
Základní bioinformatické pojmy a postupy využívané pro analýzu DNA pomocí sekvenování nové generace
Next generation sequencing: basic bioinformatic terms and analytic protocols for DNA analysis
Next generation sequencing (NGS) has become very popular both in research and clinical practice, in particular because it allows detailed and rapid insight into the patient's genome. Within the context of cancer research, NGS methods allow precise detection of germline and especially somatic mutations, which can help to diagnose a disease quickly and precisely and thus enable treatment administration based on individual patient needs. The development of novel computing methods and their application for accurate processing of NGS data is the objective of the scientific field of bioinformatics. Bioinformatic analysis is a complex process and its precise set-up is absolutely crucial for obtaining relevant results. Thus, it is necessary for bioinformaticians to understand the biological principles of the given analysis, such as the development of somatic mutations during disease course. From the perspective of a bio-analyst or physician, it is essential to understand the challenges and limits of NGS technology; basic knowledge of bioinformatics and its terminology allows for effective communication with bioinformaticians. In this review, the authors attempt to describe bioinformatic analysis with emphasis on explaining the basic concepts used in the NGS data analysis.
Key words:
next generation sequencing – bioinformatics – pipeline
Autoři:
N. Tom 1,2; F. Pardy 3; J. Kotašková 1; K. Plevová 1,2; Š. Pospíšilová 1,2
Působiště autorů:
Centrum molekulární medicíny, CEITEC (Central European Institute of Technology), Masarykova univerzita, Brno
1; Centrum molekulární biologie a genové terapie, Interní hematologická a onkologická klinika, Fakultní nemocnice Brno a Lékařská fakulta Masarykovy univerzity, Brno
2; Centrální laboratoř Genomika, CEITEC (Central European Institute of Technology), Masarykova univerzita, Brno
3
Vyšlo v časopise:
Transfuze Hematol. dnes,24, 2018, No. 3, p. 174-180.
Kategorie:
Souhrnné a edukační práce
Souhrn
Metoda sekvenování nové generace (NGS) se stala velmi populární v biomedicínském výzkumu i v klinické praxi zejména proto, že umožňuje rychlý a detailní vhled do genomu pacienta. V kontextu nádorových onemocnění umožňují metody NGS přesnou detekci jak zárodečných záměn, tak zejména somatických mutací, které mohou pomoci rychle a precizně stanovit diagnózu a přizpůsobit léčbu podle individuálních potřeb pacienta. Vývojem nových výpočetních metod a jejich aplikací za účelem precizního zpracování NGS dat se zabývá vědní obor bioinformatika. Bioinformatická analýza je komplexní proces, jehož správné nastavení je klíčové pro získání relevantních výsledků. Je proto nutné, aby bioinformatik detailně porozuměl biologické podstatě sledovaného jevu, jako je například vznik genových mutací v průběhu onemocnění. Z hlediska bioanalytika i lékaře je naopak užitečné znát jak možnosti a limity NGS technologie, tak i základní bioinformatickou terminologii, na základě které jsou pak schopni s bioinformatiky efektivně komunikovat. V této souhrnné práci se proto autoři snaží obecně popsat bioinformatickou analýzu sekvenačních dat s důrazem na vysvětlení základních pojmů používaných v oblasti analýzy NGS dat.
Klíčová slova:
sekvenování nové generace – bioinformatika – pipeline
SEKVENOVÁNÍ NOVÉ GENERACE – PRINCIP A VYUŽITÍ
Sekvenování DNA je proces určování pořadí nukleotidů v molekule nukleové kyseliny. První pokusy přečíst pořadí nukleotidů spadají do 70. let minulého století [1]. Sekvenování nové generace (NGS) je moderní způsob sekvenování, označovaný také jako “masivně paralelní sekvenování”, který umožňuje souběžné čtení milionů až miliard sekvenovaných úseků o délce desítek až tisíců nukleotidů. NGS se objevilo začátkem 21. století a v posledních letech se začalo využívat v mnoha oblastech biologie a medicíny. Svou popularitu získalo zejména díky své vysoké kapacitě, rychlosti zpracování a citlivosti analýzy. V těchto ohledech NGS zdaleka převyšuje klasickou metodu Sangerova sekvenování. Zatímco Sangerovým sekvenováním můžeme sekvenovat v jednom experimentu desítky až stovky sekvencí o délce maximálně 1 kb s citlivostí detekce alelické varianty okolo 15 % [2], pomocí technologie NGS lze analyzovat desítky milionů až miliardy sekvencí v délce až 600 bází, přičemž limit detekce lze při správném provedení snížit pod 1 % [3–5]. V důsledku značného rozšíření NGS došlo postupně i k výraznému snížení nákladů na sekvenování, které byly v počátku zavedení metody extrémně vysoké.
Na poli NGS se postupně vystřídalo několik technologických platforem, např. pyrosekvenování (Roche, 454), sekvenování pomocí ligace – SOLiD (Thermo Fisher Scientific, Inc.) nebo sekvenování založené na změně elektrického potenciálu – Ion semiconductor sequencing (Thermo Fisher Scientific, Inc.). Dnes je dominující technologií tzv. sekvenování syntézou (sequencing by synthesis – SBS) firmy Illumina. V současné době se v laboratořích setkáváme nejčastěji se sekvenátory MiSeq, NextSeq či HiSeq, které se liší svou kapacitou. Poptávka po technologiích, které mohou pracovat ještě rychleji a produkovat delší čtení, vyústila v rozvoj sekvenování tzv. “třetí” generace, které je schopno analyzovat velmi dlouhé fragmenty DNA (desítky až stovky kilobazí) v reálném čase bez potřeby PCR. Ke třetí generaci patří přístroje PacBio RSII a Sequel (Pacific Biosciences) či přenosný sekvenátor minION (Oxford Nanopore Technologies). Jejich využití v klinické praxi je však zatím omezené, zejména kvůli náročnosti na přípravu vzorku a nižší kvalitě výstupních dat.
NGS technologie přináší celou řadu možností využití. Mohou například sloužit k identifikaci zárodečných variant ve velkém počtu genů současně. Oproti klasickým metodám sekvenování, které sloužily k detekování jednonukleotidových záměn nebo krátkých inzercí a delecí, umožňují NGS technologie navíc určit také změny v počtu kopií celých genomových oblastí nebo identifikovat strukturní varianty, čímž může do jisté míry konkurovat cytogenetickým vyšetřením. Trendem poslední doby jsou tzv. genové panely. Jedná se o soubory desítek až stovek vyšetřovaných genů, jejichž výběr je definován cílem vyšetření. Tyto panely jsou vhodné pro diferenciální diagnostiku konkrétní skupiny onemocnění nebo lze využít obecnější panely např. pro analýzu genů spojených s hereditárními onemocněními. Existují i sady umožňující testování celého exomu, tzn. všech přepisovaných genů. Nejpokročilejší metodou z hlediska rozsahu sekvenovaných oblastí je potom sekvenování celých genomů, které je ale zatím v klinické praxi využíváno pouze okrajově.
Významnou oblastí využití NGS je nádorová genetika. I zde se uplatňují různé varianty genových panelů navržených pro konkrétní onemocnění (např. ClearSeq AML panel) nebo panely zahrnující stovky genů asociovaných s různými typy nádorů (např. Truseq Pan cancer panel). Vysoká citlivost, kterou NGS umožňuje, je s výhodou využívána kromě detekce zárodečných variant také k detekci somatických klonálních mutací nebo dokonce ke sledování zbytkové choroby u hematologických malignit. Tato vysoká citlivost detekce umožňuje zachytit DNA s nízkým zastoupením ve vzorku – toho se využívá u metod sledujících tzv. cirkulující DNA z rozpadajících se nádorových buněk (circulating tumor DNA; ctDNA), která je izolována z krevní plazmy; tento způsob vyšetření nádoru se označuje jako “liquid biopsy”. Mimo onkologii lze podobný postup uplatnit pro stanovení genetických aberací plodu z fetální frakce v krvi matky (cffDNA – cell free fetal DNA).
NGS analýzy produkují obrovské množství primárních dat, které je nutné vhodně upravit a správně vyhodnotit, což přispělo k dynamickému rozvoji nového vědního oboru – bioinformatiky, která se zabývá analýzou biologických dat pomocí výpočetních metod. Volba způsobu bioinformatického zpracování je kritickým krokem NGS každé analýzy. Jeho správné nastavení a provedení vyžaduje jak porozumění biologické podstatě experimentu, tak znalost relevantních softwarových nástrojů a analytických postupů.
Běžnou součástí komunikace, která se týká NGS experimentů a zpracování dat, jsou anglické pojmy označující jednotlivé kroky nebo součásti analýzy. České ekvivalenty se zatím příliš nevžily. V textu jsme se proto vyvarovali násilných překladů a dali jsme přednost původním anglickým výrazům.
NÁVRH A PŘÍPRAVA SEKVENAČNÍHO EXPERIMENTU
Považujeme za nutné pro úplnost zmínit laboratorní část NGS procesu, která je pro zpracování dat do značné míry určující. Možnosti přípravy vzorku pro NGS experiment jsou vzhledem k prudkému rozvoji této oblasti velmi široké. V první řadě je potřeba stanovit si oblasti zájmu, tzv. ROI (regions of interest), které chceme pomocí NGS analyzovat, a shromáždit příslušnou informaci o těchto oblastech do přehledného souboru (např.bed). Pokud se jedná o jednotlivé geny, popř. několik málo genů, volíme většinou přípravu vzorku založenou na PCR amplifikaci. U většího souboru genů jsou metodou volby jak PCR přístupy, tak postupy využívající tzv. enrichment probes [6], tedy hybridizační sondy, kterými je možné obohatit sekvenační knihovnu o oblasti zájmu a zároveň minimalizovat nežádoucí sekvence. Vzorek připravený k sekvenování se označuje jako sekvenační knihovna. Jedná se o směs náhodných fragmentů DNA o předem stanovené délce, jenž jsou ohraničeny známými, synteticky připravenými sekvencemi, označovanými jako tzv. adaptéry. Tyto adaptéry obsahují sekvence, které umožňují:
- a) amplifikaci knihovny pomocí PCR;
- b) identifikaci vzorků, a tedy jejich míchání do jedné sekvenační reakce, tzv. multiplexing;
- c) hybridizaci fragmentů v sekvenačním čipu.
Modifikací, která zvyšuje detekční limit pro záchyt varianty, je přidání tzv. unikátních molekulárních identifikátorů (UMI) do sekvence adaptéru. Jedná se o sekvenci 8–10 náhodných nukleotidů, která je unikátní pro každý vstupní fragment DNA [7]. Výběr vlastní sekvenační platformy závisí na typu experimentu, velikosti analyzovaného genomu, požadované hloubce pokrytí, přesnosti a ceně.
Bioinformatická analýza NGS dat
Bioinformatická analýza by měla být přizpůsobena na míru použité sekvenační technologii, sekvenačnímu kitu, experimentálnímu designu a v neposlední řadě biologické hypotéze či diagnostickým potřebám. Každé vyhodnocení NGS dat zahrnuje sekvenci kroků, výpočetní postup, který se označuje jako tzv. pipeline. Jako modelová je zde popsána pipeline sloužící k identifikaci variant na úrovni DNA (obr. 1). Běžně používané nástroje pro jednotlivé kroky jsou popsány v tabulce 1.

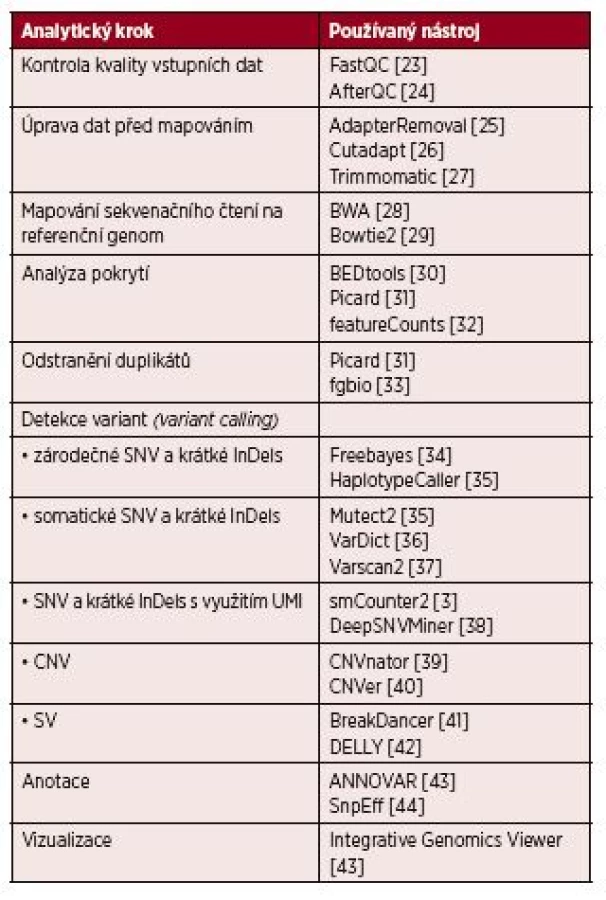
Obecná pipeline pro účely detekce mutací v DNA zahrnuje tyto základní kroky:
- Kontrola kvality vstupních dat
- Úpravy dat před mapováním
- Mapování individuálně čtených sekvencí (tzv. reads; dále sekvenační čtení) na referenční genom
- Úpravy dat po mapování
- Detekce variant
- Anotace detekovaných variant
- Vizualizace
V průběhu analýzy je využíváno několik různých datových formátů, jejichž základní přehled je uveden v tabulce 2.

Kontrola kvality (QC) vstupních dat
Po dokončení sekvenační reakce musí být zkontrolován její správný průběh a zhodnocena kvalita generovaných dat. Data jsou získána nejčastěji v podobě souborů formátu.fastq, který obsahuje sekvence analyzovaných fragmentů a informaci o jejich kvalitě [8].
Úprava dat před mapováním
Pro zvýšení kvality sekvenačních čtení se využívají některé analytické postupy [9]:
- spojování překrývajících se konců párových sekvenačních čtení tzv. merging
- párová sekvenační čtení představují fragment DNA čtený z obou jeho konců. V oblasti překryvu je tak informace o sekvenci získána ze dvou sekvenačních čtení, a proto je spolehlivější;
- odstranění nekvalitních konců sekvenačního čtení tzv. trimming
- přesnost sekvenační reakce se v jejím průběhu snižuje, a tak se snižuje i kvalita bází s rostoucí délkou sekvenačních čtení. Z tohoto důvodu je v určitých případech vhodné odstranit nekvalitní konce, které mohou být zdrojem nepřesných výsledků;
- filtrování na požadovanou délku sekvenačního čtení, tzv. length filtering
používá se zejména pro odstranění kontaminace jak adaptérovými, tak biologickými sekvencemi.
Mapování sekvenačních čtení na referenční genom
V dalším kroku jsou jednotlivá sekvenační čtení zarovnány k referenčnímu genomu. Tento krok je označován jako tzv. read alignment/read mapping [10]. Referenční genom představuje jednovláknovou konsenzuální sekvenci určitého organismu. V případě člověka obsahuje referenční genom 3 miliardy bazí. Tato část bioinformatické analýzy je stěžejní a její přesnost významně ovlivňuje celkovou kvalitu výsledků.
Účelem mapování sekvenačních čtení na referenční genom je:
- identifikovat, jakou část genomu sekvenační čtení představují;
- zjistit rozdíly mezi referenční sekvencí a sekvencí sekvenačních čtení;
- stanovit statistiku pokrytí a podíl sekvencí mimo ROI.
Výsledky mapování jsou typicky uloženy do souboru formátu .sam nebo .bam. Po mapování se často provádí další kontrola kvality, která je zaměřena na analýzu pokrytí, tzv. coverage. Cílem je ověřit, zda byly regiony zájmu (obvykle definované v souboru formátu .bed, .gff nebo .gtf) osekvenovány. Od hloubky pokrytí, tzn. počtu překrývajících se sekvenačních čtení na určité pozici, tzv. coverage depth, je odvozena citlivost detekce. Obecně platí, že čím větší hloubka pokrytí, tím větší citlivost [11]. Vysoká citlivost NGS je využívána zejména u experimentů analyzujících subklonální somatické mutace u nádorových onemocnění, případně při sledování zbytkové choroby u léčených hematoonkologických pacientů.
Úpravy po mapování sekvenačních čtení
Kvalita mapování může být vylepšena několika analytickými kroky zahrnujícími např. odstranění PCR duplikátů, které vznikají jako artefakt při amplifikaci [12]. Jejich přítomnost může zkreslit informaci o frekvenci variantních alel (VAF – „variant allele frequency“). Odstranění PCR duplikátů není vhodné provádět v případě knihoven založených na obohacení regionů zájmu pomocí PCR, jejíž produkty mohou být s PCR duplikáty zaměněny. Výjimkou jsou knihovny využívající technologii UMI, která slouží právě pro odstranění PCR duplikátů a korekci sekvenačních chyb [13].
Detekce variant
Detekce variant se označuje jako tzv. variant calling. Principem tohoto procesu je identifikace pozic, kde se nukleotidy v sekvenačních čtení liší od referenčního genomu a zejména pak ověření, zda není konkrétní nalezená nukleotidová záměna jen sekvenační či analytický artefakt, a zda se skutečně jedná o variantu v testované DNA.
Softwarový nástroj pro tyto účely se označuje jako, tzv. variant caller. Tyto programy jsou vyvíjeny specificky pro detekci konkrétních typů záměn, ať somatických, nebo zárodečných [14, 15]:
- jednonukleotidových záměn (SNV) a krátkých inzercí a delecí (InDels)
- změn počtu kopií (CNV)
- strukturních variant (SV).
Detekce zárodečných variant se soustředí zejména na genotypování, tj. určení, zda jsou alely shodné s referenční sekvencí či zda jsou variantní, a zda jsou v homozygotním, či heterozygotním stavu. V případě heterozygota by měly být obě varianty alely zastoupené přibližně stejným počtem sekvenačních čtení.
V případě somatických mutací je pozornost soustředěna na frekvenci variantní alely, která odráží zastoupení nádorového klonu ve vzorku. Pro analýzu somatických mutací se v ideálním případě používají 2 druhy materiálu: nádorová tkáň a zdravá tkáň pro odečtení zárodečných variant. U některých malignit může být obtížné definovat správný zdroj nenádorové DNA. Běžným zdrojem DNA pro určení zárodečných variant u solidních nádorů jsou jaderné krevní buňky. V případě hematoonkologických onemocnění je zdrojem nenádorové DNA stěr bukální sliznice. Nicméně v případě některých onemocnění, jako je například akutní myeloidní leukemie, může být bukální sliznice infiltrována nádorovými buňkami. Standardním výstupem detekce variant je soubor .vcf nebo .maf. Tato část analytického postupu patří spolu s mapováním sekvenačních čtení mezi klíčové a zároveň technicky nejnáročnější kroky analýzy. Optimalizace detekce variant je velmi náročná z důvodu velkého počtu dostupných programů a komplexity jejich možností nastavení.
Anotace detekovaných variant
K zjištěným záměnám jsou následně přiřazeny doplňující informace z vybraných databází, tzv. anotace, na základě kterých je možno odhadnout funkční dopad záměn a jejich klinický význam, případně varianty filtrovat a řadit podle individuálních potřeb závislých na biologické hypotéze. Mezi základní a nejčastěji používané databáze patří dbSNP [16], HapMap [17], Cosmic [18], HGMD [19], ClinVar [20], PolyPhen [21] atd.
Seznam anotovaných variant převedený do přehledné tabulky je pak nejčastější formou výsledků NGS analýz vydaných bioanalytikem lékaři či klinickému genetikovi.
Vizualizace
Důležitým krokem vyhodnocení NGS dat je vizualizace generovaných výsledků, která je obzvláště užitečná pro validaci a interpretaci získaných výsledků.
Nástroje tohoto typu nabízí vizualizaci namapovaných sekvenačních čtení [22], čehož se velmi často využívá pro kontrolu detekce inzercí a delecí, a zobrazení variant v kombinaci s anotacemi z různých veřejných databází. Jako příklad jsou na obrázku 1 uvedeny sekvenační čtení namapované na referenční genom se zvýrazněnými záměnami, které mohou představovat jak reálné varianty, tak chyby vzniklé při laboratorním zpracování nebo bioinformatické analýze.
ZÁVĚR
I přes skutečnost, že je dnes sekvenování nové generace dobře zavedenou metodou ve výzkumu, pro klinickou aplikaci nejsou stále dostatečně definovány standardy, jak na úrovni laboratorního zpracování a přípravy vzorků, tak následné bioinformatické analýzy. Pro správnou interpretaci výsledků NGS analýzy je velmi důležité, aby lékař rozuměl možnostem a limitům NGS technologie včetně základů analýzy dat. V textu jsme si kladli za cíl shrnout a objasnit nejdůležitější pojmy a kroky bioinformatické analýzy určené k detekci variant v DNA.
Seznam zkratek
AML – Acute myeloid leukemia
cffDNA – cell-free fetal DNA
CNV – Copy number variation
ctDNA – Circulating tumor DNA
DNA – dezoxyribonukleová kyselina
InDels – inzerce delece
NGS – Next-generation sequencing
PCR – Polymerase chain reaction
QC – Quality control
ROI – Region of interest
SNV – Single Nucleotide Variant
SV – Structural variation
UMI – Unique molecular identifier
VAF – Variant allele frequency
Poděkování
Tato práce byla podpořena projektem CEITEC 2020 (LQ1601) za finančního přispění Ministerstva školství, mládeže a tělovýchovy České republiky v rámci účelové podpory z prostředků Národního programu udržitelnosti II.
Čestné prohlášení autorů
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů, a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Podíl autorů na přípravě rukopisu
NT – hlavní autor práce, příprava první verze rukopisu, finalizace rukopisu
FP – spoluautor práce, příprava první verze rukopisu
JK – spoluautor práce, revize a finalizace rukopisu
KP – spoluautor práce, revize a finalizace rukopisu
ŠP – korespondující autor, spoluautor práce, schválení finální verze rukopisu
Doručeno do redakce dne 26. 4. 2018.
Přijato po recenzi dne 15. 5. 2018.
prof. RNDr. Šárka Pospíšilová, Ph.D.
Centrum molekulární medicíny, CEITEC,Masarykova univerzita
Kamenice 5
625 00 Brno
e-mail: pospisilova.sarka@fnbrno.cz
Zdroje
1. Heather JM, Chain B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016;107:1–8.
2. Tsiatis AC, Norris-Kirby A, Rich RG, et al. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations. J Mol Diagn 2010;12:425–432.
3. Malcikova J, Stano-Kozubik K, Tichy B, et al. Detailed analysis of therapy-driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia 2015;29:877–885.
4. Xu C, Gu X, Padmanabhan R, et al. smCounter2: an accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. bioRxiv 2018;281659; DOI: https://doi.org/10.1101/281659.
5. Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 2016;34:547–555.
6. Gnirke A, Melnikov A, Maguire J, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 2009;27:182–189.
7. Kivioja T, Vähärautio A, Karlsson K, et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 2011;9:72–74.
8. Cock PJA, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res 2010;38:1767–1771.
9. Fabbro CD, Scalabrin S, Morgante M, Giorgi FM. An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS One 2013; publ. elektronicky 23. 12. 2013. DOI:10.1371/journal.pone.0085024.
10. Fonseca NA, Rung J, Brazma A, Marioni JC. Tools for mapping high-throughput sequencing data. Bioinformatics 2012;28:3169–3177.
11. Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet 2014;15:121–132.
12. Ebbert MTW, Wadsworth ME, Staley LA, et al. Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinformatics 2016; publ. el. 25. 6. 2016. DOI:10.1186/s12859-016-1097-3.
13. Kou R, Lam H, Duan H, et al. Benefits and challenges with applying unique molecular identifiers in next generation sequencing to detect low frequency mutations. PLoS One 2016; publ. el. 11. 1. 2016. DOI:10.1371/journal.pone.0146638.
14. Xu C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comp Struct Biotechnol J 2018;16:15–24.
15. Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics 2013;14:S1. publ. el. 13. 9. 2013. DOI:10.1186/1471-2105-14-S11-S1.
16. Smigielski EM, Sirotkin K, Ward M, Sherry ST. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res 2000;28:352–355.
17. International HapMap Consortium. The International HapMap Project. Nature 2003;426:789–796.
18. Forbes SA, Bhamra G, Bamford S, et al. The catalogue of somatic mutations in cancer (COSMIC). Curr Protoc Hum Genet 2008; publ. el. 6. 7. 2008. DOI: 10.1002/0471142905.hg1011s57.
19. Stenson PD, Mort M, Ball EV, Shaw K, Phillips AD, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 2014;133:1–9.
20. Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014; publ. el. 14. 11. 2013. DOI: 10.1093/nar/gkt1113.
21. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013; publikováno el. 25. června 2015. DOI: 10.1002/0471142905.hg0720s76.
22. Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–26.
23. Babraham Bioinformatics – FastQC A quality control tool for high throughput sequence data. Dostupné na www: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 15 Feb 2018.
24. Chen S, Huang T, Zhou Y, Han Y, Xu M, Gu J. AfterQC: automatic filtering, trimming, error removing and quality control for fastq data. BMC Bioinformatics 2017;18:80.
25. Schubert M, Lindgreen S, Orlando L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Research Notes 2016;9:88.
26. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011;17:10–12.
27. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–2120.
28. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009;25:1754–1760.
29. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–359.
30. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2.
31. Broad Institute. picard: A set of command line tools (in Java) for manipulating high-throughput sequencing (HTS) data and formats such as SAM/BAM/CRAM and VCF. Dostupné na www: http://broadinstitute.github.io/picard. Accessed 22 Dec 2017.
32. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–930.
33. Fulcrum Genomics. fgbio: Tools for working with genomic and high throughput sequencing data. Dostupné na www: https://github.com/fulcrumgenomics/fgbio. Accessed 15 May 2018.
34. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv:12073907 [q-bio] 2012. Dostupné na www: http://arxiv.org/abs/1207.3907. Accessed 15 May 2018.
35. Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;11:11.10.1–11.10.33.
36. Lai Z, Markovets A, Ahdesmaki M, et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res 2016; publ. el. 7. 4. 2016. DOI: 10.1093/nar/gkw227.
37. Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–576.
38. Andrews TD, Jeelall Y, Talaulikar D, Goodnow CC, Field MA. DeepSNVMiner: a sequence analysis tool to detect emergent, rare mutations in subsets of cell populations. PeerJ 2016; publ. el. 24. 5. 2016. DOI: 10.7717/peerj.2074.
39. Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 2011;21:974–984.
40. Medvedev P, Fiume M, Dzamba M, Smith T, Brudno M. Detecting copy number variation with mated short reads. Genome Res 2010;20:1613–1622.
41. Chen K, Wallis JW, McLellan MD, et al. BreakDancer: An algorithm for high resolution mapping of genomic structural variation. Nat Methods 2009;6:677–681.
42. Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012;28:i333–i339.
43. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; publ. el. 3. 6. 2010. DOI: 10.1093/nar/gkq603.
44. Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92.
Štítky
Hematológia Interné lekárstvo OnkológiaČlánok vyšiel v časopise
Transfuze a hematologie dnes

2018 Číslo 3
- MUDr. Lenka Klimešová: Multiodborová vizita je kľúč k efektívnejšej perioperačnej liečbe chronickej bolesti
- Nejasný stín na plicích – kazuistika
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
Najčítanejšie v tomto čísle
- Neinfekční nemaligní lymfadenopatie – sinusová histiocytóza s masivní lymfadenopatií, nemoc Rosaiova-Dorfmanova
- Základní bioinformatické pojmy a postupy využívané pro analýzu DNA pomocí sekvenování nové generace
- Neinfekční a nemaligní lymfadenopatie – idiopatická (HHV-8 negativní) multicentrická forma Castlemanovy nemoci
- Mutácie génu F8 u pacientov s ťažkým stupňom hemofílie A a výskyt inhibítorov FVIII