
Centrum pro vzácné choroby červené krevní řady v Ústavu hematologie a krevní transfuze
Centre for rare disorders of haematopoiesis at the Institute of Haematology and Blood Transfusion
The Institute of Haematology and Blood Transfusion was designated a Centre for Rare Disorders of Haematopoiesis in 2014. Most of the patients who are currently treated in the centre suffer from rare congenital or acquired disorders of erythropoiesis. This article reviews patients with congenital disorders of hematopoietic stem cells, red blood cell (RBC) membrane defects, enzymopathies and hemoglobinopathies who have been diagnosed and treated in the centre. The centre also follows patients with paroxysmal nocturnal haemoglobinuria. The increasing number of patients with congenital disorders of erythropoiesis is due to greater implementation of diagnostic molecular genetic methods, the prolonged survival of patients as well as increasing migration from regions with a high incidence of congenital RBC disorders. Diagnosis and long term follow up of patients with rare disorders of erythropoiesis are warranted because of the high incidence of tumours and complications resulting from organ iron overload in adulthood. Moreover, diagnosis of heterozygous forms in parents may preclude the birth of a homozygote with a serious form of the disease.
KEY WORDS:
erythropoiesis – rare disorders – stem cells disorders – membrane defects, enzymopathies – hemoglobinopathies – paroxysmal nocturnal haemoglobinuria
Autori:
J. Čermák; J. Valka; M. Vostrý; S. Škranc; M. Beličková
Pôsobisko autorov:
Ústav hematologie a krevní transfuze, Praha
Vyšlo v časopise:
Transfuze Hematol. dnes,23, 2017, No. Supplementum1, p. 76-85.
Kategória:
ZVLÁŠTNÍ VYDÁNÍ SUPLEMENTU K 65. VÝROČÍ ÚHKT
Súhrn
V roce 2014 byl ÚHKT jmenován Centrem pro vzácné choroby krvetvorby. V současné době se centrum zaměřuje zejména na vrozené a získané choroby červené krevní řady. Článek podává přehled nemocných s vrozenými poruchami kmenové krvetvorné buňky, poruchami struktury membrány erytrocytu či jeho enzymatického vybavení a poruch tvorby hemoglobinu, kteří byli diagnostikováni a jsou sledováni v ÚHKT, dále jsou v centru léčeni nemocní s paroxysmální noční hemoglobinurií. Zvyšující se počet nemocných s vrozenými poruchami erytropoézy je dán jednak zavedením molekulárně genetických metod do diagnostiky, jednak prodlužující se délkou přežití nemocných a rapidně se zvyšující migrací z oblastí s vysokou incidenci těchto onemocnění. Diagnostika a dispenzarizace nemocných s vrozenými chorobami erytropoézy je nutná vzhledem ke zvýšené incidenci nádorových onemocnění v dospělém věku u těchto nemocných a možnosti vzniku orgánového postižení nadbytkem železa a dále jako prevence vzniku závažných homozygotních forem onemocnění při sňatku dvou heterozygotů.
KLÍČOVÁ SLOVA:
erytropoéza – vzácná onemocnění – poruchy kmenové buňky, membránové defekty – enzymopatie – hemoglobinopatie – paroxysmální noční hemoglobinurie
Vzácná onemocnění postihují méně než 50 jedinců na 100 000 obyvatel. Hematologie zaujímá mezi ostatními obory poněkud zvláštní postavení vzhledem k tomu, že více než 80 % chorob krvetvorby patří podle výše uvedené definice mezi vzácná onemocnění. Ústav hematologie a krevní transfuze (ÚHKT) byl již v 90. letech minulého století jmenován Centrem vysoce specializované péče pro nádorová onemocnění krvetvorby. Věstník MZd 3/14 z května 2014 ustanovuje ÚHKT rovněž Centrem pro poruchy hemostázy a Centrem pro vzácné vrozené a získané poruchy krvetvorby. Centrum se zabývá především poruchami kmenové krevní buňky a erytropoézy a jsou zde dispenzarizováni zejména nemocní s velmi vzácnými poruchami, pacienti s častěji se vyskytujícími chorobami, jakými jsou např. dědičná sférocytóza a heterozygotní formy beta talasemie, jsou většinou sledováni na regionálních hematologických pracovištích a do ÚHKT docházejí pouze v případě nutné konzultace. V následujícím textu budou zmíněny některé vzácnější poruchy kmenové krvetvorné buňky, defekty tvorby hemoglobinu, poruchy membrány erytrocytu a jeho enzymatického vybavení, které byly nalezeny u nemocných sledovaných v ÚHKT, ze získaných vzácných chorob pak bude zmíněna paroxysmální noční hemoglobinurie. Počty sledovaných nemocných jsou uvedeny v tabulce 1.

VROZENÉ PORUCHY KMENOVÉ KRVETVORNÉ BUŇKY
Diamond Blackfanův syndrom (DBA)
DBA projevující se kongenitální aplazií červené krevní řady patří mezi vrozené syndromy selhání kostní dřeně (IBMFS – Inherited Bone Marrow Failure Syndromes) a je řazena mezi tzv. ribozomopatie. Jako základní patogenetický moment je u DBA uvažována mutace některého ze 14 genů pro ribozomální proteiny (RP genů), důsledkem mutace RP genů je defekt maturace ribozomální RNA a poškození tvorby ribozomů, onemocnění je dominantně dědičné. Souvislost poškození ribozomů s rozvojem erytroblastopenie není zcela jasná, ribozomální proteiny hrají roli v indukci degradace p53 proteinu a jejich deficit vede k nárůstu hladiny p53 proteinu, jež může ve zvýšené míře indukovat apoptózu erytroidních prekurzorů. Krom toho se může uplatňovat i defekt translace pro GATA1 gen, jenž je hlavním transkripčním faktorem pro erytropoézu a v poslední době se významná úloha v patogenezi DBA přisuzuje snížené hladině HSP70 (heat schock protein 70), jenž se uplatňuje při reparaci porušené erytroidní proliferace a diferenciace. Různá hladina HSP 70 pozorovaná u mutací různých RP genů může být příčinou genotypové diverzity choroby, která je stejně jako fenotypová diverzita u DBA poměrně veliká. Základním klinickým projevem DBA je makrocytární anémie, u cca poloviny nemocných mohou být přítomny malformace skeletu (palce, horní končetiny) a orgánů (srdce, urogenitální systém), diagnostika se opírá o nález v krevním obraze (anémie, makrocytóza, retikulocytopenie) a chybění erytropoézy v kostní dřeni [1]. Je zvýšena hladina HbF a ADA (adenosin deaminázy), mutace některého z RP genu bývá přítomna u 60–65 % nemocných. Krom substituce transfuzemi erytrocytů se v léčbě DBA uplatňuje podávání kortikosteroidů, jež mohou zmírnit transfuzní dependenci u cca 80 % nemocných, je zkoušeno podávání leucinu. Důležité je monitorování zásob Fe a efektivní chelatační léčba bránící rozvoji přetížení železem. Alogenní transplantace krvetvorných buněk (SCT) je možnou léčebnou metodou, je však zatížena poměrně velkým rizikem komplikací zejména u nemocných, kde již vzniklo orgánové přetížení železem. V ÚHKT je sledováno 8 nemocných s DBA, u 1 nemocného byla DBA prokázána již v minulosti u matky, u ostatních se dědičnost prokázat nepodařilo, mutace některého z RP genů byla prokázána u 5 nemocných (62 %). Klinické projevy kolísají od mírného stupně anémie (Hb = 95–115 g/l) bez potřeby transfuzí u 3 nemocných až po dependenci na 2–3 TU erytrocytů měsíčně u dalších 2 nemocných, 4 nemocní jsou trvale léčeni kortikoidy, u 2 z nich je tato léčba kombinována s podáváním leucinu, 5 nemocných závislých na podávání transfuzí dostává trvale chelatační léčbu (deferasirox či kombinaci deferasirox + deferoxamin). Na DBA je třeba myslet u každé nově vzniklé makrocytární anémie v období dospívání, kdy může dojít k pozdní manifestaci lehčích forem řady vrozených poruch krvetvorby [2]. Extrémním příkladem může být případ 1 naší nemocné, jež byla vyšetřována po dobu 12 let s nejasnou makrocytární anémií s opakovaně prokázanou erytroblastopenií ve dřeni pod různými diagnózami a do ÚHKT byla doporučena k vyšetření jako myelodysplastický syndrom (MDS). Trvalá dispenzarizace nemocných s DBA je důležitá i vzhledem k tomu, že onemocnění se může vyvíjet do MDS, jak pozorujeme u 2 našich nemocných, kde je v kostní dřeni přítomno narůstající procento erytropoézy s dysplastickými rysy a zvýšeným počtem retikulocytů v periferní krvi, další komplikací může být rozvoj solidních nádorů v mladém věku [3], jak jsme bohužel rovněž zjistili u 1 naší nemocné.
Fanconiho anémie (FA)
Jedná se o vzácnou vrozenou formu hypoplazie dřeně s pancytopenií v krevním obraze. Příčinou je mutace některého ze skupiny 19 FA genů, tyto geny jsou aktivovány poškozením buňky a produkované FA proteiny formují finální FANCD2 protein, jenž funguje jako nosič BRCA1 proteinu, tento protein je nutný k homologní reparaci poškozené DNA [4]. Onemocnění je autozomálně recesivní. Anémie je makrocytární, je přítomna zvýšená hodnota HbF a zvýšená fragilita chromozomů po inkubaci s deoxybutyrátem, k potvrzení diagnózy slouží molekulárně genetické vyšetření. Anémie může být spojena s malformacemi skeletu (mikrocefalie, defekty paprsku radia), poškozením ledvin a kožními skvrnami. Postupně dochází k rozvoji progredujícího selhání kostní dřeně s pancytopenií. Komplikací choroby může být nejen poměrně častý přechod do MDS, ale i rozvoj akutní leukemie či solidních nádorů, zejména gynekologických, v ORL oblasti a v dutině ústní. Těžší formy choroby jsou indikovány k transplantaci krvetvorných buněk, u lehčích forem se uplatňuje podpůrná léčba, je studována možnost genové terapie. V ÚHKT jsou sledovány 4 nemocné s FA. U 2 nemocných s prokázanou mutací FANCA genu došlo během třetí dekády života k progredující pancytopenii se známkami rozvíjející se dysplazie v kostní dřeni a s nálezem monozomie 7. chromozomu u jedné z nich. Obě podstoupily alogenní transplantaci od HLA shodné sestry a přežívají 112, respektive 17 měsíců po SCT bez závažnějších komplikací. Zbývající dvě nemocné mají známky jen mírné pancytopenie a jsou dispenzarizovány s pravidelnými hematologickými a onkologickými kontrolami.
Shwachman-Diamondův syndrom (SDS)
Onemocnění se projevuje těžkou granulocytopenií a různě závažným stupněm anémie a trombocytopenie. Může být doprovázeno insuficienci exokrinní části pankreatu, kostními malformacemi a anomáliemi jater. Příčinou je mutace SBDS genu, jenž se zřejmě podílí na přenosu některých signálů mezi cytoplazmou a jádrem buňky a je nutný pro normální funkci RNA, funkci a vazbu ribozomálních podjednotek. V léčbě je metodou volby transplantace krvetvorných buněk. Do ÚHKT byl předán ke sledování 1 nemocný s SDS s výraznými malformacemi skeletu, u něhož byla prokázána mutace SBDS genu. V krevním obraze byla přítomna progredující pancytopenie s vyplavováním nezralých forem myeloidní řady do periferní krve a vyšetření kostní dřeně potvrdilo přechod onemocnění do MDS EB-2 (RAEB- 2) s mnohočetnými aberacemi karyotypu. Nemocný byl léčen kombinovanou chemoterapií a po dosažení kompletní remise byl transplantován od HLA shodného nepříbuzného dárce. Po 14 měsících však došlo k relapsu choroby a přechodu do akutní leukemie (AML) rezistentní na léčbu.
Poruchy regulačních genů krvetvorby
V poslední době se ukazuje, že některé kongenitální cytopenie mohou být způsobeny mutací některých důležitých regulačních genů. GATA-2 představuje regulátor efektivní genové exprese v hemopoetických kmenových buňkách, což významně ovlivňuje úroveň proliferace a diferenciace kmenové krvetvorné buňky. Genetický defekt GATA-2 je spojen s imunodeficiencí a zvýšenou incidencí rozvoje MDS a AML. Tzv. MonoMAC syndrom je autozomálně dominantní onemocnění vznikající na podkladě mnohočetných mutací GATA-2 genu projevující se monocytopenií, defektní funkcí NK a B lymfocytů, recidivujícími plicními infekcemi a rozvojem alveolární proteinázy. U nemocného s tímto syndromem, jenž byl do ÚHKT předán ke sledování z Dětské hematologické a dětské onkologické kliniky FN v Motole, došlo během 6 měsíců k rozvoji těžké pancytopenie s transfuzní závislostí a recidivujícími plicními infekcemi. V kontrolní sternální punkci byla přítomna poměrně výrazná dysplazie ve všech řadách společně s nálezem monozomie 7. chromozomu. U nemocného byla provedena alogenní SCT od nepříbuzného dárce a v současné době přežívá 25 měsíců bez známek choroby.
Kongenitální dyserytropoetické anémie (CDA)
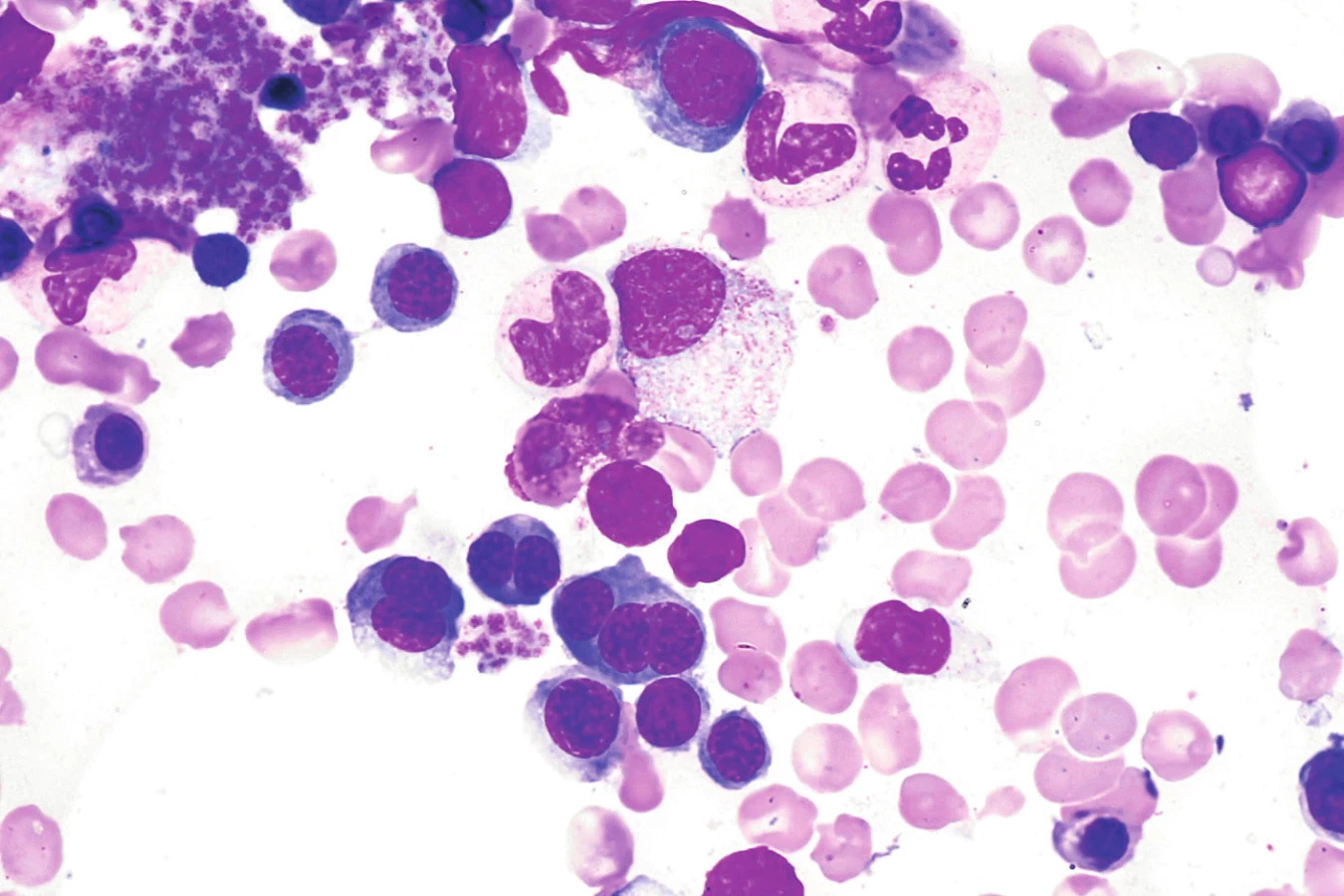
Kongenitální dyserytropoetické anémie vznikají v důsledku mutace některých genů, které hrají různým způsobem důležitou roli v tvorbě erytrocytů, důsledkem této poruchy je tvorba abnormálních erytrocytů [5]. Rozeznáváme 4 typy CDA, v ÚHKT jsou sledováni 3 nemocní s CDA II. typu. U této choroby je přítomna mutace SEC23B genu, jenž je zodpovědný za nitrobuněčný transport proteinů. Klinicky se jedná o různě těžký typ hemolytické anémie se splenomegalií a ikterem, typickým morfologickými změnami hyperplastické erytroidní řady v kostní dřeni (dvoj- či vícejaderné polychromatofilní normoblasty s jadernými můstky a dvojitou plazmatickou membránou) – obrázek 1 a pozitivní reakcí v Hamově testu s dárcovskými erytrocyty (tzv. HEMPAS). Potvrzení diagnózy je možné jednak z elfo membránových proteinů (změny rychlosti migrace proteinu pásu 3 a některých dalších proteinů), jednak molekulárně genetickým vyšetřením SEC23B genu. U 2 našich nemocných se závažnějším průběhem choroby byla potvrzena mutace SEC23B genu, u třetí nemocné byla diagnóza postavena na pozitivitě HEMPAS. Dvě nemocné s hodnotami Hb mezi 72–90 g/l jsou závislé na transfuzích erytrocytů (2–3 TU za 3 měsíce) a dostávají trvale chelatační léčbu, u 1 nemocného bylo dosaženo po splenektomii transfuzní nezávislosti, nicméně je nadále chelatován vzhledem ke zvýšeným zásobám Fe v důsledku trvající přítomnosti inefektivní erytropoézy.
PORUCHY MEMBRÁNY ČERVENÉ KRVINKY
Defekty membrány erytrocytu vedou k její zvýšené propustnosti pro některé ionty a pro vodu. Nejčastěji se vyskytující jednotkou je dědičná sférocytóza (HS), defekt různých genů, které se podílejí na tvorbě skeletu erytrocytární membrány (SPT1,ANK1 a dalších) vedou k deficitu ankyrinu, spektrinu či dalších proteinů membrány červené krvinky [6]. Důsledkem je zvýšená propustnost membrány pro ionty Na, jež klade zvýšené nároky na sodíkovou pumpu, vede k postupnému energetickému vyčerpání krvinky, změně jejího tvaru a ke snížené deformovatelnosti a k zániku ve slezině – extravaskulární hemolýze. Většina nemocných s HS je diagnostikována již v dětském věku a jsou sledováni na regionálních pediatrických a následně na hematologických pracovištích, kde je u těžších forem indikována splenektomie. V ÚHKT jsou většinou diagnostikováni dospělí nemocní, kteří nebyli blíže vyšetřeni v dětství, nemocní s nejasnou hemolýzou či nemocní s mírnou nebo jen laboratorní hemolýzou v důsledku malé expresivity genu pro HS, i když jde většinou o autozomálně dominantní onemocnění. Během posledních 3 let bylo na základě vyšetření osmotické rezistence, autohemolýzy a průtokové cytometrie (EMA – eozin maldeimidový test) diagnostikováno 5 nových dospělých nemocných s HS, u 3 z nich byl prokázán při elfo membránových proteinů pomocí SDS-PAGE deficit spektrinu a ankyrinu, či proteinu pásu 4.2 nebo pásu 3.
Do skupiny nesférocytárních hemolytických anémií jsou řazeni nemocní, u nichž je přítomna korpuskulární hemolýza, ale laboratorní nálezy nesvědčí ani pro HS, ani pro některou z erytrocytárních enzymopatií. V ÚHKT jsou sledovaní 3 nemocní s mírným či středně těžkým stupněm korpuskulární hemolytické anémie (Hb 90–115 g/l), u nichž se zatím dostupnými metodami nepodařilo jednoznačně určit příčinu. U dalších 3 nemocných svědčí laboratorní nálezy pro možnou dědičnou xerocytózu (dehydratovanou formu dědičné stomatocytózy). Onemocnění je způsobeno některou z mutací PIEZO-1 genu, vzácněji některých dalších genů, což vede k abnormální propustnosti membrány erytrocytu s neschopnosti zadržet K+ a následně H2O v buňce. Hyperhydratovaná forma stomatocytózy je vzácnější, vzniká v důsledku mutace Rh asociovaného glykoproteinu (RhAG), jež vede k retenci kationtů a vody uvnitř erytrocytu. Nemocní s dědičnou xerocytozou mají různý stupeň anémie (Hb 90–105 g/l u našich nemocných) se zvýšeným počtem retikulocytů (8 až 10,5 %) a zejména významně zvýšeným MCHC (375 až 391 g/l) a nálezem stomatocytů v periferní krvi. V laboratorních nálezech je krom známek hemolýzy přítomna zvýšená hladina feritinu v séru (750–950 μg/l), vyžadující intermitentní podávání chelátorů, i když nemocní nejsou závislí na transfuzích. V důsledku poruchy vnitřního prostředí erytrocytů mají nemocní s dědičnou xerocytozou známky aktivace koagulačního systému se zvýšenou incidencí trombotických komplikací, jež se výrazně zvyšuje splenektomií, která je u nich na rozdíl od dědičné sférocytózy kontraindikována. Z tohoto důvodu je nutné pečlivé odlišení nemocných s dědičnou xerocytózou a nemocných s dědičnou sférocytózou. V současnosti ověřujeme diagnózu u našich nemocných molekulárně genetickým vyšetřením PIEZO-1 genu.
PORUCHY ENZYMATICKÉHO VYBAVENÍ ERYTROCYTU
Deficit pyruvát kinázy (PK)
Pyruvát kináza je enzymem Krebsova cyklu – anaerobní glykolýzy. Tento cyklus slouží k tvorbě makroergních fosfátů a je tedy základním energetickým zdrojem erytrocytu [7]. Deficit PK je proto spojen s výrazným energetickým deficitem buňky, jenž se na rozdíl od HS neupravuje in vitro po přidání glukózy. Za tvorbu PK v erytrocytech je zodpovědný PKLR gen kódující tvorbu 2 izoenzymů L a R. Jako u všech vrozených poruch erytropoézy je mezi nemocnými velmi variabilní expresivita přítomné mutace genu, nejvýraznější klinické projevy jsou přítomny u homozygotů s mutací pro jeden typ izoenzymů, mírnější klinické příznaky mívají dvojití heterozygoti s mutací obou typů izoenzymů. Klinicky se choroba projevuje různě těžkým stupněm hemolytické anémie, jež může spojena se splenomegalií a komplikována cholelitiázou, a rozvojem přetížení železem při vysokém obratu erytropoézy a v důsledku opakovaných transfuzí erytrocytů. V ÚHKT je t. č. sledováno 11 nemocných s deficitem PK. U 2 nemocných, které jsou homozygoty pro mutaci PKLR genu, je přítomna závažná anémie s hodnotami Hb v rozmezí 65–73 g/l, obě nemocné jsou dependentní na podávání cca 2 TU erymasy za 3 měsíce a dostávají chelatační léčbu, jedna nemocná byla již v dětství splenektomována s mírným efektem na snížení transfuzní závislosti. Čtyři nemocní jsou dvojitými heterozygoty pro mutaci PKLR genu, mají mírný stupeň anemie (Hb = 95–115 g/l) nevyžadující podávání transfuzí a chelatační léčbu. Dalších 5 nemocných tvoří členové 2 rodin s mírnou anémií (Hb 110–120 g/l), která se zhoršuje při zátěži (zejména při infekci), u nemocných t. č. probíhá bližší vyšetřování povahy mutace PKLR genu.
Deficit glukózo-6-fosfát dehydrogenázy (G-6-P-D)
G-6-P-D je enzymem pentózového cyklu, jenž slouží jako zdroj energie pro systém redoxních reakcí, zabraňujících oxidaci hemoglobinu a umožňujících jeho udržení v redukovaném stavu [8]. Deficit G-6-P-D proto vede k variabilnímu stupni hemolýzy po požití oxidačních činidel kolísající opět díky variabilní expresi mutovaného genu od mírné chronické hemolýzy až po těžkou záchvatovitou hemolýzu (favismus). Vzhledem k tomu, že gen pro G-6-P-D je lokalizován na X chromozomu, postihuje onemocnění zejména muže, může však být přítomno i u žen díky nepříznivé lyonizaci favorizující mutovaný X chromozom. V ÚHKT je sledováno 6 nemocných s deficitem G-6-P-D, z toho je 5 mužů a 1 žena, nemocní mají mírný stupeň chronické hemolytické anémie s hodnotami Hb v rozmezí 99–127 g/l, u 3 nemocných bylo pozorováno opakované zvýraznění hemolýzy během infektu, u 1 nemocného se rozvinul těžší stupeň hemolýzy po požití furantoinu, ordinovaného praktickým lékařem. Dvěma nemocným je podávána chelatační léčba.
U jedné nemocné s mírným stupněm hemolýzy byl diagnostikován deficit hexokinázy, molekulárně geneticky nebyl typ defektu zatím blíže charakterizován, lze předpokládat, že by se mohlo jednat o dvojitého heterozygota pro dva různé izoenzymy s nízkou expresivitou mutace.
PORUCHY TVORBY HEMOGLOBINU
Poruchy tvorby protoporfyrinu
Kongenitální sideroblastická anémie (X-linked sideroblastic anemia-XLSA) vzniká nejčastěji v důsledku mutace genu pro delta-aminolevulát syntetázu (ALAS2), jež je klíčovým enzymem tvorby protoporfyrinu [9]. Onemocnění je vázáno na X chromozom, a proto postihuje zejména muže, může se však vyskytnout díky lyonizaci X chromozomu i u žen. Pro nemocné je typický nález středně těžkého či těžkého stupně výrazně hypochromní a mikrocytární anémie s normálním počtem retikulocytů, což může svádět k záměně za talasémii. Pro XLSA je ale typický nález dimorfní populace normálních a patologických erytrocytů s vysokou hodnotou RDW. Porucha tvorby protoporfyrinu vede k výrazné poruše metabolismu železa, charakterizované nárůstem jeho zásob se zvýšenou hladinou feritinu, vysokou saturací transferinu a přítomností prsténčitých sideroblastů v kostní dřeni. XLSA je třeba odlišit od talasemie vyšetřením elfo Hb a od MDS s prsténčitými sideroblasty (cytogenetika, cytochemie, vyšetření mutace SF3B1 genu), diagnózu potvrzuje molekulárně genetické vyšetření ALAS2 genu. Léčba XLSA spočívá v podávání transfuzí erytrocytů a chelatační léčby, u části nemocných byl popsán efekt vysokých dávek pyridoxinu. V ÚHKT je sledováno 6 mužů s XLSA s různým stupněm anémie (Hb kolísá mezi 69–115 g/l, MCH 16 až 22 pg, MCV 59–71 fl, RTC 0,4–1,5 %, RDW 30–35 %). Dva nemocní dostali v průběhu onemocnění transfuze erytrocytů, žádný nemocný však není trvale závislý na transfuzích, 4 nemocní dostávají chelatační léčbu v udržovacích dávkách.
Poruchy tvorby globinu
Poruchy tvorby globinu zahrnují mutace alfa či globinového genu, jež jsou spojeny s tvorbou abnormálního hemoglobinu v důsledku deficitu alfa či beta globinu (talasemie) a hemoglobinopatie vznikající záměnou jedné báze v některém globinovém řetězci.
Beta talasemie
Beta talasemie je autozomálně recesivní onemocnění vznikající v důsledku různého stupně snížení tvorby beta globinového řetězce [10]. Nedostatečná hemoglobinizace erytrocytu vede k hypochromii a makrocytóze, počet retikulocytů není zvýšen. U těžkých forem onemocnění je přítomna výrazná hyperplazie krvetvorby v kostní dřeni, jež může vést k deformitám skeletu, současně je přítomna extramedulární krvetvorba v játrech a ve slezině. Anémie závislá na podávání transfuzí spolu se zvýšeným obratem železa nutného pro hyperplastickou erytropoézu se podílejí na rozvoji přetížení železem již v dětském věku a je nutno tyto nemocné již v tomto věku intenzivně chelatovat k zabránění ireverzibilního orgánového přetížení železem. Kromě substituce transfuzemi a chelatační léčby je u nemocných s těžkou formou choroby poměrně častým výkonem alogenní SCT, kterou je však nutno provést ještě v dětském věku před rozvojem orgánového přetížení železem [11]. Díky charakteru mutace, která může zcela potlačit tvorbu beta globinu (β0talasemie), či jí do určité míry zachovat (β+talasemie) a velmi variabilní expresivitě mutovaných genů vzniká široké spektrum klinických projevů od němých forem s nálezem hypochromiích mikrocytů a drepanocytů v krevním obraze až po těžkou anémii s hepatosplenomegalií, ikterem, deformitami skeletu a závislostí na podávání transfuzí erytrocytů s rozvojem přetížení železem těžkého stupně již v dětství. Beta talasemie byla dlouho považována za chorobu, která se v našich zemích nevyskytuje a nemocní byli mylně diagnostikováni jako sideropenická anémie „refrakterní na podávání železa“. Teprve koncem 80. let minulého století se díky moderním diagnostickým metodám (ELFO Hb pomocí kapilární elektroforézy, molekulárně genetická analýza globinových genů) podařilo diagnostikovat i v naší populaci poměrně značný počet nemocných s heterozygotní formou beta talasemie (do současnosti více než 400 nemocných), kteří mají klinický obraz thalassemia minima s pouhými laboratorními změnami v krevním obraze nebo formu thalassemia minor s mírnou anémií nevyžadující podávání transfuzí [12], jedná se o heterozygoty pro β+ talasemii, nejčastěji v důsledku bodové mutace postihující intron 1 a vedoucí k poruše sestřihu RNA (IVS-I 1 G>A, IVS-I 110 G>A) či exon 3 a vedoucí k poruše štěpení RNA a tzv. polyA signálu T>G (AATAAA → GATAAA). V ÚHKT jsou sledování 2 rodiny (celkem 4 nemocní) s obrazem thalassemia minor až intermedia se závažnější anémií (Hb 75–105 g/l) a mírným zvýšením hladiny feritinu v séru, kteří však jen výjimečně dostali transfuzi a nejsou léčeni chelátory železa. U nemocných se jedná o heterozygotní formu β0talasemie v důsledku mutace exonu 1 CD121 GAA → TAA, vedoucí ke vzniku nesmyslného kodonu. Dále jsou v ÚHKT sledováni 2 nemocní s kombinací heterozygotních forem beta talasemie s Hb E (CD 26 GAG>AAG [Glu>Lys]) s klinickým obrazem thalassemia intermedia až major. Nemocní mají těžký stupeň anémie (Hb 50–86 g/l), jsou trvale závislí na podávání transfuzí erytrocytů a podávání chelatačních látek v poměrně vysokých dávkách, u obou nemocných jsou přítomny malformace skeletu a retardace růstu. U obou byla již v dětství provedena splenektomie, ale s poměrně malým efektem na redukci počtu transfuzí. Bohužel u těchto nemocných již dnes nelze bez neúnosného rizika provést alogenní SCT od nepříbuzného dárce a v době, kdy by u nich byla v dětství indikována, nebyla SCT ještě standardním terapeutickým postupem stejně tak, jako dnes zatím není genová terapie. Za poslední 3 roky byla na základě výsledku kapilární elfo Hb vyslovena suspekce na beta talasemii u 214 nemocných, z toho bylo 70 nemocných českého původu, diagnóza byla ověřena molekulárně geneticky u 49 nemocných (z toho 15 nemocných českého původu, přičemž u 4 nemocných již byla beta talasémie diagnostikována u rodičů). U 1 nemocné byla zjištěna mutace v exonu 3 beta globinového genu společně s triplikací genu pro alfa globin α (anti-3.7), která zhoršuje klinické projevy choroby díky poten-ciaci nadbytku alfa globinových řetězců, nemocná má projevy thalassemia minor s mírnou mikrocytární anémií s hodnotami Hb mezi 10–110 g/l.
U 13 nemocných byla prokázána přítomnost Hb E (CD 26 GAG> AAG [Glu>Lys]), jde o mutaci beta globinového genu, vedoucí k alternativnímu sestřihu mRNA. U 6 nemocných českého původu se jedná o heterozygotní formu s hypochromií a makrocytózou bez výraznější anémie, klinicky přitom jde většinou o obraz obdobný thalassemii minor či minima. U zbylých nemocných byla ve 3 případech prokázána homozygotní forma choroby (HbE disease), u ostatních heterozygotní forma HbE trait), jde o cizince (Thajsko, Srí Lanka, Vietnam), kteří nejsou v ÚHKT sledováni. U 1 nemocného z Indie byla prokázána kombinace HbE s Hb D Punjab (CD 121 GAA>CAA [Glu>Gln];), nemocný bohužel nebyl v ÚHKT klinicky vyšetřen.
U 3 českých nemocných byl prokázán Hb Lepore v heterozygotní formě. Jedná se o hemoglobin se 2 alfa řetězci a 2 fuzními delta/beta řetězci vzniklými v důsledku zkřížení genů pro delta a beta globin. Nemocní mají mírnou hypochromní mikrocytární anémii (Hb= 103–115 g/l) s klinickým obrazem thalassaemia minor. Obdobný klinický nález s hodnotami Hb mezi 105–119 g/l je přítomen i u 3 českých nemocných s heterozygotní formou Hb Knossos, (hetero CD 27 GCC>TCC [Ala>Ser]) variantní formou beta talasémie.
Alfa talasemie
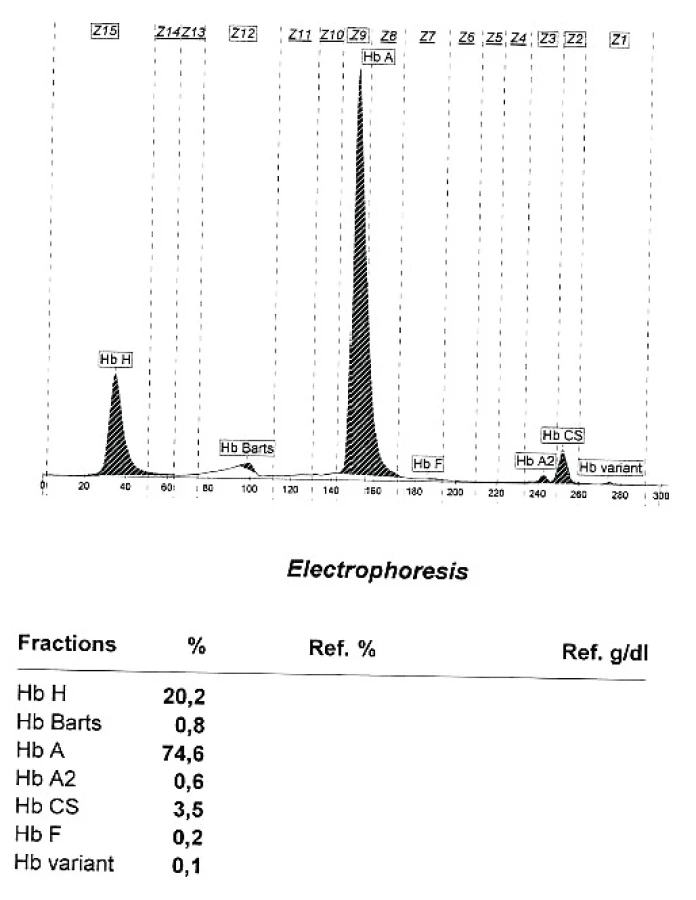
Alfa talasemie je rovněž autozomálně dědičné onemocnění charakterizované poruchou tvorby alfa globinu v důsledku mutace postihující jeden či více alfa globinových genů [13]. Anémie má obdobný charakter jako u beta talasemie (hypochromie s mikrocytózou a nezvýšeným počtem retikulocytů), na rozvoji anémie se podílí i výraznější hemolytická složka daná nestabilitou vznikajících tetramerů β řetězců (HbH). Klinické projevy závisí na počtu postižených genů a v zásadě se neliší od projevů beta talasemie, stejně tak jako charakter komplikací a léčebný přístup. Mutace 1 alfa globinového genu vede většinou pouze k laboratornímu nálezu mírné hypochromie a mikrocytózy bez klinických příznaků. Elfo Hb nebývá přínosná, může být lehce snížena hodnota HbA2 a u novorozenců přítomny stopy Hb Barts (tetrametr γ řetězců), nutné je molekulárně genetické vyšetření. V posledních 3 letech byla v ÚHKT diagnostikováno celkem 19 nemocných s mutací 1 alfa globinového genu, z tohoto počtu je 11 pacientů vietnamského původu, 4 nemocní jsou ze Středního východu či z oblasti Kavkazu a tzv. tiché nosičství alfa talasemie při mutaci 1 alfa globinového genu bylo nalezeno i u 4 nemocných ze 3 českých rodin. U 1 vietnamského nemocného byla diagnostikována přítomnost Hb Constant Spring (hetero CD142 TAA>CAA [Stop>Gln]), s abnormálně elongovaným alfa globinovým řetězcem díky mutaci terminálního stop kodonu. Mutace 2 alfa globinových genů vede k obrazu thalassemia minor s lehce sníženými hodnotami Hb, hypochromií a makrocytózou (tzv. alpha thalassemia trait). V ÚHKT bylo diagnostikováno na podkladě elfo Hb a molekulárně genetického vyšetření 10 nemocných s tímto postižením (6 vietnamských nemocných, 2 pacienti ze Středního východu a 2 české nemocné). U všech nemocných byl při elfo Hb nalezena přítomnost HbH (3,7–9,9 %) a malé množství Hb Barts (0,2–1,1 %). U 9 nemocných byla přítomna heterozygotní mutace 2 různých alfa globinových genů, u 1 nemocné se jednalo u homozygotní mutaci genu od jednoho rodiče. Mutaci 3 alfa globinových genů vedoucí k obrazu HbH disease jsme nepozorovali, nicméně u dcery vietnamských rodičů s kombinací mutace 2 alfa globinových genů od matky s Hb Constant Spring od otce jsou přítomny určité projevy HbH/Constant Spring disease (obr. 2). Nemocná má středně těžký stupeň anémie s hodnotami Hb mezi 82–90 g/l, mírnou splenomegalií a hraničními hodnotami feritinu v séru. U jednoho nemocného z Thajska byla zjištěna kombinace heterozygotních mutací pro 1 alfa globinový gen a pro Hb Constant Spring s přítomností HbE v heterozygotní formě, nemocný však nebyl v ÚHKT klinicky vyšetřen.
HEMOGLOBINOPATIE
Srpkovitá anémie (SCD)
Srpkovitá anémie je nejznámější hemoglobinopatií, mutace na 6. pozici exonu 1 beta globinového genu (CD 6 GAG> GTG [Glu>Val]) vede u homozygotů v důsledku náhrady polární aminokyseliny nepolárním valinem ke snížené rozpustnosti Hb v redukovaném stavu, snížené deformovatelnosti krvinky a zvýšené autooxidaci Hb, hemolýza může být jak extravaskulární ve slezině, tak intravaskulární (při hemolytických krizích) [14]. Hemolytické krize mohou být vyprovokovány infekci, dehydratací, prochladnutím. Takzvaná vazo-okluzivní krize vzniká obstrukcí kapilár rigidními srpky (obr. 3), obdobným mechanismem vzniká obraz akutní respirační insuficience (acute chest syndrom), komplikací může být aplastická krize, často vyvolaná přítomností parvovirové infekce. Diagnostiku srpkovité anémie lze provést již z kapilární ELFO Hb, u homozygotů nejdeme více než 50 % HbS, heterozygoti (tzv. sickle cell trait) mají méně než 50 % HbS. V ÚHKT je sledováno 11 nemocných s nálezem HbS, z toho 8 z nich pochází přímo či má rodiče ze subsaharské nebo rovníkové Afriky. U 2 nemocných se jedná u homozygotní formu SCD s hodnotami HbS mezi 71–85 %. Celkem 4krát bylo třeba tyto nemocné hospitalizovat pro počínající či probíhající hemolytickou krizi při infektu, dehydrataci či prochlazení s nutností provedení opakované erytrocytaferézy, náhrady tekutin a podávání antibiotik a analgetik. Další nemocný má rovněž homozygotní formu SCD, nicméně příznaky onemocnění jsou velmi mírné díky současné přítomnosti vysokých hodnot HbF v důsledku přítomnosti tzv. arabsko-indického haplotypu beta globinu s omezeným přesmykem gama/beta globinových genů, nemocný je z Kuvajtu. Sedm nemocných jsou heterozygoti s hodnotami HbS 36–44 %, ve 3 případech jde o děti ze smíšených manželství českých matek s černošskými otci. U 2 nemocných byla prokázána kombinovaná hemoglobinopatie HbS/C. HbC, jenž vzniká odlišnou mutací na 6. pozici exonu 1 beta globinového genu (CD 6 GAG> AAG [Glu>Lys]), vede rovněž ke zhoršení plasticity erytrocytu s tvorbou mikrosférocytů s krystalky. Přítomnost HbC zhoršuje díky svému nepříznivému účinku na některé membránové transportéry iontů projevy přítomnosti HbS v heterozygotní formě a nemocní mohu mít hemolytické krize a obdobné komplikace jako nemocní se SCD včetně proliferativní retinopatie. U našich nemocných (oba jsou z rovníkové Afriky) je přítomna jen velmi mírná anémie bez výraznějších epizod hemolýzy, nemocné je však třeba pravidelně sledovat. Nemocní s homozygotní formou HbC většinou mají známky jen velmi mírné hemolytické anémie, v ÚHKT je sledován 1 homozygot s HbC.

Nestabilní hemoglobiny a další hemoglobinopatie
V ÚHKT jsou sledováni 4 nemocní z 2 rodin s Hb Köln (CD 98 GTG>ATG [Val>Met]). Jedná se o nestabilní Hb se zvýšenou afinitou k O2. Nemocní mají známky hemolýzy a klinické projevy anémie již při mírně snížených hodnotách Hb (Hb 115–125 g/l u našich nemocných) díky poruše uvolňování O2. Anémie bývá hypochromní, ale nikoli mikrocytární, je přítomna výrazná retikulocytóza a v nátěrech lze pozorovat Heinzova tělíska. U všech našich nemocných byla provedena splenektomie, která zvýšila hodnoty Hb, a odstranila tak projevy hypoxie. Recentně byla u českého novorozence diagnostikována heterozygotní forma Hb Seattle (CD 70 GCC>GAC [Ala>Asp];), jde o nestabilní Hb vedoucí většinou ke středně těžké chronické hemolytické anémii, která je však dobře tolerována vzhledem ke snížené afinitě Hb Seattle k O2. U jedné nemocné vietnamského původu byl v heterozygotní formě prokázán Hb New York (CD 113 GTG>GAG [Val>Glu];), tato varianta HbK vede díky své nestabilitě k mírné chronické hemolytické anémii.
U 3 českých nemocných z 2 rodin byla diagnostikována přítomnost Hb Doha (CD 1 GTG>GAG [Val>Glu]) v heterozygotní formě, onemocnění nemá klinické projevy a diagnóza byla učiněna na základě nálezu přítomnosti abnormálního Hb interferujícího s vyšetřením glykosylovaného Hb. U 1 nemocného z Bulharska byl diagnostikován HbO Arab (CD 121 GAA>AAA [Glu>Lys];) v heterozygotní formě. U 1 nemocného z dětské kliniky se známkami cyanózy byl prokázán v heterozygotní formě HbM Milwaukee-2 (CD 92 CAC>TAC [His>Tyr]) s přítomnosti cca 5 % HbM. U dospělých nemocných je přítomno v krvi cca 15–30 % HbM, který má však sníženou afinitu k O2, takže onemocnění většinou i přes přítomnost methemoglobinemie nemá závažnější klinické projevy. Onemocnění je dominantně dědičné.
Paroxysmální noční hemoglobinurie (PNH)
Paroxysmální noční hemoglobinurie je získaným klonálním onemocněním krvetvorby. Deficit inhibičních systémů komplementu (CD59 a CD55 antigeny) na povrchu krvinek vede ke zvýšené citlivosti na lýzu buněk aktivovaným komplementem. Intravaskulární hemolýza s hemoglobinurií, selhání kostní dřeně a zvýšené riziko trombotických komplikací při aktivaci koagulačního systému jsou základními projevy choroby [15]. Podle velikosti PNH klonu a převažujících klinických projevů se PNH dělí do několika podskupin, v současnosti se pro klinickou praxi a léčebné přístupy nejvíce užívá rozdělení na hemolytickou a hypoplastickou formu, přičemž pro rozhodování o léčbě má zásadní význam i přítomnost či vysoké riziko vzniku trombotických komplikací [16]. V ÚHKT je v současnosti sledováno 35 nemocných s PNH, 15 nemocných má projevy závažné hemolýzy s transfuzní dependencí a u 12 z nich vznikly během průběhu nemoci závažné trombotické či infekční komplikace nebo je přítomen závažný stupeň chronického renálního selhání. Všichni tito nemocní s komplikacemi jsou léčeni eculizumabem, inhibitorem C5 složky aktivovaného komplementu, u žádného z nich nebyla během léčby pozorována recidiva trombózy a u 6 z nich bylo dosaženo trvalé nezávislosti na podávání transfuzí. U 8 nemocných je přítomna mírná či středně těžká hemolýza bez komplikací, nemocní dostávají profylaktickou antikoagulační léčbu, 6 z nich dostává či bylo v minulosti léčeno imunosupresí (prednison + cyklosporin A). U 5 nemocných je přítomna jen minimální hemolýza, většina z nich je pouze sledována, 4 nemocní mají pouze minimální klon s < 2 % CD59 kompletně deficitních erytrocytů. U 3 nemocných je přítomna hypoplastická forma PNH, 1 nemocný byl léčen imunosupresí, další byl úspěšně transplantován a třetí nemocný je připravován na alogenní SCT.
Závěrem je nutno říci, že nemocných se vzácnými chorobami červené krvetvorby neustále přibývá. Příčin je několik, jednak se zpřesnila diagnostika těchto onemocnění zavedením molekulárně genetických metod, dále přibývá nemocných předaných mezi dospělé díky komplexní péči dětských hematologů, jež předchází rozvoji závažných komplikací již v dětském věku, dále se výrazně změnila péče o druhou a další generaci vietnamské populace u nás, která již absolvuje běžné preventivní zdravotní prohlídky a zásadně se zvýšil počet migrující populace z oblastí s vysokým výskytem vrozených poruch červené řady. Správná diagnostika a dispenzarizace nemocných s vrozenými poruchami erytropoézy je důležitá pro prevenci vzniku závažných forem těchto onemocnění při sňatku dvou heterozygotů. Kromě toho se s prodlužující délkou života nemocných díky moderní péči zvyšuje i riziko možných pozdních komplikací, jakými jsou rozvoj MDS a AML u nemocných s vrozenými poruchami kmenové krvetvorné buňky či rozvoj komplikací spojených s orgánovým přetížením železem u vrozených hemolytických chorob.
Podíl autorů na přípravě rukopisu
JČ – příprava rukopisu, léčba pacientů
JV – zpracování dat do registru nemocných
MV – molekulárně genetická vyšetření
SŠ – laboratorní hematologické a molekulárně genetické vyšetření
MB – zpracování dat do databáze molekulárně genetických vyšetření
Prohlášení autorů
Autoři práce prohlašují, že s v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Poděkování
Autoři děkují spolupracujícím kolegům a pracovištím: prof. MUDr. Dagmar Pospíšilové, Ph.D., z Dětské kliniky FN Olomouc, doc. RNDr. Vladimíru Divokému, CSc., z Ústavu buněčné biologie a genetiky LF Univerzity Palackého v Olomouci a prof. MUDr. Janu Starému, DrSc., z kliniky Dětské hematologie a dětské onkologie FN v Praze Motole.
doc. MUDr. Jaroslav Čermák, CSc.
Ústav hematologie a krevní transfuze
U Nemocnice 1
128 00 Praha 2
e-mail: cermak@uhkt.cz
Zdroje
1. Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol 2008;142:859–876.
2. Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev 2010;24:101–122.
3. Vlachos A, Rosenberg PS, Atsidaftos E, et al. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood 2012;119:3815–3819.
4. D’Andrea AD, Grompe M. Molecular biology of Fanconi anemia: implications for diagnosis and therapy. Blood 1997;90:1725–1736.
5. Iolascon A, Esposito MR, Russo R. Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: from morphology to molecular approach. Haematologica 2012;97:1786–1794.
6. Bolton-Maggs PHB, Langer JC, Iolascon A et al. Guidelines for the diagnosis and management of hereditary spherocytosis – 2011 update. Br J Haematol 2004;156:37–49.
7. Grace RF, Zanella A, Neufeld EJ, et al. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am J Hematol 2015;90:825–830.
8. Beutler E. Glucose-6-phosphate dehydrogenase deficiency: a historical perspective. Blood 2008;111:16–24.
9. Camaschella C. Hereditary sideroblastic anemias: pathophysiology, diagnosis, and treatment. Semin Hematol 2009;46:371–377.
10. Rund D, Rachmilewitz E. β thalassemia. N Engl J Med 2005;353:1135–1146.
11. Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood 2011;118:3479–3478.
12. Divoka M, Partschova M, Kucerova J, et al. Molecular chracteristics of β-thalassemia in the Czech and Slovak populations: Mediterranean, Asian and unique mutations. Hemoglobin 2016;40:156–162.
13. Harteveld CR, Higgs CR. α-thalassemia. Orphanet J Rare Dis 2010;5:13–34.
14. Stuart MJ, Nagel RL. Sickle cell disease. Lancet 2004;364:1343–1360.
15. Parker CJ. Paroxysmal nocturnal hemoglobinuria. Curr Opin Hematol 2012;19:141–148.
16. Brodsky RA. How I treat paroxysmal nocturnal hemoglobinuria. Blood 2009;113:6522–6527.
Štítky
Hematológia Interné lekárstvo OnkológiaČlánok vyšiel v časopise
Transfuze a hematologie dnes

2017 Číslo Supplementum1
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
- Nejasný stín na plicích – kazuistika
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
Najčítanejšie v tomto čísle
- Dysfibrinogenemie a afibrinogenemie v České republice
- Centrum pro vzácné choroby červené krevní řady v Ústavu hematologie a krevní transfuze
- Transfuze a imunohematologie v ÚHKT
- Funkční následky mutací v genu pro nukleofosmin u akutní myeloidní leukemie