
Guidelines EAU pro léčbu mužské infertility
4. Genetické poruchy u infertilních mužů
Autori:
A. Jungwirth; T. Diemer; G. R. Dohle; A. Giwercman; Z. Kopa; C. Krausz; H. Tournaye
Vyšlo v časopise:
Urol List 2012; 10(4): 52-59
Kategória:
Guidelines
© European Association of Urology 2012
3. Testikulární nedostatečnost (selhání spermatogeneze)
4. Genetické poruchy u infertilních mužů
4.1 Úvod
Znalost genetických abnormalit souvisejících s infertilitou je nezbytná pro každého urologa zabývajícího se andrologií, aby mohl poskytnout adekvátní informace párům hledajícím léčbu mužské infertility.
In vitro fertilizace (IVF), ICSI a extrakce spermií z epididymis nebo varlete (v případě azoospermie) nabízejí mužům s velmi nízkým počtem spermií solidní šanci dosáhnout otcovství. Druhou stranou mince je skutečnost, že spermie infertilních mužů vykazují zvýšený výskyt aneuploidie, dalších genetických abnormalit a poškození DNA a také zvyšují pravděpodobnost přenosu genetických abnormalit na další generace. Přestože existuje šance, že v budoucnu budeme moci provádět screening ejakulátu [1,2], v současné urologické praxi se užívá screening vzorků periferní krve.
4.2 Chromozomální abnormality
Chromozomální abnormality mohou být numerické (jako je například trisomie) nebo strukturální (jako inverze či translokace) [3,4]. V přehledu údajů nashromážděných z 11 publikací a zahrnujících 9 766 infertilních mužů byla zaznamenána 5,8% incidence chromozomálních abnormalit [3], z níž 4,2 % činila incidence abnormalit pohlavního chromozomu a 1,5 % incidence autozomálních abnormalit.
Pro srovnání – incidence abnormalit v údajích nashromážděných ze tří studií čítajících 94 465 novorozenců mužského pohlaví představovala 0,38 %, ze kterých 131 (0,14 %) tvořily abnormality pohlavního chromozomu a 232 (0,25 %) autozomální abnormality [4].
Frekvence chromozomálních abnormalit se zvyšuje spolu se závažností testikulární insuficience. Pacienti s < 5 mil. spermatozoi/ml vykazují (ve srovnání s obecnou populací) desetinásobně vyšší incidenci (4 %) zejména autozomálních strukturálních abnormalit [5]. Nejvyšší riziko hrozí mužům se sekreční azoospermií.
Na základě frekvence chromozomálních aberací u pacientů s různou koncentrací spermií je analýza karyotypu indikována u všech mužů s azoospermií a mužů s oligozoospermií s < 5 mil. spermatozoi/ml [5]. U párů s anamnézou opakované potratovosti, malformací nebo mentální retardace by měla být analýza karyotypu požadována bez ohledu na koncentraci spermií pacienta.
4.2.1 Chromozomální abnormality spermií
Analýza vícebarevné fluorescenční in situ hybridizace (FISH) umožňuje vyšetřit spermie z pohledu normální chromozomové výbavy.
Aneuploidie spermií, zejména aneuploidie pohlavního chromozomu, souvisí se závažným poškozením spermatogeze [3,6–10] a vyskytuje se rovněž u mužů s translokacemi [11].
FISH analýzu spermatozoí je zatím třeba považovat pouze za experimentální metodu, ovšem je vhodná pro vyšetření spermatozoí u mužů s definovanou andrologickou poruchou [6]. Je zapotřebí vyvinout nové techniky, jež umožní odlišit populaci geneticky abnormálních spermií od normálních spermií, a techniky pro bezpečný screening jednotlivých spermatozoí před IVF a ICSI.
4.2.2 Chromozomální abnormality pohlavních hormonů (Klinefelterův syndrom a jeho varianty [47,XXY; 46,XY/47, XXY mozaiky])
Klinefelterův syndrom je nejčastější abnormalitou pohlavních chromozomů [3,12]. Dospělí muži trpící Klinefelterovým syndromem mají malá varlata postrádající zárodečné buňky. Fenotyp se může lišit od normálních znaků virilizace až po znaky androgenní nedostatečnosti, která se vyznačuje ženským typem ochlupení, minimálním tělesným ochlupením celkově a dlouhými pažemi i dolními končetinami – důsledkem pozdního uzavření epifýzy. Funkce Leydigových buněk je u mužů s Klinefelterovým syndromem obvykle narušena [13]. Hladina testosteronu může být normální nebo nízká, hladina estradiolu normální nebo zvýšená a hodnota hormonu stimulujícího folikuly (FSH) je zvýšená. Navzdory nízkým hodnotám testosteronu je libido překvapivě normální, ale v průběhu stárnutí může být zapotřebí substituce androgenů. Schopnost produkce spermií a výskyt zárodečných buněk se může u mužů s Klinefelterovým syndromem a mozaikou 46,XY, 47,XXY lišit. Jedna kazuistika popisuje zhoršování spermatogeneze u pacienta s Klinefelterovým syndromem; autoři doporučují zvážit včasný odběr spermií [14].
Vzhledem k tomu, že FISH analýza spermatických buněk vykazuje zvýšenou frekvenci abnormalit pohlavních hormonů a vyšší incidenci autozomální aneuploidie (disomie u chromozomů 13, 18 a 21), existují obavy, zda embrya vzniklá pomocí techniky ICSI nevykazují abnormální charakteristiky [15].
U pacientů s Klinefelterovým syndromem byla zaznamenána tvorba 24,XY spermií u 0,9 % a 7,0 % mužů [16–18] a u 1,36–25 % mužů se somatickým karyotypem 47,XXY [19–22].
U mužů s azoospermií lze jako terapeutickou modalitu doporučit TESE nebo MicroTESE, protože spermatozoa lze odebrat přibližně u 30 % jedinců. Dosud se pomocí techniky ICSI narodilo 49 zdravých dětí bez preimplantační genetické diagnostiky (PDG) a jeden případ 47,XXY [12]. Studie popisující ICSI v kombinaci s PDG u 113 embryí však uvádí (ve srovnání s kontrolní skupinou) významný pokles počtu normálních embryí u párů s Klinefelterovým syndromem (54 vs 77,2 %) [15]. Vzhledem k významně vyšší incidenci abnormalit pohlavního hormonu a autozomálních abnormalit u pacientů s Klinefelterovým syndromem se rozhodně doporučuje preimplantační diagnostika nebo amniocentéza a vyšetření karyotypu.
U mužů s Klinefelterovým syndromem je nezbytné pravidelné sledování (v ideálním případě každý rok) a v případě, že se hladina testosteronu pohybuje v rozmezí hypogonadizmu, je třeba zahájit androgenní nahrazovací terapii. U všech mužů s Klinefelterovým syndromem, kteří podstoupili biopsii varlete pro extrakci spermií, je důležité dlouhodobé sledování endokrinního stavu.
4.2.3 Autozomální abnormality
Genetické poradenství by mělo být nabídnuto všem párům hledajícím léčbu infertility (včetně IVF/ICSI), u nichž je muž nositelem autozomální karyotypové abnormality.
Mezi nejběžnější druhy autozomálních karyotypových abnormalit patří Robertsonovy translokace, reciproční translokace, paracentrická inverze a marker chromozomy. Vzhledem k vyššímu riziku aneuploidie a nevyvážené chromozomální výbavy plodu je nutné testování těchto strukturálních chromozomálních anomálií. Stejně jako u pacientů s Klinefelterovým syndromem umožní FISH analýza spermií přesnější odhad rizika těchto poškození u potomka.
Pokud přistoupíme k IVF/ICSI u pacientů s translokacemi, měla by být stanovena preimplantační genetická diagnóza nebo provedena amniocentéza a analýza karyotypu. Embrya s prokázanou nevyváženou translokací by se neměla používat k implantaci.
4.3 Genetické defekty
4.3.1 Genetické poruchy vázané na chromozom X a mužská infertilita
Muž má pouze jediný chromozom X. To znamená, že recesivní porucha vázaná na chromozom X se projeví u mužů a tento defekt se přenese na jejich dcery, nikoli syny.
4.3.2 Kallmannův syndrom
Nejobvyklejší poruchou vázanou na chromozom X v oblasti infertility je Kallmanův syndrom.
Predominantní formou je recesivní porucha vázaná na chromozom X způsobená mutací na KALIG-1 genu na Xp22.3 [23]. Kallmannův syndrom může mít na svědomí i několik dalších nově identifikovaných autozomálních mutací genů [24].
Pacienti s Kallmanovým syndromem mají hypogonadotropní hypogonadizmus a anosmii, ale mohou se u nich projevit i další klinické znaky jako obličejová asymetrie, rozštěp patra, barvoslepost, hluchota, nesestouplá varlata a renální abnormality. Vzhledem k tomu, že spermatogenezi je možné poměrně snadno stimulovat pomocí hormonální léčby [25], doporučuje se před zahájením terapie rozhodně provést genetický screening.
Léčba pomocí gonadotropinů vede ve většině případů k přirozenému početí, a to dokonce u mužů s poměrně nízkým počtem spermií. Identifikace postiženého genu (vázaného na chromozom X, autozomálně dominantního nebo recesivního) může pomoci stanovit přesnější genetickou informaci, tj. odhad rizika přenosu na potomka.
4.3.3 Mírná forma syndromu androgenní insenzitivity
AR gen je lokalizovaný na dlouhém rameni chromozomu X. Mutace AR genu mohou způsobovat částečnou až úplnou androgenní insenzitivitu [26]. Fenotypově se syndrom úplné androgenní insenzitivity (CAIS) vyznačuje ženským zevním genitálem a absencí pubického ochlupení (Morrisův syndrom). Pro syndrom parciální androgenní insenzitivity je charakteristických několik různých fenotypů – od predominantně ženského fenotypu přes obojaké genitálie až po predominantně mužský fenotyp s mikropenisem, perineální hypospadií a kryptorchizmem. Poslední zmiňovaný fenotyp se rovněž označuje termínem Reifensteinův syndrom. U výše uváděných závažných forem androgenní rezistence nehrozí riziko přenosu na dítě, neboť ani pomocí současných technologií nemohou mít tito muži vlastní biologické potomky. U pacientů s mírnou formou AIS je primárním (nebo v některých případech dokonce jediným) symptomem infertilita. Poruchy androgenního receptoru způsobující infertilitu při současné absenci jakékoli abnormality genitálu jsou velmi vzácné, u infertilních mužů bylo zaznamenáno pouze několik mutací [26–30].
4.3.4 Další poruchy vázané na chromozom X
Na chromozomu X byl identifikován neočekávaně velký počet genů typu specifického pro varlata a typu s obohacenou expresí, zejména jsou hojně zastoupeny na chromozomu X premeiotické geny (ve srovnání s autozomálními chromozomy) [31,32]. Doposud byly u poměrně malé populace testovány pouze dva geny – USP26 a TAF7L – a u žádného z nich nebyla prokázána korelace s mužskou infertilitou [33,34].
4.4 Chromozom Y a mužská infertilita
4.4.1 Úvod
První souvislost mezi azoospermií a mikroskopicky detekovatelnými delecemi na dlouhém rameni chromozomu Y odhalili Tiepolo a Zuffardi v roce 1976 [35]. První případy mikrodelecí chromozomu Y a mužské infertility byly zaznamenány v roce 1992 [36] a od této doby byla publikována řada kazuistik s touto poruchou. Mikrodelece se nalézají ve třech navzájem se nepřekrývajících oblastech chromozomu Y: AZFa+b+c [37].
Několik let po objevu tří oblastí AZF bylo díky přesným znalostem struktury chromozomu Y v Yq11 zjištěno, že oblasti AZFb a AZFc se navzájem překrývají a že oblast AZFd ve skutečnosti neexistuje [38]. Klinicky významné delece odstraňují, částečně anebo ve většině případů úplně, jednu nebo více oblastí AZF a současně představují nejčastější molekulárně genetickou příčinu závažné oligozoospermie a azoospermie [39].
Ve všech AZF oblastech existuje řada kandidátních genů, jejich funkce v rámci spermatogeneze však nebyla zatím prokázána [40].
Vzhledem k tomu, že se delece vyskytují v bloku (tj. odstraňují více než jeden gen), nelze posoudit roli jediného AZF genu ve fenotypu AZF delecí, a není tedy jasné, zda se tyto podílejí na spermatogenezi, nebo nikoli. Delece specifické pro určitý gen, které odstraňují pouze jediný gen, byly popsány pouze v oblasti AZFa. Tyto studie nasvědčují tomu, že gen USP9Y není nezbytný pro spermatogenezi a pravděpodobně slouží pouze k „jemnému doladění“ tvorby spermií [41].
V oblasti AZFc byl popsán nový typ Yq delecí, označovaný jako gr/gr delece [42]. Tyto delece odstraňují polovinu genů v AZFc oblasti a ovlivňují v této oblasti genetické mapování (např. DAZ, CDY1, BPY2).
4.4.2 Klinické implikace mikrodelecí chromozomu Y
O klinickém významu Yq delecí se vzhledem k velké rozmanitosti v četnosti Yq delecí u „fertilních“ mužů již dlouhou dobu diskutuje. Po více než deseti letech klinického výzkumu máme k dispozici následující fakta o delecích na chromozomu Y:
- Tyto se nevyskytují u mužů s normospermií, což jasně prokazuje příčinnou souvislost mezi delecemi na chromozomu Y a selháním spermatogeneze [43].
- Nejvyšší četnost výskytu delecí na chromozomu Y byla zaznamenána u mužů s azoospermií (8–12 %) a oligozoospermií (3–7 %).
- Výskyt delecí je extrémně vzácný u mužů s koncentrací spermií > 5 mil. spermatozoí/ml (přibližně 0,7 %).
- Nejčastější jsou delece v oblasti AZFc (přibližně 65–70 %), následně v oblastech AZFb a AZFb+c nebo AZFa+b+c (25-30 %). Výskyt delecí v oblasti AZFa je extrémně vzácný (5 %).
- Úplné odstranění oblasti AZFa souvisí se závažným testikulárním fenotypem (syndrom pouze ze Sertoliho buněk), zatímco úplná absence oblasti AZFb souvisí se zástavou spermatogeneze. Úplné odstranění oblasti AZFc se projevuje různými fenotypy, od azoospermie až po oligozoospermii.
- Klasické AZF delece nejsou spojeny s rizikem vzniku kryptorchizmu ani testikulárního karcinomu [39].
Výše uváděná specifita a korelace genotypu/fenotypu v praxi znamená, že analýza delecí na chromozomu Y má pro odběr spermií z varlete jak diagnostický, tak prognostický význam. V případě delecí gr/gr nelze hovořit o podobně striktní korelaci genotyp/fenotyp. Tento typ parciální delece AZFc se může vyskytovat rovněž u mužů s normozoospermií, ovšem významně méně často (0,5–1 %) než u mužů s abnormální spermatogenezí (3–5 %). Ve studii zahrnující zatím největší vzorek bělošské populace (> 1 000 mužů) měli nositelé gr/gr delece sedminásobně vyšší riziko, že se u nich rozvine oligozoospermie [44]. Manifestace fenotypu se může u různých etnik lišit v závislosti na pozadí chromozomu Y [45,46]. V nedávné době provedená metaanalýza zahrnující pouze studie nezatížené žádnými metodologickými ani selekčními chybami prokázala u nositelů gr/gr delecí riziko 2,4násobně menší produkce spermií [47]. Jiná studie uvádí gr/gr deleci jako potenciální rizikový faktor pro vznik testikulárního tumoru ze zárodečných buněk [48]. Tyto údaje je však nutné dále potvrdit v kontrolních studiích zahrnujících etnicky a geograficky odpovídající populaci.
Vzhledem k tomu, že delece na chromozomu Y se automaticky přenáší na potomka mužského pohlaví, je nezbytné genetické poradenství. Ve většině případů je u otce a syna přítomna stejná mikrodelece [49–52], ve výjimečných případech však může mít syn rozsáhlejší mikrodeleci [53]. Předpokládá se, že parciální delece AZFc (gr/gr a b2/b3) mohou být predispozicí pro výskyt úplné delece AZFc v následující generaci [54]. Fenotyp syna se může manifestovat řadou způsobů, a rozsah selhání spermatogeneze (v rozmezí azoo-/oligozoospermie) tedy nelze vzhledem k rozdílné genetické výbavě a přítomnosti/absenci environmentálních faktorů, jež mohou mít potenciálně toxický vliv na reprodukční funkci, zcela predikovat. Velký podíl spermatozoí u mužů s úplnou delecí úseku AZFc neobsahuje žádný pohlavní chromozom [55,56]. Tito muži mají potenciální riziko, že se u jejich potomka vyskytne 45,X0 Turnerův syndrom nebo jiná fenotypová anomálie související s mozaikou pohlavního chromozomu, včetně obojakého genitálu.
Screening mikrodelecí chromozomu Y u nositelů karyotypu mozaiky 46,XY/45,X0 s obojakým pohlavím a/nebo znaky Turnerova syndromu prokázal poměrně vysokou incidenci delecí AZFc (33 %) [57]. Existují údaje prokazující spojitost mezi Yq mikrodelecemi a celkovou nestabilitou chromozomu Y, jež vede ke vzniku 45,X0 buněčných linií [58,59]. Navzdory tomuto teoretickému riziku mají děti narozené otcům s Yq mikrodelecemi normální fenotyp [39,60], což lze pravděpodobně vysvětlit nižší mírou implantace a zřejmě vyšším rizikem spontánní potratovosti embryí s karyotypem 45,X0.
V případě, že ICSI podstoupí muž s mikrodelecemi chromozomu Y, je třeba u jeho synů dlouhodobě monitorovat stav fertility a zvážit kryoprezervaci spermatozoí v mladším věku. Dosud však byl popsán jediný případ [48] vyššího rizika vzniku testikulárního karcinomu ze zárodečných buněk u nositelů gr/gr delece. Preventivní opatření (např. ultrazvukové vyšetření varlat) bude tedy u synů nositelů gr/gr delecí nutné zvážit pouze v případě, že bude toto riziko potvrzeno v dalších studiích.
4.4.2.1 Vyšetření mikrodelecí na chromozomu Y
Screening delecí AZF je indikován na základě počtu spermií a provádí se u mužů s azoospermií a závažnou oligozoospermií (< 5 mil. spermatozoí/ml). Díky guidelines Evropské andrologické akademie (EAA) [60] a programu pro externí kontrolu kvality EAA/EMQN (European Molecular Genetics Quality Network) (http://www.emqn.org/emqn/) bylo testování Yq více standardizováno a poskytuje spolehlivý výsledek v různých genetických laboratořích. EAA guidelines uvádí seznam primerů umožňujících detekovat > 95 % klinicky relevantních delecí [60]. Každý primer zahrnuje dva markery pro každou oblast a kontrolní markery z Yp a chromozomu X. Významná variabilita ve frekvenci výskytu delecí (uváděná v prvních studiích) byla pravděpodobně důsledkem technických problémů a nespolehlivých markerů spíše než odrazem skutečného rozdílu v četnosti výskytu u různých etnik.
4.4.2.2 Chromozom Y: gr/gr delece
V oblasti AZFc byl popsán nový typ Yq delecí označovaný jako gr/gr delece [42]. Tyto delece odstraňují polovinu genů v AZFc oblasti, což ovlivňuje genetické mapování v této oblasti. Studie zahrnující dosud největší počet bělošské populace prokázala, že nositelé gr/gr delece mají téměř osminásobně vyšší riziko oligozoospermie (OR = 7,9, 95% CI: 1,8–33,8; p < 0,001) [43]. Četnost výskytu gr/gr delece u pacientů s oligozoospermií činí přibližně 4 %. Čtyři metaanalýzy prokázaly, že gr/gr delece představuje významný rizikový faktor pro narušení funkce produkce spermií [61,62].
Frekvence výskytu gr/gr delece a odpovídající fenotypová reprezentace se však mohou mezi jednotlivými etniky lišit v závislosti na pozadí chromozomu Y. Například v některých haploskupinách chromozomu Y je výskyt delece korigován a pravděpodobně nemá žádný negativní vliv na spermatogenezi. Rutinní provádění screeningu gr/gr delece je stále předmětem debaty zejména v laboratořích obsluhujících z etnického a geografického hlediska rozmanitou populaci.
4.4.2.3 Závěry
Testování mikrodelecí není nezbytné u mužů s obstrukční azoospermií, kteří podstupují ICSI, neboť spermatogeneze by u těchto mužů měla být normální.
Pro muže s vážně poškozenou spermatogenezí (< 5 mil. spermatozoí/ml) je ovšem testování Yq mikrodelecí žádoucí jak z diagnostických, tak prognostických důvodů. Mikrodelece na Yq mají rovněž význam pro genetické vyšetření (viz níže).
V případě detekce kompletních mikrodelecí v oblasti AZFa nebo AZFb nestojí za to indikovat mikrotestikulární extrakci spermií vzhledem k téměř nulové šanci detekce jakýchkoli spermií.
Bylo prokázáno, že gr/gr delece představují signifikantní rizikový faktor pro poruchu spermatogeneze, je třeba však nashromáždit další důkazy týkající se prognostického významu gr/gr delece a vzniku TCGTs.
Pokud si muž s mikrodelecí a jeho partnerka přejí přistoupit k ICSI, je třeba je upozornit, že mikrodelece se přenáší na syny a nikoli na dcery.
Syn, který zdědí mikrodeleci po svém otci, bude mít abnormální spermatogenezi, protože u mužů s normozoospermií nebyl zaznamenán výskyt kompletních AZF delecí.
4.4.3 Autozomální defekty se závažnými abnormalitami fenotypu a infertilita
Některé dědičné poruchy souvisejí se závažnými nebo generalizovanými abnormalitami a infertilitou (tab. 6). Lékaři jsou o těchto defektech informováni často již od útlého dětství pacienta. Na infertilitu je třeba u těchto pacientů pohlížet v kontextu s celkovým zdravotním stavem a schopností párů vychovávat potomka.
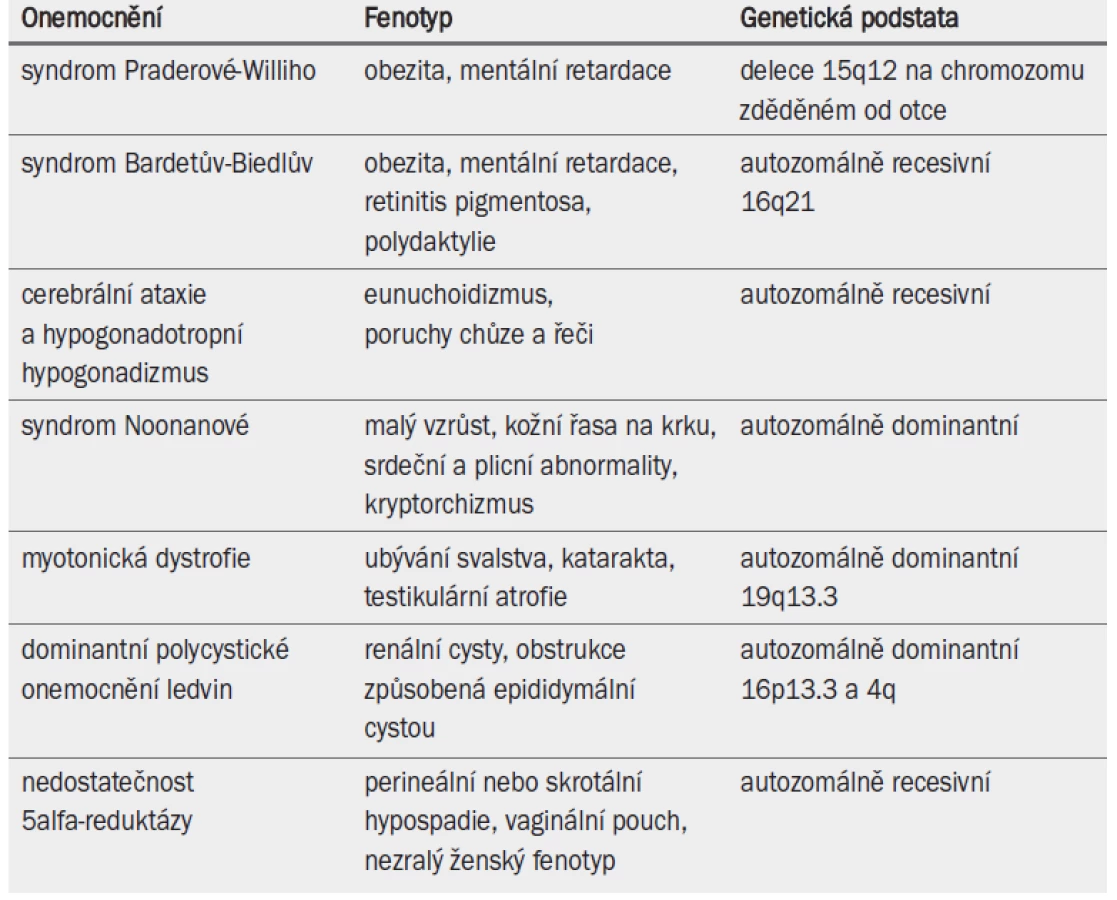
4.5 Mutace cystické fibrózy a mužská infertilita
Cystická fibróza (CF) je fatální autozomálně recesivní porucha. Jedná se o nejběžnější genetické onemocnění bělochů; 4 % pacientů jsou nositelé genových mutací zahrnujících gen transmembránového regulátoru vodivosti cystické fibrózy (CFTR). Tento gen se nachází na krátkém rameni chromozomu 7 a kóduje membránový protein, který funguje jako iontový kanál, a dále ovlivňuje tvorbu ejakulačního vývodu, semenného váčku, chámovodu a distálních dvou třetin nadvarlete.
Vrozená bilaterální absence chámovodu (CBAVD) souvisí s mutacemi genu CFTR a trpí jí přibližně 2 % mužů s obstrukční azoospermií, kteří navštěvovali kliniku v Edinburgu [63]. Incidence u mužů s obstrukční azoospermií se však v jednotlivých zemích liší. Chybějící chámovod je snadné přehlédnout, a proto by měli být všichni muži s azoospermií – zvláště muži s objemem ejakulátu < 1,5 ml a pH nižším než 7,0 – důkladně vyšetřeni, aby bylo možné vyloučit CBAVD.
V současné době je v databázi CFTR uvedeno přibližně 1 500 mutací (http://www.genet.sickkids.on.ca/cftr/). Bylo publikováno mnoho studií zabývajících se muži s CBAVD, kteří byli testováni na různý počet mutací. Obecně platí, že čím více mutací je testováno, tím vyšší podíl pacientů s mutacemi bude zjištěn. V přehledu publikovaných studií zahrnujícím 449 mužů s CBAVD byla u 244 mužů detekována delta F508 mutace, u 54 mužů R117H mutace a u 37 mužů W1282X mutace; u 1–9 mužů bylo nalezeno 63 jiných mutací, ve všech kazuistikách však nebyly testovány všechny mutace [64]. Zdá se, že čím více mutací bude definováno a testováno, tím více se procento mužů s CBAVD, u kterých budou mutace objeveny, blíží 100 %. V současnosti není praktické testovat muže na všechny známé mutace, neboť některé mají velmi nízký výskyt v určité populaci. Testování je obvykle omezeno na mutace vyskytující se nejčastěji v určité populaci.
Mutace se mohou vyskytovat v obou kopiích genu CFTR, ale u většiny mužů s CBAVD se vyskytují pouze v jedné kopii. V některých pravděpodobně heterozygotních případech může být přítomna neznámá druhá mutace, existuje zde však další mechanizmus. U dvou třetin mužů s CBAVD může být detekována varianta DNA-pátá alela – v nekódující oblasti CFTR [65]. Vzhledem k tomu, že varianta 5t je považována spíše za mírnou formu mutace CFTR než za polymorfizmus, doporučuje se testovat ji u všech pacientů s CBAVD.
Muži s CBAVD mají často méně závažné klinické znaky cystické fibrózy (např. anamnézu plicních infekcí). Je proto velmi důležité sledovat děti narozené po ICSI, jejichž otec má CBAVD (ať heterozygotní, nebo homozygotní).
Pokud má muž CBAVD, je důležité testovat jeho i jeho partnerku na mutace cystické fibrózy. Pokud je i žena nositelkou, musí pár velmi důkladně zvážit, zda přistoupit k ICSI s použitím mužových spermií, neboť šance, že dítě bude mít cystickou fibrózu, činí 25 %, pokud je muž heterozygotní, a 50 %, pokud je homozygotní. V případě, že má partnerka negativní výsledek testování známých mutací, je pravděpodobnost, že bude nositelkou neznámých mutací, přibližně 0,4 %.
4.6 Unilaterální nebo bilaterální absence/abnormalita chámovodu a renální anomálie
Unilaterální absence chámovodu je obvykle spojena s ipsilaterální absencí ledviny [66] a má zřejmě různé genetické příčiny. Muži s unilaterální absencí chámovodu jsou obvykle fertilní a tento stav je nejčastěji odhalen incidentálně při vazektomii.
Přesto může být u mužů s unilaterální absencí chámovodu mutace cystické fibrózy zapříčiněna stejným primárním genetickým onemocněním jako u mužů se skutečnou CBAVD. U mužů s bilaterální absencí chámovodu a renálními abnormalitami nebyly abnormality genu CFTR prokázány [67].
Testování na mutace cystické fibrózy se doporučuje u mužů s unilaterální absencí chámovodu a normálními ledvinami i u mužů s bilaterální absencí nebo bilaterálními abnormalitami. V případě, že jsou výsledky negativní a nebyla definována renální anatomie, stojí za to provést abdominální ultrazvukové vyšetření. Nálezy mohou zahrnovat unilaterální absenci chámovodu s ipsilaterální absencí ledviny až bilaterální cévní abnormality a renální abnormality, jako je pánevní dystopie ledviny.
4.7 Neznámé genetické poruchy
Pokud uvážíme vysoký odhadovaný počet genů podílejících se na gametogenezi u mužů, je pravděpodobné, že většina „idiopatických“ forem poruch spermatogeneze je zapříčiněna mutacemi nebo polymorfizmem genů spermatogeneze [34]. Navzdory intenzivnímu výzkumu zaměřenému na nové genetické faktory nebyly dosud identifikovány žádné klinicky relevantní genové mutace ani typy polymorfizmu (kromě mutací vázaných na chromozom Y) [34,68,69]. Příslibem významnějších pokroků v této oblasti může být implementace nových analytických přístupů [70,71].
ICSI zprostředkovává otcovství mužům s vážně poškozenou spermatogenezí v situacích, které byly dříve považovány za beznadějné, a v případech, kdy lze získat pouze malé množství spermatozoí.
To vede k obavám, že se narodí dítě s abnormalitou, neboť ICSI může obejitím selektivních procesů ženského genitálního ústrojí umožnit defektním spermiím oplodnit vajíčko. A naopak – mohou být oplodněna i ta vajíčka, která by za normálních okolností nebyla oplodnění schopna.
Statistiky plodových abnormalit z ICSI center však naštěstí neprokazují v porovnání s normální populací zvýšený výskyt vrozených malformací. Na druhou stranu však děti počaté pomocí techniky ICSI mají vyšší riziko vzniku de novo aberací pohlavního chromozomu (přibližně trojnásobně vyšší riziko ve srovnání s dětmi počatými přirozenou cestou) a strukturálních abnormalit zděděných z otcovy strany [72–74]. Indikace pro ICSI se neustále rozšiřují a zahrnují v současné době i techniky oplodnění nezralými spermiemi. Je tedy nesmírně důležité monitorovat nadále míru výskytu plodových abnormalit s detailní podskupinovou analýzou podle klinické a molekulární diagnózy otce.
4.8 Fragmentace DNA u spermatozoí
U mužů s oligospermií obsahují spermatozoa významnější poškození DNA, jež s sebou nese nižší šanci na přirozené početí a v menší míře i nižší šanci na početí pomocí IVF/ICSI a častější výskyt potratovosti v časném stadiu těhotenství [75,76]. Ke zmírnění poškození DNA může dojít po operaci varikokély [77,78].
4.9 Genetické poradenství a ICSI
Nejlepším postupem je zvolit léčbu společně s pacientem a jeho partnerkou a poskytnout jim kompletní informace o genetických rizicích. Hned na počátku je třeba pár důkladně informovat o rizicích, která potenciálně hrozí jejich potomkovi, aby se mohli lépe rozhodnout, zda k ICSI přistoupit, nebo nikoli. V případě konfliktu mezi přáním páru a zájmem budoucího dítěte je z etického hlediska správné terapii odmítnout. V případě, kdy jsou oba partneři nositeli defektů (tj. mutace cystické fibrózy), existuje až 50% pravděpodobnost narození dítěte s klinickým onemocněním a časného úmrtí po řadě let morbidity. Mnoho lékařů a zdravotnický personál na klinikách pro léčbu infertility se může domnívat, že jejich povinnost péče o budoucí dítě a zájmy společnosti převáží přání páru samého a že není etické k léčbě přistoupit. Pokud dojde ke konfliktu, který není možno vyřešit dohodou, zájmy budoucího dítěte mají obvykle přednost před zájmy páru. Dvojice musí navíc zvážit provedení preimplantační diagnózy a náhradu pouze normálních embryí.
4.10 Závěry a doporučení pro infertilní pacienty s genetickými poruchami
Nové poznatky týkající se genetického hlediska infertility a zavádění ICSI vyžadují porozumění základům genetiky jak na straně lékařů, tak široké veřejnosti.
Pokroky v diagnostických metodách umožní identifikovat genetické příčiny u více onemocnění a zároveň detekovat známé poruchy za nižší cenu; u některých onemocnění může být vhodná genová terapie.
Všem mužům s poškozenou spermatogenezí (< 10 mil. spermatozoí/ml), kteří mají zájem o léčbu pomocí in vitro fertilizace/intracytoplazmatické injekce spermií (ICSI), by měla být nabídnuta standardní analýza karyotypu [2] (stupeň doporučení: B).
Muži s Klinefelterovým syndromem mohou vyžadovat v průběhu stárnutí androgenní substituční terapii (stupeň doporučení: B).
U všech mužů s Klinefelterovým syndromem, kteří podstoupili biopsii varlete pro extrakci spermií, je důležité dlouhodobé sledování endrokrinního stavu (stupeň doporučení: B).
U mužů s vážně poškozenou spermatogenezí (< 5 mil. spermatozoí/ml) je velmi žádoucí testování mikrodelecí Yq [39,60] (stupeň doporučení: B).
V případě, že jsou u muže přítomny strukturální abnormality chámovodu (bilaterální a unilaterální absence chámovodu), je důležité provést u něj i jeho partnerky testování mutací genu CF [64] (stupeň doporučení: A).
Genetické poradenství je nezbytné u párů, u nichž byla při klinickém nebo genetickém vyšetření zjištěna genetická abnormalita, a u pacientů, kteří jsou nositeli (potenciálně) dědičného onemocnění [1] (stupeň doporučení: A).
5. Obstrukční azoospermie
Zdroje
1. Griffin DK, Finch KA. The genetic and cytogenetic basis of male infertility. Human Fertil Mar 2005; 8(1): 19–26. http://www.ncbi.nlm.nih.gov/pubmed/15823847.
2. Carrell DT. The clinical implementation of sperm chromosome aneuploidy testing: pitfalls and promises. J Androl 2008; 29(2): 124–133. http://www.ncbi. nlm.nih.gov/pubmed/17881765.
3. Johnson MD. Genetic risks of intracytoplasmic sperm injection in the treatment of male infertility: recommendations for genetic counseling and screening. Fertil Steril 1998; 70(3): 397–411. http://www.ncbi.nlm.nih.gov/pubmed/9757865.
4. van Assche EV, Bonduelle M, Tournaye H et al. Cytogenetics of infertile men. Hum Reprod 1996; 11 (Suppl 4): 1–24. http://www.ncbi.nlm.nih.gov/pubmed/9147109.
5. Vincent MC, Daudin M, De MP et al. Cytogenetic investigations of infertile men with low sperm counts: a 25-year experience. J Androl 2002; 23(1): 18–22. http://www.ncbi.nlm.nih.gov/pubmed/11780918.
6. Tempest HG, Martin RH. Cytogenetic risks in chromosomally normal infertile men. Curr Opin Obstet Gynecol 2009; 21(3): 223–227. http://www.ncbi.nlm.nih.gov/pubmed/19424064.
7. Clementini E, Palka C, Iezzi I et al. Prevalence of chromosomal abnormalities in 2078 inferitle couples referred for assisted reproduction techniques. Hum Reprod 2005; 20(2): 437–442. http://www.ncbi.nlm.nih.gov/pubmed/15567875.
8. Gianaroli L, Magli MC, Cavallini G et al. Frequency of aneuploidy in sperm from patients with extremely severe male factor infertility. Hum Reprod 2005; 20(8): 2140–2152. http://www.ncbi.nlm.nih.gov/pubmed/15845594.
9. Pang MG, Kim YJ, Lee SH et al. The high incidence of meiotic errors increases with decreased sperm count in severe male factor infertilities. Hum Reprod 2005; 20(6): 1688–1694. http://www.ncbi.nlm.nih.gov/pubmed/15734753.
10. Machev N, Gosset P, Viville S. Chromosome abnormalities in sperm from infertile men with normal somatic karyotypes: teratozoospermia. Cytogenet Genome Res 2005;111 (3–4): 352–357. http://www.ncbi.nlm.nih.gov/pubmed/16192715.
11. Baccetti B, Collodel G, Marzella R et al. Ultrastructural studies of spermatozoa from infertile males with Robertsonian translocations and 18, X, Y aneuploidies. Hum Reprod 2005; 20(8): 2295–2300. http://www.ncbi.nlm.nih.gov/pubmed/15878922.
12. Lanfranco F, Kamischke A, Zitzmann M et al. Klinefelter’s syndrome. Lancet 2004; 364(9430): 273–283. http://www.ncbi.nlm.nih.gov/pubmed/15262106.
13. Wang C, Baker HW, Burger HG et al. Hormonal studies in men with Klinefelter’s syndrome. Clin Endocrinol (Oxf) 1975; 4(4): 399–411. http://www.ncbi.nlm.nih.gov/pubmed/1157343.
14. Ichioka K, Utsunomiya N, Kohei N et al. Adult onset of declining spermatogenesis in a man with nonmosaic Klinefelter’s syndrome. Fertil Steril 2006; 85(5): 1511.e1–2. http://www.ncbi.nlm.nih.gov/pubmed/16616747.
15. Staessen C, Tournaye H, Van Assche E et al. PGD in 47,XXY Klinefelter’s syndrome patients. Hum Reprod Update 2003; 9(4): 319–330. http://www.ncbi.nlm.nih.gov/pubmed/12926526.
16. Chevret E, Rousseaux S, Monteil M, et al. Increased incidence of hyperhaploid 24 XY spermatozoa detected by three-colour FISH in a 46,XY/47,XXY male. Hum Genet 1996; 97(2): 171–175. http://www.ncbi.nlm.nih.gov/pubmed/8566948.
17. Martini E, Geraedts JP, Liebaers I et al. Constitution of semen samples from XYY and XXY males as analysed by in-situ hybridization. Hum Reprod 1996; 11(8): 1638–1643. http://www.ncbi.nlm.nih.gov/pubmed/8921108.
18. Lenz P, Luetjens CM, Kamischke A et al. Mosaic status in lymphocytes of infertile men with or without Klinefelter syndrome. Hum Reprod 2005; 20(5): 1248–1255. http://www.ncbi.nlm.nih.gov/pubmed/15665007.
19. Cozzi J, Chevret E, Rousseaux S et al. Achievement of meiosis in XXY germ cells: study of 543 sperm karyotypes from an XY/XXY mosaic patient. Hum Genet 1994; 93(1): 32–34. http://www.ncbi.nlm.nih.gov/pubmed/8270252.
20. Guttenbach M, Michelmann HW, Hinney B et al. Segregation of sex chromosomes into sperm nuclei in a man with 47,XXY Klinefelter’s karyotype: a FISH analysis. Hum Genet 1997; 99(4): 474–477. http:// www.ncbi.nlm.nih.gov/pubmed/9099836.
21. Estop AM, Munne S, Cieply KM et al. Meiotic products of a Klinefelter 47,XXY male as determined by sperm fluorescence in-situ hybridization analysis. Hum Reprod 1998; 13(1): 124–127. http://www.ncbi.nlm.nih.gov/pubmed/9512242.
22. Foresta C, Galeazzi C, Bettella A et al. High incidence of sperm sex chromosomes aneuploidies in two patients with Klinefelter’s syndrome. J Clin Endocrinol Metab 1998; 83(1): 203–205. http://www.ncbi.nlm.nih.gov/pubmed/9435442.
23. Franco B, Guioli S, Pragliola A et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 1991; 353(6344): 529–536. http://www.ncbi.nlm.nih.gov/pubmed/1922361.
24. Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol 2009; 5(10): 569–576. http://www.ncbi.nlm.nih.gov/pubmed/19707180.
25. Miyagawa Y, Tsujimura A, Matsumiya K et al. Outcome of gonadotropin therapy for male hypogonadotropic hypogonadism at university affiliated male infertility centers: a 30-year retrospective study. J Urol 2005; 173(6): 2072–2075. http://www.ncbi.nlm.nih.gov/pubmed/15879837.
26. Gottlieb B, Beitel LK, Wu JH et al. The androgen receptor gene mutations database (ARDB): 2004 update. Hum Mutat 2004; 23(6): 527–533. http://www.ncbi.nlm.nih.gov/pubmed/15146455.
27. Tincello DG, Saunders PT, Hargreave TB. Preliminary investigations on androgen receptor gene mutations in infertile men. Mol Hum Reprod 1997; 3(11): 941–943. http://www.ncbi.nlm.nih.gov/pubmed/ 9433918.
28. Gottlieb B, Lombroso R, Beitel LK et al. Molecular pathology of the androgen receptor in male (in) fertility. Reprod Biomed Online 2005; 10(1): 42–48. http://www.ncbi.nlm.nih.gov/pubmed/15705293.
29. Ferlin A, Vinanzi C, Garolla A et al. Male infertility and androgen receptor gene mutations: clinical features and identification of seven novel mutations. Clin Endocrinol (Oxf) 2006; 65(5): 606–610. http://www.ncbi.nlm.nih.gov/pubmed/17054461.
30. Rajender S, Singh L, Thangaraj K. Phenotypic heterogeneity of mutations in androgen receptor gene. Asian J Androl 2007; 9(2): 147–179. http://www.ncbi.nlm.nih.gov/pubmed/17334586.
31. Wang PJ, McCarrey JR, Yang F et al. An abundance of X-linked genes expressed in spermatogonia. Nat Genet 2001; 27(4): 422–426. http://www.ncbi.nlm.nih.gov/pubmed/11279525.
32. Wang PJ. X chromosomes, retrogenes and their role in male reproduction. Trends Endocrinol Metab 2004; 15(2): 79–83. http://www.ncbi.nlm.nih.gov/pubmed/15036254.
33. Stouffs K, Tournaye H, Liebaers I et al. Male infertility and the involvement of the X chromosome. Hum Reprod Update 2009; 15(6): 623–637. http://www.ncbi.nlm.nih.gov/pubmed/19515807.
34. Nuti F, Krausz C. Gene polymorphisms/mutations relevant to abnormal spermatogenesis. Reprod Biomed Online 2008; 16(4): 504–513. http://www.ncbi.nlm.nih.gov/pubmed/18413059.
35. Tiepolo L, Zuffardi O. Localization of factors controlling spermatogenesis in the nonfluorescent portion of the human Y chromosome long arm. Hum Genet 1976; 34(2): 119–124. http://www.ncbi.nlm.nih.gov/pubmed/1002136.
36. Ma K, Sharkey, A, Kirsch S et al. Towards the molecular localisation of the AZF locus: mapping of microdeletions in azoospermic men within 14 subintervals of interval 6 of the human Y chromosome. Hum Mol Genet 1992; 1(1): 29–33. http://www.ncbi.nlm.nih.gov/pubmed/1301132.
37. Vogt P, Edelmann A, Kirsch S et al. Human Y chromosome azoospermia factors (AZF) mapped to different subregions in Yq11. Hum Mol Genet 1996; 5(7): 933–943. http://www.ncbi.nlm.nih.gov/pubmed/8817327.
38. Repping S, Skaletsky H, Lange J et al. Recombination between palindromes P5 and P1 on the human Y chromosome causes massive deletions and spermatogenic failure. Am J Hum Genet 2002; 71: 906–922. http://www.ncbi.nlm.nih.gov/pubmed/12297986.
39. Krausz C, Degl’Innocenti S. Y chromosome and male infertility: update, 2006. Front Biosci 2006; 11: 3049–3061. http://www.ncbi.nlm.nih.gov/pubmed/16720375.
40. Skaletsky H, Kuroda-Kawaguchi T, Minx PJ et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 2003; 423(6942): 825–837. http://www.ncbi.nlm.nih.gov/pubmed/12815422.
41. Tyler-Smith C, Krausz C. The will-o’-the-wisp of genetics--hunting for the azoospermia factor gene. N Engl J Med 2009; 360(9): 925–927. http://www.ncbi.nlm.nih.gov/pubmed/19246366.
42. Repping S, Skaletsky H, Brown L et al. Polymorphism for a 1.6-Mb deletion of the human Y chromosome persists through balance between recurrent mutation and haploid selection. Nat Genet 2003; 35(3): 247–251. http://www.ncbi.nlm.nih.gov/pubmed/14528305.
43. Krausz C, Forti G, McElreavey K. The Y chromosome and male fertility and infertility. Int J Androl 2003; 26(2): 70–75. http://www.ncbi.nlm.nih.gov/pubmed/12641824.
44. Giachini C, Laface I, Guarducci E et al. Partial AZFc deletions and duplications: clinical correlates in the Italian population. Hum Genet 2008; 124(4): 399–410. http://www.ncbi.nlm.nih.gov/pubmed/18807255.
45. Vogt PH. AZF deletions and Y chromosomal haplogroups: history and update based on sequence. Hum Reprod Update 2005; 11(4): 319–336. http://www.ncbi.nlm.nih.gov/pubmed/15890785.
46. Krausz C, Giachini C, Xue Y et al. Phenotypic variation within European carriers of the Y-chromosomal gr/gr deletion is independent of Y-chromosomal background. J Med Genet 2009; 46(1): 21–31. http://www.ncbi.nlm.nih.gov/pubmed/18782837.
47. Visser L, Westerveld GH, Korver CM et al. Y chromosome gr/gr deletions are a risk factor for low semen quality. Hum Reprod 2009; 24(10): 2667–2673. http://www.ncbi.nlm.nih.gov/pubmed/19602516.
48. Nathanson KL, Kanetsky PA, Hawes R et al. The Y deletion gr/gr and susceptibility to testicular germ cell tumor. Am J Hum Genet 2005; 77(6): 1034–1043. http://www.ncbi.nlm.nih.gov/pubmed/16380914.
49. Mulhall JP, Reijo R, Alagappan R et al. Azoospermic men with deletion of the DAZ gene cluster are capable of completing spermatogenesis: fertilization, normal embryonic development and pregnancy occur when retrieved testicular spermatozoa are used for intracytoplasmic sperm injection. Hum Reprod 1997; 12(3): 503–508. http://www.ncbi.nlm.nih.gov/pubmed/9130751.
50. Silber SJ, Alagappan R, Brown LG et al. Y chromosome deletions in azoospermic and severely oligozoospermic men undergoing intracytoplasmic sperm injection after testicular sperm xtraction. Hum Reprod 1998; 13(12): 3332–3337. http://www.ncbi.nlm.nih.gov/pubmed/9886509.
51. Kamischke A, Gromoll J, Simoni M et al. Transmisson of a Y chromosomal deletion involving the deleted in azoospermia (DAZ) and chromodomain (CDYI) genes from father to son through intracytoplasmic sperm injection: case report. Hum Reprod 1999; 14(9): 2320–2322. http://www.ncbi.nlm.nih.gov/pubmed/10469702.
52. Mau Kai C, Juul A, McElreavey K et al. Sons conceived by assisted reproduction techniques inherit deletions in the azoospermia factor (AZF) region of the Y chromosome and the DAZ gene copy number. Hum Reprod 2008; 23(7): 1669–1678. http://www.ncbi.nlm.nih.gov/pubmed/18440997.
53. Stuppia L, Gatta V, Calabrese G et al. A quarter of men with idiopathic oligo-azospermia display chromosomal abnormalities and microdeletions of different types in interval 6 of Yq11. Hum Genet 1998; 102(5): 566–570. http://www.ncbi.nlm.nih.gov/pubmed/9654206.
54. Zhang F, Lu C, Li Z et al. Partial deletions are associated with an increased risk of complete deletion in AZFc: a new insight into the role of partial AZFc deletions in male infertility. J Med Genet 2007; 44(7): 437–444. http://www.ncbi.nlm.nih.gov/pubmed/17412880.
55. Siffroi JP, Le Bourhis C, Krausz C et al. Sex chromosome mosaicism in males carrying Y chromosome long arm deletions. Hum Reprod 2000; 15(12): 2559–2562. http://www.ncbi.nlm.nih.gov/pubmed/11098026.
56. Jaruzelska J, Korcz A, Wojda A et al. Mosaicism for 45,X cell line may accentuate the severity of spermatogenic defects in men with AZFc deletion. J Med Genet 2001; 38(11): 798–802. http://www.ncbi.nlm.nih.gov/pubmed/11732492.
57. Patsalis PC, Sismani C, Quintana-Murci L et al. Effects of transmission of Y chromosome AZFc deletions. Lancet 2002; 360(9341): 1222–1224. http:// www.ncbi.nlm.nih.gov/pubmed/12401251.
58. Patsalis PC, Skordis N, Sismani C et al. Identification of high frequency of Y chromosome deletions in patients with sex chromosome mosaicism and correlation with the clinical phenotype and Y-chromosome instability. Am J Med Genet A 2005; 135(2): 145– 149. http://www.ncbi.nlm.nih.gov/pubmed/15880425.
59. Le Bourhis C, Siffroi JP, McElreavey K et al. Y chromosome microdeletions and germinal mosaicism in infertile males. Mol Hum Reprod 2000; 6(8): 688–693. http://www.ncbi.nlm.nih.gov/pubmed/10908277.
60. Simoni M, Bakker E, Krausz C. EAA/EMQN best practice guidelines for molecular diagnosis of y-chromosomal microdeletions. State of the art 2004. Int J Androl 2004; 27(4): 240–249. http://www.ncbi.nlm.nih.gov/pubmed/15271204.
61. Stouffs K, Lissens W, Tournaye H et al. What about gr/gr deletions and male infertility? Systematic review and meta-analysis. Hum Reprod Update 2011; 17(2): 197–209. http://www.ncbi.nlm.nih.gov/pubmed/20959348.
62. Navarro-Costa P, Goncalves J, Plancha CE. The AZFc region of the Y chromosome: at the crossroad between genetic diversity and male infertility. Hum Reprod Update 2010; 16(5): 525–542. http://www.ncbi.nlm.nih.gov/pubmed/20304777.
63. Donat R, McNeill AS, Fitzpatrick DR et al. The incidence of cystic fibrosis gene mutations in patients with congenital bilateral absence of the vas deferens in Scotland. Br J Urol 1997; 79(1): 74–77. http://www.ncbi.nlm.nih.gov/pubmed/9043501.
64. De Braekeleer M, Ferec C. Mutations in the cystic fibrosis gene in men with congenital bilateral absence of the vas deferens. Mol Hum Reprod 1996; 2(9): 669–677. http://www.ncbi.nlm.nih.gov/pubmed/ 9239681.
65. Chillon M, Casals T, Mercier B et al. Mutations in cystic fibrosis gene in patients with congenital absence of the vas deferens. New Engl J Med 1995; 332(22): 1475–1480. http://www.ncbi.nlm.nih.gov/ pubmed/7739684.
66. Drake MJ, Quinn FM. Absent vas deferens and ipsilateral multicystic dysplastic kidney in a child. Br J Urol 1996; 77(5): 756–757. http://www.ncbi.nlm.nih.gov/pubmed/8689131.
67. Augarten A, Yahav Y, Kerem BS et al. Congenital bilateral absence of the vas deferens in the absence of cystic fibrosis. Lancet 1994; 344(8935): 1473–1474. http://www.ncbi.nlm.nih.gov/pubmed/7968122.
68. Krausz C, Giachini C. Genetic risk factors in male infertility. Arch Androl 2007; 53(3): 125–133. http://www.ncbi.nlm.nih.gov/pubmed/17612870.
69. Tuttelmann F, Rajpert-De Meyts E, Nieschlag E et al. Gene polymorphisms and male infertility—a meta-analysis and literature review. Reprod Biomed Online 2007; 15(6): 643–658. http://www.ncbi.nlm.nih.gov/pubmed/18062861.
70. Aston KI, Carrell DT. Genome-wide study of single-nucleotide polymorphisms associated with azoospermia and severe oligozoospermia. J Androl 2009; 30(6): 711–725. http://www.ncbi.nlm.nih.gov/pubmed/19478329.
71. Carrell DT, De Jonge C, Lamb DJ. The genetics of male infertility: a field of study whose time is now. Arch Androl 2006; 52(4): 269–274. http://www.ncbi.nlm.nih.gov/pubmed/16728342.
72. Van Steirteghem A, Bonduelle M, Devroey P et al. Follow-up of children born after ICSI. Hum Reprod Update 2002; 8(2): 111–116. http://www.ncbi. nlm.nih.gov/pubmed/12099626.
73. Bonduelle M, Van Assche E, Joris H et al. Prenatal testing in ICSI pregnancies: incidence of chromosomal anomalies in 1586 karyotypes and relation to sperm parameters. Hum Reprod 2002; 17(10): 2600–2614. http://www.ncbi.nlm.nih.gov/pubmed/12351536.
74. ESHRE Capri Workshop group Intracytoplasmic sperm injection (ICSI) in 2006: evidence and evolution. Hum Reprod Update 2007; 13(6): 515–526. http://www.ncbi.nlm.nih.gov/pubmed/17630396.
75. Zini A, Meriano J, Kader K et al. Potential adverse effect of sperm DNA damage on embryo quality after ICSI. Hum Reprod 2005; 20(12): 3476–3480. http://www.ncbi.nlm.nih.gov/pubmed/16123087.
76. Zini A, Sigman M. Are tests of sperm DNA damage clinically useful? Pros and cons. J Androl 2009; 30(3): 219–229. http://www.ncbi.nlm.nih.gov/pubmed/19059901.
77. Zini A, Blumenfeld A, Libman J et al. Beneficial effect of microsurgical varicocelectomy on human sperm DNA integrity. Hum Reprod 2005; 20(4): 1018–1021. http://www.ncbi.nlm.nih.gov/pubmed/15608026.
78. Smit M, Romijn JC, Wildhagen MF et al. Decreased sperm DNA fragmentation after surgical varicocelectomy is associated with increased pregnancy rate. J Urol 2010; 183(1): 270–274. http://www.ncbi.nlm.nih.gov/pubmed/19913801.
Štítky
Detská urológia UrológiaČlánok vyšiel v časopise
Urologické listy

2012 Číslo 4
- Vyšetření T2:EGR a PCA3 v moči při záchytu agresivního karcinomu prostaty
- Lék v boji proti benigní hyperplazii prostaty nyní pod novým názvem Adafin
Najčítanejšie v tomto čísle
- Vedlejší účinky hormonální substituční léčby testosteronem
- Adenomatoidní tumor varlete – diagnostika a doporučené operační postupy
-
Guidelines EAU pro léčbu mužské infertility
13. Poruchy ejakulace -
Guidelines EAU pro léčbu mužské infertility
5. Obstrukční azoospermie