibbons R, Steensma D. Unusual Types of αThalassemia. In Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. (Eds). Disorders of hemoglobin: genetics, pathophysiology and clinical management, 2nd edition. Cambridge: Cambridge University Press, 2009, s. 296–320.
13. Higgs DR. The molecular basis of α thalassemia. In Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. (Eds). Disorders of hemoglobin: genetics, pathophysiology and clinical management, 2nd edition. Cambridge: Cambridge University Press, 2009, s. 241–265.
14. Lam KW, Jeffreys AJ. Processes of de novo duplication of human α-globin genes. Proc Natl Acad Sci USA 2007; 104(26): 10950–10955.
15. Indrak K, Fei YJ, LI HW, et al. A Czechoslovakian teenager with Hb E-ß0-thalassemia [IVS-I-1 (G→A)] complicated by the presence of an a-globin gene triplication. Ann Hematol 1991; 63(1): 42–44.
16. Indrak K, Gu YC, Novotny J, Huisman TH. A new alpha-thalassemia-2 deletion resulting in microcytosis and hypochromia and in vitro chain imbalance in the heterozygote. Am J Hematol, 1993; 43(2): 144–145.
17. De Gobbi M, Viprakasit V, Hughes JR, et al. A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science 2006; 312(5777): 1215–1217.
18. Clegg JB, Weatherall DJ. Haemoglobin synthesis in alpha-thalassaemia (haemoglobin H disease). Nature 1967; 215: 1241–1243.
19. Chong SS, Boehm CD, Higgs DR, Cutting GR. Single-tube multiplex-PCR screen for common deletional determinants of α-thalassemia. Blood 2000; 95(1): 360–362.
20. Harteveld CL, Voskamp A, Phylipsen M, et al. Nine unknown rearrangements in 16p13.3 and 11p15.4 causing alpha- and beta-thalassaemia characterised by high resolution multiplex ligation-dependent probe amplification. J Med Genet 2005; 42(12): 922–931.
21. Sulovska L, Holub D, Zidova Z, et al. Characterization of iron metabolism and erythropoiesis in erythrocyte membrane defects and thalassemia traits. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2015. DOI: 10.5507/bp.2015.054. [Epub ahead of print]
22. Indrak K, Fei Y-J, Li H-W, et al. A Czechoslovakian teenager with Hb E- β0-thalassemia [IVS-I-1 (G→A)] complicated by the presence of an α-globin gene triplication. Ann Hematol 1991; 63(1): 42–44.
23. Orkin SH, Goff SC, Hechtman RL. Mutation in an intervening sequence splice junction in man. Proc Natl Acad Sci USA 1981; 78(8): 5041–5045.
Štítky
Hematológia Interné lekárstvo OnkológiaČlánok vyšiel v časopise
Transfuze a hematologie dnes

2016 Číslo 3
- Nejasný stín na plicích – kazuistika
- Těžké menstruační krvácení může značit poruchu krevní srážlivosti. Jaký management vyšetření a léčby je v takovém případě vhodný?
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
Najčítanejšie v tomto čísle
- Alfa-talasemie u 45 českých rodin a 37 rodin cizinců žijících v České republice: přehled literatury a molekulárně-genetická diagnostika
- Diagnostika a prognostické faktory lymfomu z buněk pláště
- Imunogenetické faktory ovlivňující aloimunizaci proti antigenům krevních skupinových systémů
- Analýza zhody vyšetrenia inhibítorov FVIII Bethesda metódou a modifikovanou Nijmegen metódou a vplyv hraničných titrov inhibítorov na farmakodynamiku a farmakokinetiku FVIII u pacientov s hemofíliou A