
25 let vývoje metod molekulární biologie a jejich uplatnění v hemato(onko)logii
Development of molecular biology methods and their applications in haemato(onco)logy in the last 25 years
The field of molecular biology aims to explain the fundamentals governing living processes at molecular level by identifying mechanisms that produce such processes. Central discoveries describing the innate systems of living nature helped derive molecular technologies that work in vitro. The application of these methods has not only improved understanding of molecular biology but has also helped to improve prevention, diagnosis and treatment of human diseases.
This review focuses on the key technologies of molecular genetics and their development over the past 25 years. The application of these methods in the diagnosis and monitoring of haemato(oncolo)gical patients that have helped significantly to improve prognosis and treatment approaches, is described.
Keywords:
polymerase chain reaction (PCR) – next generation sequencing (NGS) – microarray – minimal residual disease (MRD)
Autori:
K. Machová Poláková 1; N. Čuřík 1; H. Votavová 1; J. Trka 2
Pôsobisko autorov:
Ústav hematologie a krevní transfuze, Praha
1; CLIP – Childhood Leukaemia Investigation Prague a Klinika dětské hematologie a onkologie 2. LF UK a FN Motol, Praha
2
Vyšlo v časopise:
Transfuze Hematol. dnes,25, 2019, No. 1, p. 34-42.
Kategória:
Souhrnné/edukační práce
Súhrn
Obor molekulární biologie má za cíl vysvětlovat podstatu fungování živých procesů na úrovni molekul identifikováním mechanismů, které produkují takové procesy. Fundamentální objevy popisující přirozené systémy živé přírody vedly k odvození molekulárních technologií, které fungují in vitro. Jejich vlastní použití nejenže dále prohlubuje poznání v oboru molekulární biologie, ale je klíčové pro zlepšování prevence, diagnostiky a léčby lidských onemocnění.
Tento souhrnný článek pojednává o zásadních molekulárně genetických metodách, jejichž vývoj v uplynulých 25 letech umožnil jejich uplatnění v diagnostice a monitorování hemato(onko)logických onemocnění, a které zásadním způsobem přispěly ke zlepšení prognózy pacientů a léčebných přístupů.
Klíčová slova:
polymerázová řetězová reakce (PCR) – sekvenování nové generace (NGS) – microarray – minimální zbytková nemoc (MRN)
SEKVENOVÁNÍ DNA A SEKVENOVÁNÍ NOVÉ GENERACE – NGS
První molekulárně genetické technologie (RFLP – restriction fragment length polymorphism; SSCP single strand conformation polymorphism) umožňující screening nukleotidových záměn byly založeny na restrikčních endonukleázách, které jsou přirozeně produkovány bakteriemi pro destrukci cizorodé DNA bakteriofágů. Informační hodnota těchto analýz však byla limitovaná z důvodu neznalosti cílových genů a oblastí genomu. Sekvenování DNA je vysoce informativní technologie, která umožňuje identifkovat definitivní nukleotidové záměny v cílových genech i nekódujících oblastech genomu. Obrázek 1 ve zjednodušené formě představuje přirozenou předlohu principu sekvenování a jeho vývoj jako technologie pro in vitro použití. Sekvenování (uváděno též jako sekvencování), je biochemický proces, který umožňuje „číst DNA“, tedy pořadí nukleových bází A (adenin), C (cytosin), G (guanin) a T (tymin).
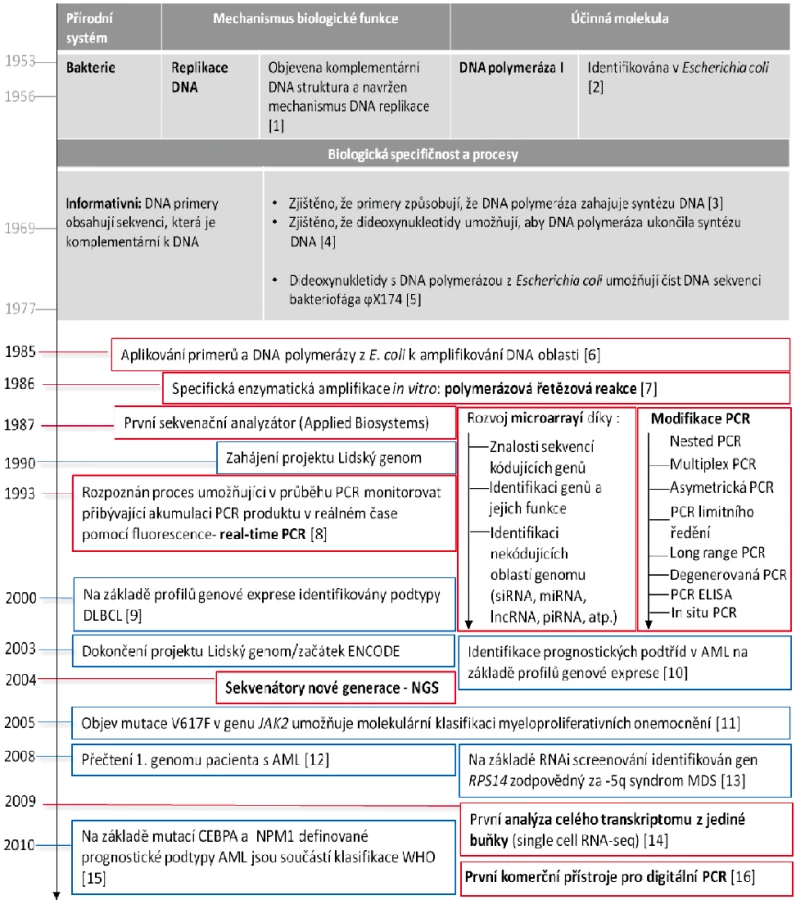
S uvedením prvního automatizovaného sekvenátoru (rok 1987) přišel i celosvětový projekt lidského genomu (The Human Genome Project; rok 1990), který umožnil prozkoumat lidskou genetickou výbavu a její změny asociované s různorodými onemocněními. Veškeré validní informace z prozkoumaných oblastí lidského genomu jsou shromažďovány v různých veřejně dostupných databázích. Nejvíce používanou a nejznámější databází je National Center for Biotechnology Information (U.S. National Library of Medicine, 8600 Rockville Pike, Bethesda MD, 20894 USA). Znalost lidského genomu umožnila prudký rozvoj vysokokapacitních analýz na bázi čipů (anglicky „microarray“; o využití čipů v hematologii pojednává následující kapitola). Praktické využití si sekvenování a následné analýzy porovnávající získanou sekvenci z vyšetřovaného vzorku se známou referenční sekvencí zdravého lidského genomu nachází ve vyšetřování somatických a germinálních mutací v cílových genech, které jsou spjaty s prognózou hematologických onemocnění. Dlouhé úseky sekvencí cílových genů, které mohou na různých pozicích nukleotidů nést záměny, je vhodné analyzovat současně, tedy v rámci jedné analýzy. Klasické sekvenování využívající Sangerovy metody [5] a sekvenátory na bázi automatizovaných kapilárních elektroforéz umožňuje v jedné analýze detekovat mutace napříč celou cílovou sekvencí. Klasické sekvenování je například stále zlatým standardem pro vyšetření cca 1000 bp dlouhé kódující oblasti kinázové domény BCR-ABL1, která u pacientů s chronickou myeloidní leukemií (CML) rezistentní k léčbě inhibitory tyrozinových kináz (TKI) může nést jednobodové mutace, které způsobují selhání léčby [17]. Nevýhodou klasického sekvenování pro vyšetřování mutací je nízká citlivost, která limituje celé vyšetření minimálně 15–20% zastoupením mutované sekvence v analyzovaném vzorku. Klinická praxe ukázala, že citlivost vyšetření klasickým sekvenováním limituje časné odhalení mutací v případech rychle postupující progrese onemocnění. Sekvenování nové generace (NGS) umožňuje díky své vysoké citlivosti na úrovni pohybující se mezi 1–3 % odhalit mutace mnohem dříve v průběhu léčby [18–20]. Navíc se ukázalo, že spolehlivě detekuje tzv. „low-level“ mutace, které mají prognostický význam, jelikož jsou spjaté s rizikem progrese CML [21]. NGS rovněž umožňuje postihnout biologii klonálního vývoje mutovaných CML klonů, protože rozkrývá celou jejich komplexitu, odlišuje polyklonální mutace od kompozitních, tj. v případě vícečetných mutací NGS určí, zda mutace pocházejí z více klonů nebo z jednoho klonu. Kompozitní mutace určují odlišný stupeň citlivosti CML buňky k TKI oproti situaci, kdy se mutace nacházejí v odlišných klonech, což je důležitá informace pro následnou volbu léčebné strategie pacienta. Podobně lze pomocí NGS analýzy mutací určit přesnou klonální hierarchii rozvoje AML a molekulární kinetiku relapsu tohoto onemocnění [22].
Zmíněné sekvenování nové generace neboli správněji ale méně často používané „Massive Parallel Sequencing“ je metoda, která využívá možnosti sekvenovat paralelně mnoho jednotlivých molekul nukleových kyselin v jediném experimentu. Díky pomocí PCR předem připravených tzv. knihoven NGS umožňuje sekvenovat celý genom člověka v jediné reakci („Whole Genome Sequencing“ – WGS). Další aplikace NGS jsou určovány typem, resp. druhem přípravy sekvenačních knihoven. Můžeme tak omezit sekvenování na přepisované části genomu („Whole Exome Sequencing“ – WES), nebo si vybrat jako cíl sekvenování již přepsanou RNA („Whole Transcriptome Sequencing“ – WTS nebo také RNASeq). „Capture-based Sequencing“ je metoda založená na vychytávání a sekvenování určitých úseků nukleové kyseliny na základě částečné předchozí znalosti cílové sekvence.
Metody NGS jsou masivně využívány ve výzkumu a čím dál více i v rutinní diagnostice vrozených i získaných geneticky podmíněných chorob, ale i nemocí infekčních. Výhodou NGS je tzv. škálovatelnost („scalability“), neboli variabilita hloubky sekvenování – můžeme si totiž zvolit, kolik molekul daného úseku nukleové kyseliny bude sekvenováno. Tato tzv. hloubka pokrytí určuje, zda budeme schopni odhalit i malé subklony v případě krevních malignit, či chiméry při vyšetření zárodečného genomu. Postupy založené na WGS jsou zatím v diagnostice spíše výjimkou kvůli velkému objemu získaných dat a nejasnému významu řady genetických změn v nepřepisované části genomu. Oproti tomu WES či RNASeq jsou již běžně využívané a nahrazují postupně v diagnostice jednotlivé cílené přístupy vycházející z PCR či metod molekulární cytogenetiky. Například u imunodeficiencí je dnes WES metodou první volby z hlediska pracnosti, času i ceny [23]. RNASeq umožňuje identifikaci známých i dosud nepopsaných fúzních genů, určení exprese jednotlivých genů a jejich variant, i přítomnost exprimovaných mutací a malých insercí či delecí („indels“), a spojuje tak výhody molekulárně genetického a cytogenetického vyšetření. Navíc přináší možnost vytvoření celkového expresního profilu sekvenovaných buněk, který se využívá pro identifikaci zajímavých subtypů např. u leukemií – nejlépe známým příkladem jsou tzv. BCR-ABL1-like akutní lymfoblastické leukemie (ALL), neboli leukemie s expresním profilem podobným Ph-pozitivním ALL [24].
Individuální návrh knihoven pak umožňuje zvolit si jen určitou oblast genomu, několik oblastí, genů či jen jednotlivých exonů – jednotlivé amplikony. Pak mluvíme o amplikonovém NGS, v případě kombinace řady amplikonů do jedné reakce pak o tzv. panelovém sekvenování. Panelové sekvenování dnes dominuje diagnostice pro svou relativně nižší cenu, jednoduchost provedení a analýzy, ale i pro vyšší hloubku pokrytí. Příkladem je panelové sekvenování u myeloidních a myeloproliferativních onemocnění, která poměrně často nesou mutace v kritických genech ovlivňujících vyzrávání myeloidní řady v průběhu krvetvorby. Během jediné analýzy získáváme vysoké množství dat, které umožňuje provést stratifikaci pacientů a rozhodovat o léčbě [25]. Musíme ale mít na paměti, že vybraný „panel“ genů je omezený a neumožňuje popsat nové genetické změny ani se k případně nově popsaným genům vrátit – jedná se tedy o limitovaný diagnostický nástroj ve srovnání s WES, WGS či WTS.
Specifickou oblastí aplikace amplikonového NGS je detekce reziduální nemoci. V posledních letech se rychle rozvíjí sekvenování přestaveb imunoreceptorových genů pro sledování reziduální nemoci u lymfoidních malignit. Konsorcium EuroClonality-NGS zavedlo a validovalo tuto metodu pro identifikaci cílů pro monitorování zbytkové nemoci a v současnosti pracuje na metodách kvantifikace. Zdá se, že NGS zvýší specifitu vyšetření a umožní tak zpřesnit prognostické odhady [26].
Správná analýza výsledků NGS (s výjimkou omezených panelů) vyžaduje přiměřenou počítačovou i bioinformatickou podporu; příprava a skladování dat, stejně jako detekce technických chyb a jejich případná korekce, je zejména v případě větších objemů analyzovaných vzorků velmi důležitá. Nicméně stále platí, že bioinformatický algoritmus je jen přípravným krokem pro skutečnou analýzu dat, kterou musí provádět zkušený lékař s biologickými znalostmi či biolog s medicínskými znalostmi, odborník na danou chorobu.
DNA A RNA ČIPY
(„microarrays“)
DNA/RNA čip („microarray“) je miniaturizovaná testovací jednotka nesoucí na svém povrchu v přesně definovaném uspořádání imobilizované fragmenty DNA/RNA (sondy), ke kterým hybridizují hledané cílové sekvence ve vzorku. Vzhledem k vysoké hustotě sond na čipu lze současně testovat tisíce až desetitisíce cílových sekvencí, což umožnilo vědcům globální pohled na genom a transkriptom. K masivnímu rozšíření čipů došlo nejdříve v oblasti genové exprese a následně byly vyvinuty platformy pro studium polymorfismů a mutací, molekulárně cytogenetické analýzy, a nakonec i pro nově objevené nekódující RNA (ncRNA). V hematologii byly čipy široce použity pro charakterizaci genových expresních profilů specifických nejen pro daná onemocnění, ale i pro jejich různé podtypy či stadia. Golub et al. (1999) publikovali jednu z prvních studií, která demonstrovala použitelnost expresních profilů pro spolehlivou klasifikaci akutních leukemií [27]. Obdobný přístup byl úspěšně použit například pro subklasifikaci difuzního velkobuněčného B-lymfomu (DLBCL) [9] či pro stanovení prognostických podtříd akutní myeloidní leukemie (AML) [10]. Stejná diskriminační schopnost byla prokázána i u expresních profilů ncRNA, jako jsou mikroRNA (miRNA) a dlouhé ncRNA (lncRNA) [28, 29]. V neposlední řadě byly v hematologii expresní čipy využity pro hledání vhodných diagnostických/prognostických markerů, genů predikujících odpověď na léčbu a potenciálních terapeutických cílů. V tomto kontextu lze zmínit například prognostický marker ZAP-70 (dnes slouží jako doprovodný faktor), jehož zvýšená genová exprese je asociována s agresivnějším průběhem chronické lymfocytární leukemie (CLL) [30]. U dětské ALL byl pomocí čipů identifikován gen CASP8AP2, jehož expresní hladina v době diagnózy predikovala odpověď na léčbu [31]. Ve studii Hofmann et al. (2002) definovali panel 56 odlišně exprimovaných genů, které byly asociovány s rozvojem sekundární rezistence na imatinib u pacientů s Ph-pozitivní ALL [32].
V oblasti DNA aplikací byla pro sledování změn v počtu kopií DNA úseků (amplifikace, delece, tzv. CNV – „copy number variations“) vyvinuta tzv. arrayCGH (array komparativní genomová hybridizace) založená na kohybridizaci rozdílně značené nádorové a kontrolní DNA k sondám na čipu. V současné době jsou však více používány tzv. „single nucleotide polymorphism“ (SNP) čipy, které umožňují stejnou analýzu CNV včetně detekce SNP. Metoda SNP čipů využívá sondy schopné rozlišit jednotlivé alely konkrétních jednonukleotidových polymorfismů, které jsou ve velkém počtu rozmístěny po celém genomu každého jedince. Rozsáhlou studii využívající SNP čipy pro detekci genetických aberací u dětí s ALL provedli například Mullighan et al. (2007) [33]. V hematologii patří čipy bezesporu mezi užitečné analytické nástroje nejenom pro výzkumné účely, ale našly své uplatnění i v diagnostických postupech (např. SNP čipy).
POLYMERÁZOVÁ ŘETĚZOVÁ REAKCE – PCR
PCR je výchozí metodou pro většinu molekulárně genetických analýz. Umožňuje množení cílových úseků DNA na taková množství výsledného produktu syntézy, která lze vizualizovat pomocí různých zobrazovacích technologií. K vývoji PCR vedla řada na sebe navazujících objevů, zjištěných v systémech živé přírody, které zobrazuje obrázek 1. V průběhu času se zdokonalovaly PCR termocyklery, což také přispělo k odvození mnoha modifikací PCR.
V hematoonkologických onemocněních se v diagnostice nejvíce uplatňují tyto aplikace PCR:
- multiplex PCR,
- real-time qPCR, a do popředí zájmu se dostává
- digitální PCR.
1. Multiplex PCR (M-PCR)
M-PCR představuje modifikaci metody PCR. Funguje na stejných základních principech, ale využívá většího počtu sad PCR primerů a případně i většího počtu templátů zároveň. To umožňuje souběžnou amplifikaci většího počtu specifických úseků DNA v jedné reakční směsi, což vede k materiálové, finanční a časové úspoře oproti jednotlivým molekulárním vyšetřením příslušných oblastí genomu. Metoda si našla uplatnění v řadě oborů medicínského výzkumu, mezi něž se v průběhu 90. let minulého století zařadila i hematoonkologie.
AML a ALL dětí i dospělých představují vysoce heterogenní onemocnění způsobené, resp. provázené řadou různých chromozomových aberací (translokací), genových fúzí a genových mutací. Cytogenetické vyšetření FISH (fluorescenční in situ hybridizace) představuje základní metodu umožňující detekci genových fúzí při diagnostice těchto onemocnění, ale jde o poměrně pracnou metodu. Potenciálem M-PCR je možnost rychlého screeningu většího počtu uvažovaných chromozomových aberací, což je příhodné pro diagnostiku a prognostiku u akutních leukemií. Koncem 90. let byla pomocí osmi paralelních multiplex PCR reakcí provedena úspěšná retrospektivní detekce 29 chromozomových aberací (80 variant zlomů) na 164 vzorcích pacientů s AML, resp. ALL, a ve vysokém procentu vzorků odhalila submikroskopické delece nebo skryté přestavby, které se předtím nepodařilo identifikovat cytogenetickými metodami [34]. Metoda byla opět v paralelním uspořádání s úspěchem využita pro detekci KMT2A (MLL) fúzních genů u primárních vzorků pacientů s AML a ALL i u buněčných linií s tímto typem přestavby [35] a pro detekci přestaveb BCR-ABL1 a PML/RARA (PML-RARα) typických pro CML, resp. akutní promyelocytární leukemii [36]. Na základě dosažených výsledků si metoda M-PCR postupně nacházela cestu do rutinního využití v klinických laboratořích. Jedním z milníků byla první multicentrická validační studie pro detekci 12 typů fúzních genů asociovaných s AML, ALL a CML, která ukázala, že metoda umožňuje spolehlivou a citlivou detekci molekulárních markerů těchto leukemických onemocnění [37].
Při současném využití v diagnostice, prognostice a monitorování hematologických chorob lze M-PCR vhodně propojovat také s dalšími metodami. Multiplex PCR byla v kombinaci s mikroelektronickým hybridizačním čipem a fluorescenční sondou využita k rychlému a přesnému detekování fúzních transkriptů přítomných v ALL, CML nebo AML bez nutnosti srovnávat velikost PCR produktů pomocí gelové elektroforézy [38]. Ve spojení s kapilární elektroforézou byla M-PCR využita k detekci mutací v genu MPL u pacientů s esenciální trombocytemií a primární myelofibrózou [39]. Jako perspektivní se jeví kombinace M-PCR připravující amplikony pro následné sekvenování umožňující např. detekci velkého počtu mutací zároveň [40]. V současné době existují platformy jako například AmpliSeq umožňující najednou amplifikovat až 24 tisíc úseků DNA (reprezentujících stovky genů) a vznik DNA knihoven pro následné masivní sekvenování metodou NGS.
2. Real-time qPCR
Real-time qPCR (kvantitativní PCR v reálném čase) umožňuje sledovat amplifikaci cílové sekvence v reálném čase díky snímání emitované fluorescence v průběhu PCR. Tento proces byl rozpoznán v roce 1993 [8]. Co je zcela převratné u tohoto typu PCR, je přesná kvantifikace PCR produktů v průběhu exponenciální fáze amplifikačního procesu na rozdíl od odhadovaného množství produktu na konci klasické PCR. Pro snímání fluorescence, která umožňuje kvantifikaci cílového úseku sekvence v průběhu PCR, se nejhojněji využívají tzv. TaqMan hydrolyzační sondy. Exonukleázová aktivita Taq polymerázy v průběhu syntézy DNA řetězce próbu hydrolyzuje a díky tomu dochází k emitování fluorescence, kterou zaznamenávají detektory real-time qPCR přístrojů.
Monitorování minimální zbytkové/reziduální nemoci (MRN) je významným prognostickým markerem a nedílnou součástí řady léčebných protokolů hematologických malignit. V poslední době se termín MRN stále častěji interpretuje jako měřitelná zbytková nemoc, což vychází ze zavedení vysoce citlivých molekulárních technik detekce reziduálních leukemických buněk, jejichž přítomnost je spojena s horší prognózou [41]. MRN je zatím stále nejčastěji kvantifikována prostřednictvím real-time qPCR cílových fúzních genů, fúzních transkriptů či onkogenů nesoucích aberace v sekvenci DNA nebo majících narušenou expresi.
Příkladem MRN u lymfoidních malignit je kvantifikace přeuspořádaní genů pro imunoglobulin (IGH) a pro receptor T buněk (TCR). IGH/TCR přestavby jsou unikátní sekvence, u kterých se má za to, že jsou jedinečné pro každou lymfoidní prekurzorovou buňku, tedy i maligní lymfoidní prekurzorovou buňku. Kvantifikace Ig/TCR přestaveb se využívá především u onemocnění postihující B liniové prekurzorové buňky, tedy u dětských a dospělých pacientů s B-ALL, u pacientů s B-CLL, s B-lymfomem a mnohočetným myelomem. Vyšetření se rovněž provádí u onemocnění postihující i T liniové prekurzorové buňky u dětských a dospělých pacientů s T-ALL a u pacientů s T-lymfomy. Určení Ig/TCR přestavby není technicky jednoduchá záležitost, stejně tak jako následující kvantifikace se zajištěním požadované citlivosti a specifičnosti analýzy [42]. Pouze specializované laboratoře, které jsou součástí konsorcií zabývajících se standardizací metodiky, jako je například EURO-MRD konsorcium (http://euromrd.org), zaručují kvalitní MRN analýzu založenou na kvantifikaci Ig/TCR.
Chromozomové aberace jsou výborným nádorově-specifickým markerem pro sledování MRN. PCR primery leží na opačných stranách oblasti fúze genů a vysoce specificky a s výraznou citlivostí detekují množství MRN. U folikulárního lymfomu se využívá kvantifikace přestavby chromozomů 14 a 18 zasahující geny BCL2 a IGH [43]. U plášťového lymfomu se kvantifikuje přestavba genů CCND1(BCL1) a IGH; t(11;14) [44]. Jedná se o pacient-specifické fúze, kdy DNA zlomy postihují oblast genů v rozsahu do 2 kb. U chromozomových aberací, které postihují geny, v kterých se zlomy vytvářejí v rozsáhlejších (více jak 200 kb), především intronových oblastech, se jako markery pro MRN monitorování využívají fúzní transkripty. Většina fúzí na úrovni transkriptů je pro pacienty shodná a je možné pracovat s úzkým setem primerů a prób. Typickým případem monitorování fúzního transkriptu je kvantifikace BCR-ABL1 u CML a Ph+ ALL [45-46], NPM1/ALK u anaplastického velkobuněčného lymfomu s přestavbou t(2;5) a ETV6/RUNX1 (TEL-AML1), PML/RARA, CBFB/MYH11 a RUNX1/RUNX1T1 (AML1-
-ETO) u akutních leukemií [44].
Další využití pro sledování průběhu hematologických onemocnění má real-time qPCR při poškozeních genů na úrovni jejich sekvence či míry jejich exprese. Dobře známým příkladem jsou mutace v genu NPM1, které představují nejčastější molekulární lézi u pacientů s AML s cytogeneticky normálním nálezem. Přítomnost NPM1 transkriptů nesoucích mutace se ukázala být velmi silným nezávislým prediktivním faktorem relapsu onemocnění [47]. Příkladem využití aberantní genové exprese pro monitorování MRN v praxi jsou transkripty genů WT1, jejichž množství je v krvetvorných buňkách AML pacientů obsaženo v přemíře oproti zdravým jedincům. Měření exprese těchto genů slouží jako alternativní marker MRN u pacientů, u kterých nebyla zjištěna chromozomová změna vedoucí k fúzi genů [48].
Kvantifikace MRN přispěla k zásadním změnám v léčebných postupech hematologických malignit, kdy tyto změny významně zlepšily celkové prospívání pacientů. Například u CML mohly být uskutečněny klinické studie vysazující léčbu TKI, které se opírají právě o spolehlivé, standardizované monitorování hluboké molekulární odpovědi podle zbytkových hladin BCR-ABL1 [45].
3. Digitální PCR
V současné době se v souvislosti s měřením MRN dynamicky rozvíjí využití digitální PCR. Ovšem pojem digitální PCR byl poprvé použitý již v roce 1999 autory Vogelsteinem a Kinzlerem, kteří popisovali kvantifikaci mutace v RAS genu ve vzorku, který byl rozdělen pro provedení PCR reakcí v mikrodestičce obsahující 384 PCR jamek [49]. Termín digitální PCR byl pro tento typ analýzy velmi výstižný, jelikož zachycoval povahu reakce a ducha té doby [50]. Nicméně popisovaná metoda nebyla zcela nová, jelikož již o dekádu dříve byly používány termíny PCR jediné molekuly nebo PCR limitního ředění. Vůbec první publikace využívající digitální PCR pro kvantifikaci cílové sekvence se zabývala virem HIV [51]. Autoři se zaměřili na zjištění genetické diverzity HIV populace infikující lymfocyty ve vzorku krve HIV pozitivních pacientů. Rozpoznali, že analýza celé masy vzorku by neumožňovala zjistit sekvenční odlišnosti mezi individuálními provirálními molekulami. Použili proto limitní ředění vzorků, následnou PCR replikátů a sekvenování získaných PCR produktů. Díky analýzám limitního ředění se zjistilo, že lze na základě míry ředění a frekvence pozitivních amplifikací odvozenou Poissonovou distribucí dosti přesně spočítat množství provirálních molekul HIV. V analogii s touto prací byla PCR limitního ředění využita pro charakterizaci a kvantifikaci Ig/TCR klonální přestavby v době diagnózy u dětských ALL [52]. Metodika limitního ředění byla s příchodem real-time qPCR opuštěna, a to především z důvodu toho, že real-time qPCR představuje uzavřený systém s minimalizací kontaminace, která se vyskytovala v případě PCR limitního ředění, což byla významná nevýhoda metody v té době. V současné době zažívá PCR limitního ředění velký comeback díky vývoji přístrojů umožňujících v uzavřeném systému s minimem rizika kontaminace provádět digitální PCR. Komerčně dostupné přístroje pro digitální PCR se datují k roku 2011 [16]. Výhoda digitální PCR oproti real-time qPCR spočívá především v tom, že umožňuje s daleko vyšší přesností detekovat a kvantifikovat cílové molekuly, které jsou v analyzovaném vzorku obsaženy velmi málo. Zatím nejrozšířenější platforma kapkové digitální PCR (droplet digital PCR) distribuuje PCR reakci do několika desítek tisíc individuálních PCR reakcí před samotnou amplifikací. Míra signálu fluorescence vysílaného z jednotlivých kapek po skončení PCR slouží pro zjištění počtu kapek nesoucích cílový analyt, a tím umožňuje kvantifikovat jeho množství přítomné ve vzorku. Pojem absolutní kvantifikace se s oblibou užívá právě v souvislosti s digitální PCR a intuitivně poukazuje na výhodnosti této metody, která nevyžaduje plazmidové standardy pro odvození počtu kopií cílové sekvence, jak je tomu v případě real-time qPCR. Digitální PCR má mnoho dalších výhod, jako je například nižší citlivost k různým látkám inhibujícím PCR reakci.
Digitální PCR nalezne své uplatnění v lékařské praxi hemato(onko)logických onemocnění, u kterých bude zapotřebí s vysokou přesností a spolehlivostí kvantifikovat MRN. Zajisté uslyšíme o digitální PCR v souvislosti s kvantifikací zbytkových molekul BCR-ABL1 v případě pacientů s CML, kteří budou splňovat kritéria pro vysazení léčby TKI, a u kterých se léčba bude v praxi vynechávat.
ZÁVĚR
Postupný rozvoj metod založených na PCR, následovaný objevem sekvenování nové generace, zcela změnil paradigma diagnostiky a sledování zbytkové nemoci u hematologických malignit i diagnostiku poruch krvetvorby či imunity. V současné době musí každé diagnostické pracoviště pečlivě zvažovat, jakou kombinaci laboratorních přístupů zvolí. Hledisek je řada: především jsou to klinické požadavky na minimální nutný rozsah diagnostiky, dále finanční možnosti, přístrojové vybavení, tradice příslušné laboratoře, a zejména objem vzorků procházejících diagnostickým procesem.
Je zřejmé, že postupně dojde k omezení jednotlivých cílených metod (PCR nebo FISH na detekci jednotlivé aberace) ve prospěch širokozáběrových metod typu čipů a nejrůznějších variant NGS. Využití těchto metod, přinášejících množství nových informací, s sebou však nese i určitá úskalí. Mezi ně patří např. potřeba velmi čistých populací buněk k analýzám, která může být zaručena jen sortováním buněk, stejně jako nutnost vyšetřit spolu se somatickým i zárodečný genom. Vývoj také směřuje k analýzám na úrovni jednotlivých buněk. Ty umožní získat informace o „nezprůměrované“ expresi genů (např. markerů MRN) nebo o ko-lokalizaci genových mutací, což bude hrát důležitou roli ve volbě individualizovaných terapeutických strategií, jak tomu již začíná být v translační medicíně solidních nádorů.
Hlavní změnou paradigmatu diagnostiky je ale fakt, že namísto převažující laboratorní práce a jednoduché interpretace výsledků vyšetření jsme konfrontováni s relativním úbytkem práce v laboratoři a výrazným nárůstem analytické a interpretační části vyšetření. Tato změna postupně povede k výrazným změnám v personálním obsazení laboratoří i změně komunikace mezi laboratorními pracovníky a lékaři-kliniky.
Podíl autorů na přípravě rukopisu
KMP – koncept, psaní a finální revize rukopisu
NČ – psaní rukopisu, revize rukopisu a souhlas s finální verzí
HV – psaní rukopisu, revize rukopisu a souhlas s finální verzí
JT – psaní rukopisu (NGS, Závěr), revize rukopisu a souhlas s finální verzí.
Čestné prohlášení
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Poděkování
Autoři Machová Poláková K., Čuřík N. a Votavová H. byli podporováni MZČR 00023736.
Doručeno do redakce dne 8. 11. 2018.
Přijato po recenzi dne 14. 12. 2018.
Mgr. Kateřina Machová Poláková, Ph.D.
Ústav hematologie a krevní transfuze
U Nemocnice 1
128 00 Praha 2
e-mail: katerina.machova@uhkt.cz
Zdroje
1. Watson JD, Crick FHC. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953;171:737–738.
2. Kornberg A, Lezhman I, Simms E. Polydesoxyribonucleotide synthesis by enzymes from Escherichia coli. Fed Proc 1956b;291–292.
3. Kornberg A, Lehman IR, Bessman MJ, Simms ES. Enzymic synthesis of deoxyribonucleic acid. Biochim Biophys Acta 1956a;21:197–198.
4. Atkinson MR, Deutscher MP, Kornberg A, Russell AF, Moffatt JG. Enzymatic synthesis of deoxyribonucleic acid. XXXIV. Termination of chain growth by a 2‘,3‘-dideoxyribonucleotide. Biochemistry 1969;8:4897–4904.
5. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 1977;74:5463–5467.
6. Saiki RK, Scharf S, Faloona F, et al. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985;230:1350–1354.
7. Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 1986;51:263–273.
8. Higuchi R, Fockler C, Dollinger G, Watson R. Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology (N Y) 1993;11:1026–1030.
9. Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000;403:503–511.
10. Bullinger L, Döhner K, Bair E, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med 2004;350:1605–1616.
11. James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434:1144–1148.
12. Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008;456:66–72.
13. Ebert BL, Pretz J, Bosco J, et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 2008;451:335–339.
14. Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 2009;6:377–382.
15. Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010;115:453–474.
16. Hindson CM, Chevillet JR, Briggs HA, et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods 2013;10:1003–1005.
17. Klamová H, Žižková H, Burda P, et al. Současné trendy v léčbě a diagnostice chronické myeloidní leukemie. Transfuze Hematol dnes 2017;23(Supl 2):34–42.
18. Soverini S, De Benedittis C, Machova Polakova K, et al. Unraveling the complexity of tyrosine kinase inhibitor-resistant populations by ultra-deep sequencing of the BCR-ABL kinase domain. Blood 2013;122:1634–1648.
19. Machova Polakova K, Kulvait V, Benesova A, et al. Next-generation deep sequencing improves detection of BCR-ABL1 kinase domain mutations emerging under tyrosine kinase inhibitor treatment of chronic myeloid leukemia patients in chronic phase. J Cancer Res Clin Oncol 2015;141:887–899.
20. Musilova M, Razga F, Jurcek T, et al. BCR-ABL1 kinase domain mutational analysis of CD34+ stem/progenitor cells in newly diagnosed CML patients by next-generation sequencing. Am J Hematol 2014;89:1016–1017.
21. Soverini S, De Benedittis C, Papayannidis C, et al. Clinical impact of low-burden BCR-ABL1 mutations detectable by amplicon deep sequencing in Philadelphia-positive acute lymphoblastic leukemia patients. Leukemia 2016;30:1615–1619.
22. Čulen M, Kosařová Z, Ježíšková I, et al. Sekvenování nové generace u akutní myeloidní leukemie: nový pohled na patogenezi a vývoj leukemických klonů. Transfuze Hematol dnes 2017;23:185–191.
23. Maffucci P, Filion CA, Boisson B, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol 2016;7:220.
24. Sanchez R, Morgades M, Ayala R, et al. Targeted RNA-Seq identify a subset of adolescent and adult patients with acute lymphoblastic leukemia with BCR-ABL1-like characteristics. Blood 2017;130:2710.
25. Yang F, Press RD. Next-generation sequencing multi-gene mutation panels in myeloid malignancies. The Hematologist 2016;13:6–7.
26. Kotrova M, Muzikova K, Mejstrikova E, et al. Next generation amplicon sequencing of immunoglobulin heavy chain gene rearrangaments for minimal residual disease (MRD) stratification in childhood acute lymphoblastic leukemia (ALL): a comparison with classical qPCR-based technique. Blood 2014;124:2395.
27. Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999;286:531–537.
28. Dostalova Merkerova M, Krejcik Z, Votavova H, Belickova M, Vasikova A, Cermak J. Distinctive microRNA expression profiles in CD34+ bone marrow cells from patients with myelodysplastic syndrome. Eur J Hum Genet 2011;19:313–319.
29. Garzon R, Volinia S, Papaioannou D, et al. Expression and prognostic impact of lncRNAs in acute myeloid leukemia. Proc Natl Acad Sci U S A 2014;111:18679–18684.
30. Wiestner A, Rosenwald A, Barry TS, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood 2003;101:4944–4951.
31. Flotho C, Coustan-Smith E, Pei D, et al. Genes contributing to minimal residual disease in childhood acute lymphoblastic leukemia: prognostic significance of CASP8AP2. Blood 2006;108:1050–1057.
32. Hofmann WK, de Vos S, Elashoff D, et al. Relation between resistance of Philadelphia-chromosome-positive acute lymphoblastic leukaemia to the tyrosine kinase inhibitor STI571 and gene-expression profiles: a gene-expression study. Lancet 2002;359:481–486.
33. Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007;446:758–764.
34. Pallisgaard N, Hokland P, Riishøj DC, Pedersen B, Jørgensen P. Multiplex reverse transcription-polymerase chain reaction for simultaneous screening of 29 translocations and chromosomal aberrations in acute leukemia. Blood 1998;92:574–588.
35. Andersson A, Höglund M, Johansson B, et al. Paired multiplex reverse-transcriptase polymerase chain reaction (PMRT-PCR) analysis as a rapid and accurate diagnostic tool for the detection of MLL fusion genes in hematologic malignancies. Leukemia 2001;15:1293–1300.
36. Salto-Tellez M, Shelat SG, Benoit B, et al. Multiplex RT-PCR for the detection of leukemia-associated translocations: validation and application to routine molecular diagnostic practice. J Mol Diagn 2003;5:231–236.
37. Gocke CD, Mason J, Brusca L, et al. Risk-based classification of leukemia by cytogenetic and multiplex molecular methods: results from a multicenter validation study. Blood Cancer J 2012;2:e78.
38. Corradi B, Fazio G, Palmi C, Rossi V, Biondi A, Cazzaniga G. Efficient detection of leukemia-related fusion transcripts by multiplex PCR applied on a microelectronic platform. Leukemia 2008;22:294–302.
39. Pettersson Cheng A, Viskari A, Odén U, et al. Improved MPL mutation screening with multiplex PCR and capillary electrophoresis. Br J Haematol 2017;179:838–840.
40. Park N, Vassiliou G. Design and application of multiplex PCR seq for the detection of somatic mutations associated with myeloid malignancies. Methods Mol Biol 2017;1633:87–99.
41. Goldman JM, Gale RP. What does MRD in leukemia really mean? Leukemia 2014;28:1131.
42. Van der Velden VHJ, Hochhaus A, Cazzaniga G., Szczepanski T, Gabert J, van Dongen JJM. Detection of minimal resicual disease in hematologic malignancies by real-time quantitative PCR: principles, approaches, and laboratory aspects. Leukemia 2003;17:1013–1034.
43. Yunis JJ, Oken MM, Kaplan ME, Ensrud KM, Howe RR, Theologides A. Distinctive chromosomal abnormalities in histologic subtypes of non-Hodgkin’s lymphoma. N Engl J Med 1982;307:1231–1236.
44. Tsujimoto Y, Yunis J, Onorato-Showe L, Erikson J, Nowell PC, Croce CM. Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14), chromosome translocation. Science 1994;224:965–977.
45. Zemanová K, Žižková H, Jurček T, et al. Chronická myeloidní leukemie – standardizace molekulárního monitorování hladiny transkriptů BCR-ABL1 v České republice. Transfuze Hematol dnes 2016;22:56–64.
46. Hrabovský Š, Folber F, Šálek C, Horáček JM, Mayer J, Doubek M. Pokroky v léčbě akutní lymfoblastické leukémie dospělých. Transfuze Hematol dnes 2015;21:84–91.
47. Ivey A, Hills RK, Simpson MA, et al. Assessment of minimal residual disease in standard-risk AML. N Engl J Med 2016;374:422–433.
48. Kreuzer KA, Saborowski A, Lupberger J, et al. Flurescent 5‘-exonuclease assay for the absolute quantification of Wilms‘ tumour gene (WT1) mRNA: implications for monitoring human leukameias. Br J Haematol 2001;114:313–318.
49. Vogelstein B and Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A 1999;96:9236–9241.
50. Morley AA. Digital PCR: A brief history. Biomol Detection Quantif 2014;1:1–2.
51. Simmonds P, Balfe P, Peutherer JF, Ludlam CA, Bishop JO, Brown AJ. Human immunodeficiency virus-infected individuals conatin provirus in small nubmer of peripheral mononuclar cells and at low copy numbers. J Virol 1990;64:864–872.
52. Sykes PJ, Neoh SH, Brisco MJ, Hughes E, Condon J, Morley AA. Quantitation of targets for the polymerase chain reaction by use of limited dilution. Biotechniques 1992;13:444–449.
Štítky
Hematológia Interné lekárstvo OnkológiaČlánok vyšiel v časopise
Transfuze a hematologie dnes

2019 Číslo 1
- Nejasný stín na plicích – kazuistika
- Těžké menstruační krvácení může značit poruchu krevní srážlivosti. Jaký management vyšetření a léčby je v takovém případě vhodný?
- Využití koncentrátu protrombinového komplexu u akutních krvácivých stavů
- Co s koagulopatií u COVID-19-pozitivních pacientů?
- Statinová intolerance
Najčítanejšie v tomto čísle
- Hodgkinův lymfom – nekončící příběh
- Non-Hodgkinův lymfom v České republice
- Vývoj transfuzní služby v České republice po roce 1990
- 25 let vývoje dětské hematologie