
Kongenitální adrenální hyperplazie, defekt 17α−hydroxylázy jako vzácná příčina hypertenze a hypokalemie
Congenital adrenal hyperplasia, defect of 17α-hydroxylase as a rare cause of hypertension and hypocalaemia
Authors describe infrequently occurring case of congenital adrenal hyperplasia, partial defect of 17α-hydroxylase in female patient with hypertension, hypocalaemia, menstrual cycle disorders and sterility. Diagnosis was established on the basis of native examination of steroid spectrum together with both stimulation and suppression tests and CT examination. After the treatment with dexamethason was established, the blood pressure and electrolyte levels reached the correction. This report supports the importance of the whole steroid spectrum examination in the patients with this symptomatology in order to eliminate rare causes of endocrine hypertension.
Key words:
congenital adrenal hyperplasia – endocrine hypertension – hypocalaemia – defect of 17α-hydroxylase
Autoři:
V. Olšovská 1; J. Šulcová 2; A. Hondlová 1; H. Šiprová 1
Působiště autorů:
II. interní klinika Lékařské fakulty MU a FN u sv. Anny, Brno, přednosta doc. MUDr. Miroslav Souček, CSc.
1; Oddělení steroidních hormonů Endokrinologického ústavu, Praha, ředitel doc. MUDr. Vojtěch Hainer, CSc.
2
Vyšlo v časopise:
Vnitř Lék 2005; 51(2): 226-230
Kategorie:
Kazuistiky
Souhrn
Autoři popisují řídce se vyskytující případ kongenitální adrenální hyperplazie, parciálního defektu 17α-hydroxylázy u pacientky s hypertenzí, hypokalemií, poruchami menstruačního cyklu a sterilitou. Diagnóza byla stanovena na základě vyšetření steroidního spektra nativně a při stimulačních a supresních testech a CT-vyšetření. Po zavedené terapii dexametazonem došlo k úpravě krevního tlaku i iontogramu. Sdělení dokládá význam vyšetření celého steroidního spektra u nemocných s touto symptomatologií k vyloučení vzácnějších příčin endokrinní hypertenze.
Klíčová slova:
kongenitální adrenální hyperplazie – endokrinní hypertenze – hypokalemie – defekt 17α-hydroxylázy
Úvod
Na steroidogenezi v kůře nadledvin se podílí velké množství enzymů kódovaných příslušnými geny. Mezi nejdůležitější patří 21-hydroxyláza (P450c21, CYP 21), 11β-hydroxyláza (P450c11, CYP11B2, CYP11B1), 3β-dehydrogenáza (HSD3B2, HSD3B1), 17α-hydroxyláza (P450c17, CYP 17), desmoláza (P450scc). Při genových poruchách dochází k poškození tvorby steroidních enzymů a k rozvoji nadledvinové insuficience s deficitem cílových steroidů, především kortizolu. Tento deficit vede zpětnou vazbou ke stimulaci hypotalamu a hypofýzy s nadprodukcí ACTH. Nadbytek ACTH stimuluje sekreci steroidů kůry nadledvin před enzymovým blokem (schéma 1). Vzniká tzv. Kongenitální adrenální hyperplazie (CAH, dříve označovaná jako adrenogenitální syndrom), autozomálně recesivně dědičné onemocnění s pestrou klinickou symtomatologií, která závisí na typu a stupni (kompletní a parciální formy) enzymatického defektu. Většina steroidních prekurzorů má virilizující účinky, proto se v závislosti na míře bloku projevuje kombinace příznaků nadledvinové insuficience a virilizace (nejčastěji u deficitu 21-hydroxylázy, dále vzácněji u 3β-hydroxysteroidní dehydrogenázy). Pokud se hromadí steroidní prekurzory s mineralokortikoidními účinky, mohou navíc způsobit hypertenzi a hypokalemii (deficit 11β-hydroxylázy). V případě vzácného defektu 17α-hydroxylázy se kromě snížené tvorby kortizolu, androgenů a estradiolu uplatňuje v klinickém obraze zvýšená tvorba steroidních prekurzorů s mineralokortikoidním účinkem, zejména 11-deoxykortikosteronu, která vede k hypertenzi a hypokalemii.

Popis případu
Dnes 42letá nemocná je sledována od 28 let pro obtížně korigovatelnou hypertenzi. Menarche byla ve 14 letech, v dospělosti byla sledována a gynekologicky léčena pro nepravidelné cykly, ovariální cysty, sterilitu, opakovaně provedena abraze pro metroragie. Dvakrát neúspěšně absolvovala pokus o graviditu v IVF programu. Vzhledem k hypertenzi a hypokalemii byla pacientka v roce 1999 odeslána na endokrinologii k vyloučení primárního hyperaldosteronizmu. Hormonální vyšetření tehdy vykazovalo suprimovanou hladinu PRA nativně i po stimulaci, aldosteron nativně i po stimulaci byl mírně snížený. Snížené byly i hladiny testosteronu a DHEAS, ostatní hormonální vyšetření bylo v normě včetně estradiolu, FSH, LH a PRL. Kortizol v séru v diurnálním profilu byl v normě, vyšetření volného kortizolu v moči lehce nad horní hranicí normy, supresibilní ve 2 mg dexametazonovém testu, ACTH v normě, odpady 17-ketosteroidů v moči za 24 hodin také v normě. Kompletní vyšetření steroidů tehdy provedeno nebylo. Vyšetření katecholaminů v moči za 24 hodin vykazovalo normální hodnoty. Vzhledem k výraznému zvětšení pravé nadledviny na CT, která byla popisována velikosti 55 × 30 × 30 mm, denzitně nehomogenní, byla doporučena adrenalektomie. Levá nadledvina byla velikosti 25 × 15 × 15 mm. Pravostranná adrenalektomie byla provedena v říjnu roku 1999, bez komplikací, histologicky byl nález popsán jako enkapsulovaný adenom s ostrůvky oxyfilních buněk. Po operaci pacientka užívala přechodně hydrokortizon, který byl v lednu roku 2001 zcela vysazen. Byla sledována v endokrinologické ambulanci v místě bydliště. Podle dokumentace byly hodnoty TK uspokojivé při terapii amlodipinem v dávce 10 mg denně. Hypokalemie byla korigována tabletami kalium chloratum. Subjektivně se cítila dobře, udávala jen sklon k mírným perimaleolárním otokům.
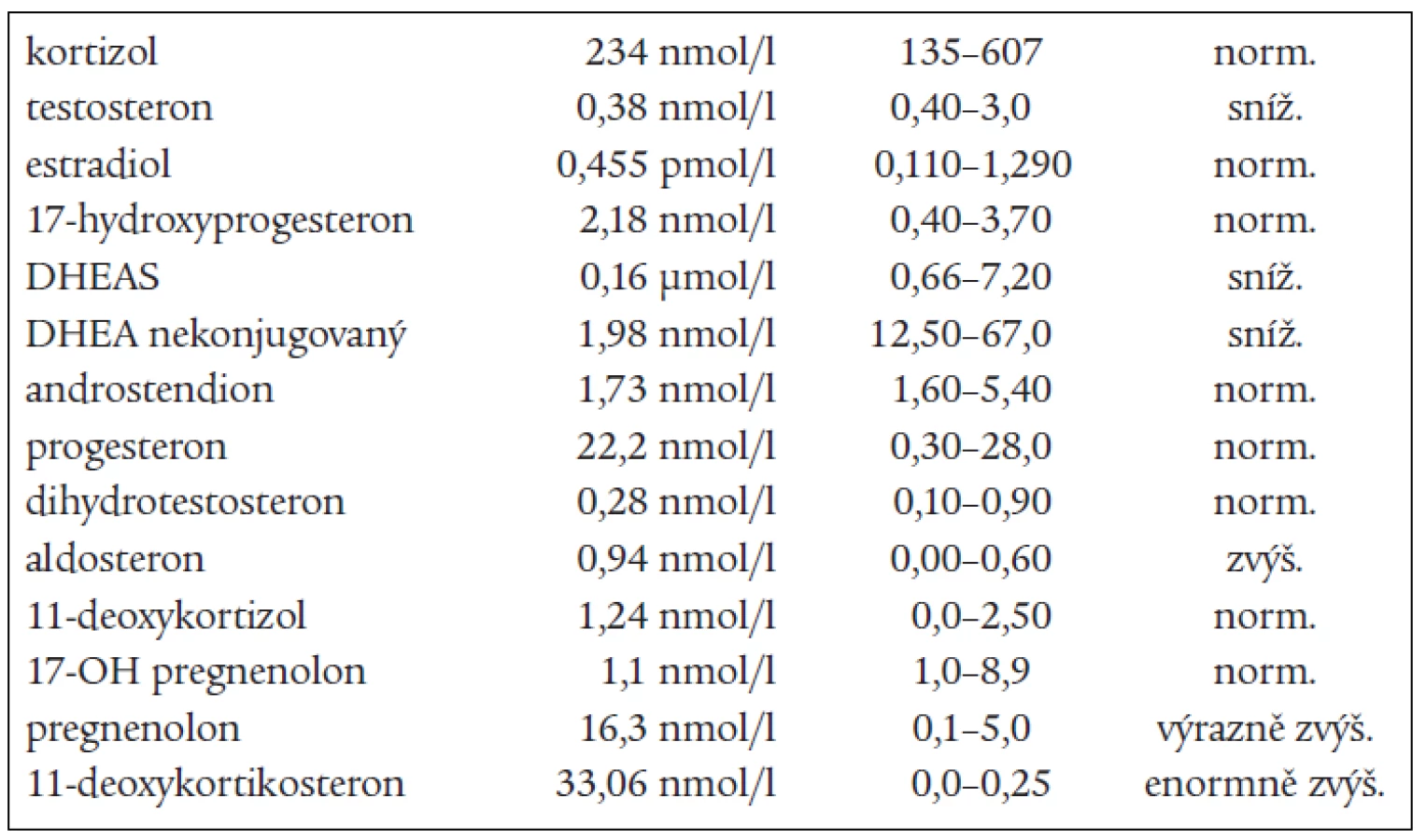
V roce 2003 byla pacientka odeslána k přešetření za hospitalizace pro trvající sklon k hypokalemii a progresi nálezu při kontrolním CT-vyšetření. Došlo ke zbytnění zbylé levé nadledviny, která byla defigurovaná s vícečetnými hypodenzními uzly o průměru 2 cm, které měly nativně lehce záporné hodnoty a po aplikaci kontrastní látky se vysycovaly asi o 20 HU. Celkový tvar byl laločnatý, rozměry 45 × 35 × 55 mm. Subjektivně byla pacientka bez výraznějších potíží, klinicky eufunkční, TK korigován monoterapií, menstruační cyklus jen lehce nepravidelný. Kalium v séru při přijetí bylo 2,9 mmol/l, ostatní základní biochemické vyšetření bylo v normě, včetně odpadů iontů v moči za 24hodin. Při hormonálním vyšetření včetně vyšetření celého steroidního spektra byly zachyceny enormně vysoké hladiny 11-deoxy-kortikosteronu a pregnenolonu, na horní hranici normy byly hodnoty aldosteronu a progesteronu, jedenkrát zachycen aldosteron mírně zvýšený. Snížené byly hladiny testosteronu,
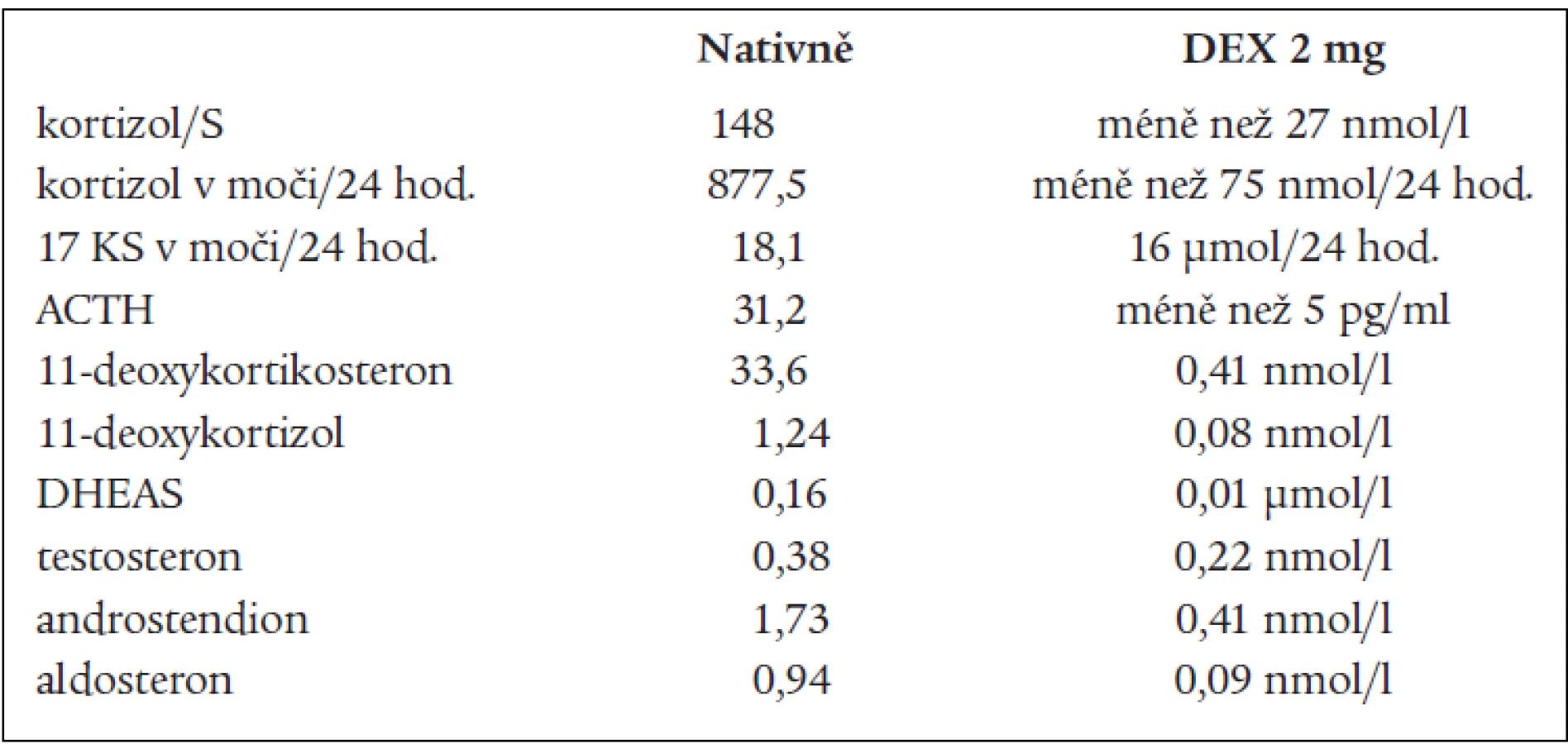
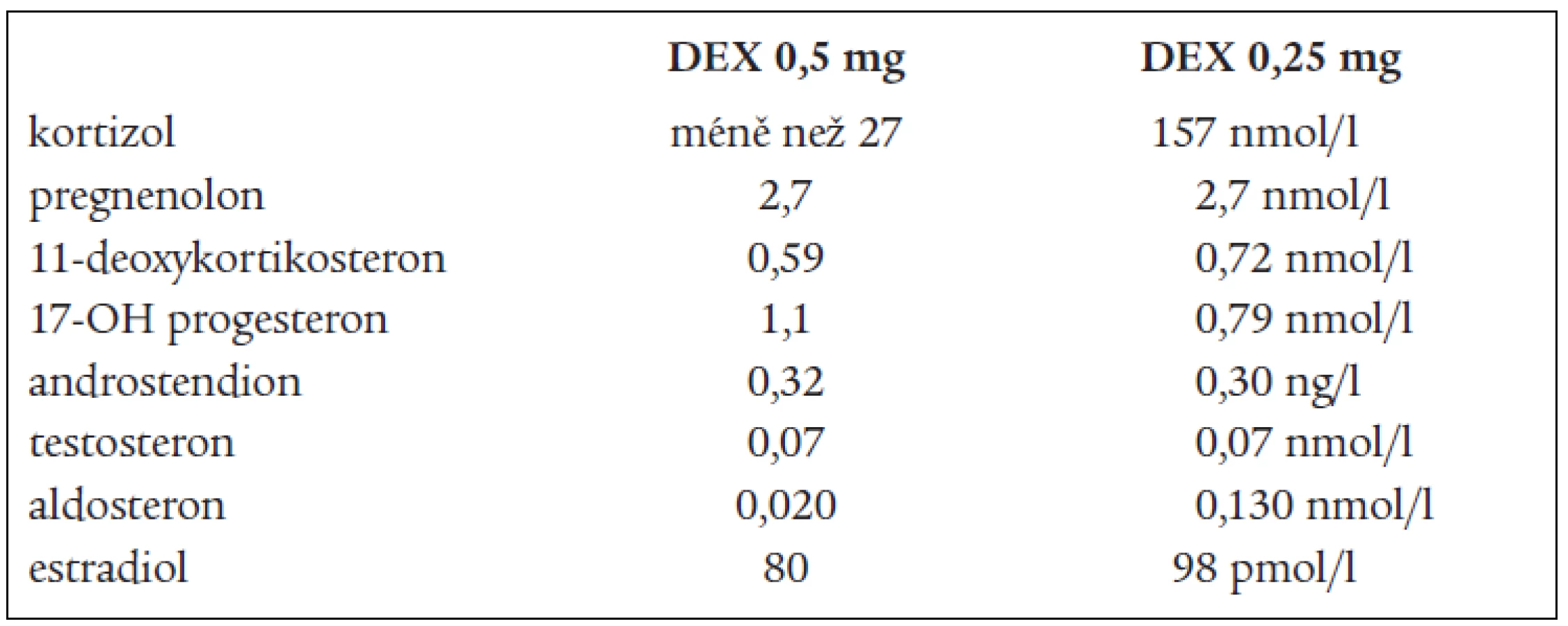
DHEAS, DHEA, androstendionu (tab. 1 a 3). Snížená byla i hladina plazmatické reninové aktivity. Ostatní steroidy v séru i v moči byly v normě, normální byly i hladiny hypofyzárních hormonů včetně ACTH. FSH a LH byly na jaře roku 2003 hraničně zvýšené. Všechny hladiny steroidů byly supresibilní v dexametazonovém testu (tab. 2). Tyto výsledky svědčily proti diagnóze tumoru a vedly k podezření na diagnózu kongenitální adrenální hyperplazie při parciálním bloku 17α-hydroxylázy, příčinou hypertenze byly vysoké hladiny 11-deoxykortikosteronu. Na genetické vyšetření byla pacientka odeslána, ale nebylo možné provést cílené vyšetření na gen pro 17α - ydroxylázu, které se v současnosti v České republice neprovádí. Proto byla diagnóza stanovena na základě biochemického vyšetření a klinického obrazu. Byla zavedena supresní terapie dexametazonem, zpočátku v dávce 0,5 mg denně. Při této dávce došlo k úpravě TK a iontogramu, bylo možno vysadit antihypertenzní medikaci i kalium, hladina 11-DOC byla jen mírně zvýšená, pregnenolon v normě, aldosteron snížený (tab. 4). Klinicky byl při této dávce v dalším průběhu sklon k hypotenzi a hyperkalemii, proto byla dávka dexametazonu snížena na 0,25 mg a přechodně byl přidán hydrokortizon 10 mg ráno, který byl později vysazen. Pacientka je subjektivně bez potíží, normotenzní, iontogram je v normě. Vyvinula se však postupně amenorea se zvýšením hladin FSH a LH a byla za kontrol gynekologa zavedena substituční terapie. Vyšetření steroidů, kromě mírně zvýšené hladiny 11-DOC, je v normě. Při kontrolním CT vyšetření nadledvin po půl roce zůstává nález stejný, bez progrese v oblasti levé nadledviny. Pacientka zůstává v dalším sledování endokrinologické ambulance.

Diskuse
Blok 17α-hydroxylázy může být vzácnou příčinou hypertenze, hypokalemie a od dětství periferního hypogonadizmu, u děvčat s amenoreou, eunuchoidním habitem a vysokými hladinami FSH a LH. Jeho incidence je velmi nízká, v literatuře jsou popisovány jen ojedinělé případy. Vázne zde přeměna pregnenolonu na 17-hydroxypregnenolon a progesteronu
na 17-hydroxyprogesteron. V kůře nadledvin i v gonádách je snížená tvorba androgenů a estrogenů, v kůře nadledvin je snížená tvorba kortizolu a pod tlakem zvýšené hladiny ACTH je zvýšená tvorba 17-deoxysteroidů, které mají mineralokortikoidní účinky. Při tomto bloku by měly být 17-hydroxylované steroidy a jejich metabolity snížené, nebo při dolním okraji normy. Jsou to z C21 steroidů 17-OH-pregnenolon, 17-OH-progesteron, kortizol, 11-deoxykortizol a z C19 steroidů DHEAS, DHEA, testosteron, androstendion a 11β-OH-androstendion. Výsledky naší pacientky splňují tuto podmínku. To, že jsou některé z těchto steroidů v mezích normy, lze vysvětlit tím, že enzymové bloky nejsou většinou úplné, a hyperplastické nadledviny příslušný deficit kompenzují. Naopak steroidní hormony, které nemají 17-OH skupinu a jejich prekurzory i metabolity, by měly být zvýšené. Je to progesteron, pregnenolon, 11-deoxykortikosteron (11-DOC) a aldosteron. Z výsledků je patrné, že i tato podmínka je splněna (tab. 1). Hladina aldosteronu může být u defektu 17α-hydroxylázy snížená, protože extrémně vysoké hladiny ACTH dependentních steroidů, hlavně deoxykortikosteronu, vedou k expanzi extracelulárního volumu a hypertenzi a k potlačení plazmatické reninové aktivity, což může vést ke sníženým hladinám aldosteronu. U naší nemocné jsme zachytili v různých obdobích hladinu aldosteronu sníženou, v normě, i lehce nad normu. Konečné hormony steroidogeneze (kortizol, dihydrotestosteron a estradiol) jsou v normě, proto nejsou u pacientky výrazné známky hypogonadizmu, ale výrazný nadbytek 11-DOC způsobuje hypertenzi a hypokalemii. Pokud se týká supresních a stimulačních testů, tak všechny steroidy reagovaly snížením v dexametazonovém testu (tab. 2) a výrazným (u 11-DOC) či mírným vzestupem v testu s ACTH (tab. 3), což svědčí proti diagnóze tumoru a potvrzuje příčinu onemocnění. V terapii je důležitá správně volená dávka supresní léčby, kdy je nutno brát v úvahu klinický stav, hodnoty TK a hladiny steroidů (tab. 4). Při zahájení terapie dexametazonem mohl být počáteční sklon k hypotenzi a hyperkalemii způsoben potlačeným renin-aldosteronovým systémem, který se může navrátit k normě až za několik měsíců. U dospělých pacientek je obvykle nutné zavedení substituční kombinované terapie estrogeny a gestageny.
V kazuistice jsme chtěli upozornit na možnost vzácného výskytu defektu 17α-hydroxylázy, který může být příčinou endokrinní hypertenze a hypokalemie a poruch menstruačního cyklu. Při podezření na tuto diagnózu je nutné vyšetření celého steroidního spektra včetně testů s ACTH a s dexametazonem.
Tato práce vznikla za podpory výzkumného záměru Ministerstva zdravotnictví ČR č. MZ 000000023761.
MUDr. Věra Olšovská
www.fnusa.cz
e-mail: vera.olsovska@fnusa.cz
Doručeno do redakce: 11. 3. 2004
Přijato po recenzi: 14. 4. 2004
Zdroje
1. Hampl R, Stárka L. Současné možnosti steroidní laboratorní diagnostiky. DMEV 1999; 4 : 206–211.
2. Šulcová J, Stárka L. Adrenální enzymopatie s pozdním nástupem. In: Stárka L et al. Aktuální endokrinologie. Praha: Maxdorf 1999 : 540–562.
3. Procházková V, Lebl J et al. Klinická diagnostika pacientů s kongenitální adrenální hyperplazií. Česko–slovenská pediatrie 1999; 10 : 544–547.
4. Costa-Santos M, Kater CE et al. Two prevalent CYP17 mutations and genotype-phenotype correlations in
24 Brazilian patients with 17-hydroxylase deficiency. Hum Genet 2004; 89 : 95–96.
5. Van Den Akker EL, Koper JW et al. Differential inhibition of 17-hydroxylase an 17, 20-lyase activities by three movel missense CYP 17 mutations identified in patients with P450c17 deficiency. J Clin Endocrinol Metab 2002; 87. 5714–5719.
6. Biglieri EG, Herron MA et al. 17 - hydroxylation deficiency in man. J Clin Invest 1966; 45 : 1946–1954.
7. Imai T, Yanase T et al. Canadian Mennonites and individuals residing in the Friesland region of The Netherlands share the same molecular basis of 17-hydroxylase deficiency. Hum Genet 1992; 89 : 95–96.
8. Costa-Santos M., Kater, CE et al. Two intronic mutations cause 17-hydroxylase deficiency by disrupting splice acceptor sites: direct demonstration of abberant splicing and absent enzyme activity by expression of the entire CYP17 gene in HEK–293 cells. J Clin Metab 2004, 89, 43–48.
9. Kater CE, Biglieri EG. Disorders of steroid 17-hydroxylase deficiency. Endocrin Metab Clin North Am 1994; 23 : 341–357.
10. Grumbach MM, Hughes IA et al. Disorders of sex differentiation. In: Larsen PR, Kronnenberg HM et al. Wiliams textbook of endocrinology. 10th ed. Philadelphia: WB Saunders 2003; 842–1002.
11. Yanase T, Simpson ER et al. 17-hydroxylase/ 17,20 lyase deficiency: from clinical investigation to molecular definition. Endocrin Rev 1991; 12 : 91–108.
12. Mendonca BB, Inacio M et al. Male pseudohermafroditism due to 17-betahydroxysteroid dehydrogenase 3 deficiency. Diagnosis, psychological evaluation and management. Medicine 2000; 79:
299–309.
13. Rabinovici J, Blankstein J et al. In vitro fertilization and primary embryonic cleabage beta estradiol, are possible in 17-hydroxylase deficiency despite extremely intrafollicular 17-beta estradiol. J Clin Metab 1989; 68 : 693––697.
14. Votava F, Kračmar P et al. Novorozenecký screening kongenitální adrenální hyperplazie. Čes Slov pediatrie 2002; 12 : 690–696.
15. Lisá L. Nové poznatky o léčbě vrozené adrenální hyperplazie. Forum medicinae 2000; 2 : 52–57.
16. Ferrari P, Bianchetti M. Juvenile hypertension, the role of genetically altered steroid metabolism. Hormone research 2001; 5 : 213–223.
17. Widimský J. Hypertenze. Praha: Triton 2002 : 368–370.
Štítky
Diabetológia Endokrinológia Interné lekárstvoČlánok vyšiel v časopise
Vnitřní lékařství

2005 Číslo 2
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vztah mezi statiny a rizikem vzniku nádorových onemocnění − metaanalýza
- Liečba bolesti po jednodňovej chirurgii
- Statinová intolerance
- Monoklonální protilátky v léčbě hyperlipidemií
Najčítanejšie v tomto čísle
- Pulzní tlak v mladé populaci stanovený 24hodinovým ambulantním monitorováním krevního tlaku a jeho vztah k metabolickým a antropometrickým parametrům
- Kongenitální adrenální hyperplazie, defekt 17α−hydroxylázy jako vzácná příčina hypertenze a hypokalemie
- Syndróm z rozpadu nádoru – tumor lysis syndrome
- Vlastní zkušenosti se vznikem a trváním spontánní remise u dospělých diabetiků typu 1