
Kyselina močová a kardiorenální riziko
Uric acid and cardiorenal risk
Hyperuricemia has long been known to be associated with cardiovascular and kidney disease, but it is unclear whether uric acid has a causal role in these comorbid disease processes or whether it is a secondary phenomenon. This review summarizes relevant studies concerning uric acid and possible links to hypertension, cardiovascular and renal diseases. Recent animal studies and epidemiologic studies have identified serum uric acid elevations as an independent risk factor for cardiovascular disease, hypertension and metabolic syndrome. Hyperuricemia has also been found to be an independent risk factor for the development of new‑onset as well as progression of chronic kidney disease. According to the current views, there is no need to treat all patients with asymptomatic hyperuricemia. Further clinical trials, aimed at determining whether lowering of uric acid levels would be of clinical benefit in the prevention and treatment of cardiovascular and renal diseases, are needed.
Keywords:
uric acid – cardiovascular risk – hypertension – metabolic syndrome – kidney disease
Authors:
V. Monhart
Authors‘ workplace:
Interní klinika 1. LF UK a ÚVN, Praha
Published in:
Kardiol Rev Int Med 2009, 11(3): 123-128
Category:
Nontraditional risk factors cardiovascular disease
Overview
I když je výskyt hyperurikemie u kardiovaskulárních a renálních onemocnění zjišťován již dlouhodobě, není zcela zřejmé, zda kyselina močová má příčinnou souvislost s těmito stavy, nebo je pouze jejich druhotným projevem. Uváděný přehled shrnuje důležité studie týkající se kyseliny močové a možných vazeb na hypertenzi, kardiovaskulární a renální onemocnění. Nedávné experimentální a epidemiologické studie prokázaly, že zvýšení sérové hladiny kyseliny močové je nezávislým rizikovým faktorem kardiovaskulárních onemocnění, hypertenze a metabolického syndromu. Kyselina močová je také nezávislým rizikovým faktorem jak vzniku, tak také progrese již stávajících chronických onemocnění ledvin. Podle současných názorů není důvod léčit všechny pacienty s asymptomatickou hyperurikemií. Je nezbytné provedení dalších klinických studií k potvrzení, zda by snížení hyperurikemie bylo přínosné pro prevenci a léčbu kardiovaskulárních a renálních onemocnění.
Klíčová slova:
kyselina močová – kardiorenální riziko – hypertenze – metabolický syndrom – onemocnění ledvin
Úvod
Výskyt zvýšené sérové koncentrace kyseliny močové (KM) u řady kardiovaskulárních, metabolických a renálních onemocnění je znám již několik desetiletí. Dříve se předpokládalo, že antioxidační vlastnosti KM by mohly přinášet určitou ochranu proti stárnutí, oxidačnímu stresu a z něj vyplývajícího buněčného poškození. Dosud získané experimentální, epidemiologické a klinické poznatky ale naznačují, že hyperurikemie může být rizikovým faktorem kardiovaskulárních (KV) a renálních onemocnění, zvláště u pacientů s hypertenzí, srdečním selháním nebo diabetes mellitus [1]. Proto se v posledních letech stále intenzivněji hledá odpověď na otázku, zda je KM příčinou či následkem kardiorenálních onemocnění [2].
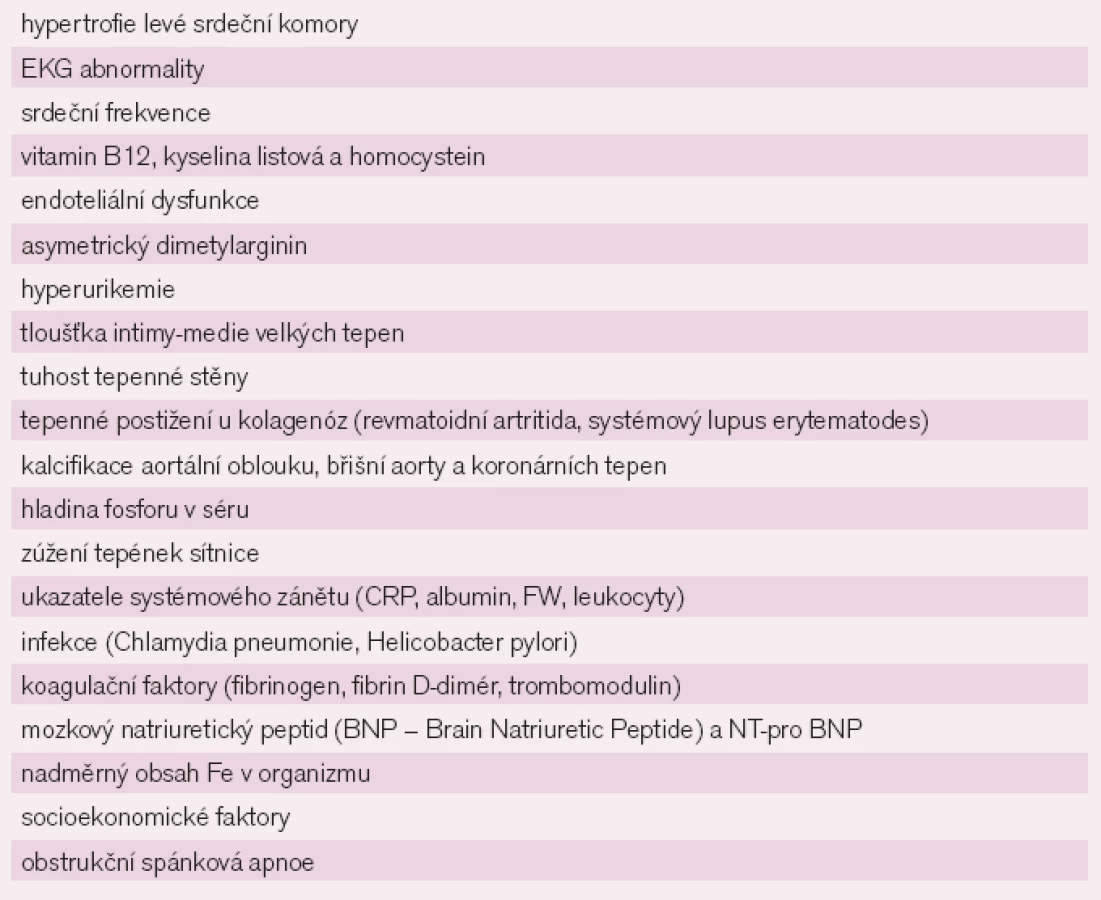
Známé klasické rizikové faktory – dyslipidemie (zvýšení LDL cholesterolu, snížení HDL cholesterolu), kouření, hypertenze, diabetes mellitus, abdominální obezita, psychosociální faktory, nedostatečná fyzická aktivita a nesprávné stravovací návyky (v konzumaci ovoce, zeleniny a alkoholu) jsou zodpovědné za více než 90% populačního rizika vzniku KV onemocnění [3–4]. KM je zařazena do početné skupiny dalších možných KV rizikových faktorů [5] (tab. 1). Ačkoliv není zcela jasné, jakým mechanizmem se KM uplatňuje v patogenezi KV onemocnění, je prokázáno, že hyperurikemie zasahuje do funkce endotelu, oxidativního metabolizmu, adhezivity a agregace krevních destiček. Zřejmý paradox mezi protektivními a toxickými účinky KM může vycházet ze zjištění, že antioxidační působky se mohou za určitých okolností změnit v prooxidační, zvláště jsou li v krvi přítomné ve vysoké koncentraci [6].
Kyselina močová a hyperurikemie
KM (2,6,8–trioxypurin) je u lidí konečným produktem metabolizmu purinů. Zdroje purinů jsou především endogenní – biosyntéza KM „de novo“ a katabolizmus nukleových kyselin (cca 600mg/den). Exogenní zdroj zahrnuje puriny přítomné v potravě (cca 100mg/den). Za vyrovnaného stavu jsou denní produkce + potravní zdroj (cca 700mg/den) zcela vyváženy odstraněním stejného množství z organizmu – 30% cestou trávicí trubice (bakteriální intestinální urikolýza) a 70% (cca 500mg/den) je vyloučeno močí [7]. Existuje čtyřstupňový renální mechanizmus vylučování KM:
- ve 100% je ultrafiltrována glomeruly,
- na začátku proximálního tubulu se 99% KM zpětně resorbuje aktivním transportním mechanizmem,
- v dalším úseku proximálního tubulu dochází k její sekreci do primární moči,
- následná tzv. postsekreční resorpce způsobí, že do definitivní moči je vyloučeno pouze 7–12% profiltrovaného množství.
Kromě primární hyperurikemie dané metabolickou poruchou existuje sekundární hyperurikemie, a to buď z nadměrné produkce, nebo snížené renální clearance urátů v důsledku některých onemocnění, působení léků či stravovacích zvyklostí (tab. 2). U většiny pacientů s primární hyperurikemií je frakční exkrece KM v ledvinách snížená. Sekundární hyperurikemie u pacientů s pokročilejším chronickým onemocněním ledvin souvisí především se sníženou hodnotou glomerulární filtrace, ale v některých případech se na ní podílejí změny tubulární resorpce, které mohou být i důsledkem podávání diuretik.

V biologickém prostředí tělesných tekutin je reaktivita KM významně závislá na hodnotě pH. V extracelulární tekutině s fyziologickou hodnotou pH 7,40 je 98% KM přítomno v ionizované formě ve formě urátů. Při dlouhotrvající hyperurikemii může dojít k ukládání nerozpustných urátů v kloubech a měkkých tkáních ve formě tofů. Hodnota pH moči má na rozdíl od krve významně vyšší variabilitu ovlivňující její saturační konstantu. Za situace kyselé moči (pH 5), kdy je saturační konstanta významně nižší než u moči alkalické (pH 7), může dojít k vysrážení urátů s následnou krystalurií či mikrolitiázou nebo k ukládání ve formě mikrotofů v dřeni ledviny.
Kyselina močová a kardiovaskulární onemocnění
Epidemiologické studie u běžné populace
Výsledky studie NHANES I (National Health And Nutrition Examination Survey) za období 1971–1987 [8] a dalších epidemiologických studií ARIC (Atherosclerosis Risk In Communities) a PIUMA (Progetto Ipertensione Umbria Monitoraggio Ambulatoriale) upozornily na spojení KM s KV morbiditou a mortalitou [9–10].
Pokračování průřezové populační studie NHANES I Epidemiological Follow up Study u 5 926 osob ve věkovém rozmezí 25–74 let s průměrnou dobou sledování 16 let prokázalo, že významné riziko KV mortality při zvýšené hladině KM platí pro obě pohlaví a je nejvyšší ve věkové skupině 45–54 let [11]. Zvýšené riziko úmrtí na ischemickou chorobu srdeční (ICHS) při porovnání nejvyššího kvartilu KM (> 416 µmol/l) oproti nejnižšímu kvartilu (< 321 µmol/l) bylo výraznější u žen (300%) než u mužů (70%) a nezáviselo na současném užívání diuretik, stupni KV rizika nebo přítomnosti menopauzy u žen. Ne všechny populační epidemiologické studie však potvrdily, že KM je rizikovým faktorem KV onemocnění. Framingham Heart Study [12] sice prokázala korelaci mezi KM a rizikem KV postižení u žen, po úpravě výsledků na ostatní ukazatele (např. hypertenzi, BMI a užívání diuretik) se však významnost tohoto vztahu nepotvrdila.
Studie u pacientů s prokázanou hypertenzí, srdečním selháním nebo ICHS
Důkazy o spojitosti KM se závažným cévním postižením přinesly především studie u hypertoniků. Pacienti s hypertenzí a hyperurikemií mají 3–5krát vyšší riziko vzniku ICHS nebo cévní mozkové příhody ve srovnání s jedinci s normální koncentrací KM.
Multivariační analýza údajů u 3 900 hypertoniků v průběhu NHANES III Epidemiological Follow up Study (1988–1994) prokázala, že zvýšená sérová koncentrace KM je spojená s významně vyšším rizikem vzniku srdečního infarktu a cévní mozkové příhody [13]. Také při sledování 8 690 osob s mírnou a středně závažnou hypertenzí v rámci Worksite Hypertension Treatment Program [14] bylo zjištěno, že zvýšení urikemie o 60 µmol/l přináší 32% zvýšení rizika KV příhod. Ve studii SHEP (Systolic Hypertension in the Elderly Program), která pět let sledovala u 4 327 hypertoniků léčených chlortalidonem nebo placebem vztah mezi KM, diuretickou léčbou a výskytem KV příhod, bylo prokázáno, že výchozí hodnota sérové hladiny KM je nezávislým prediktorem ICHS [15]. Rovněž 12 let trvající studie PIUMA u 1 720 dosud neléčených hypertoniků potvrdila významné riziko zvýšené koncentrace KM pro vznik KV příhod a celkovou mortalitu [10].
U 294 pacientů s chronickým srdečním selháním byl prokázán významný vztah mezi zvýšenou hladinou KM a mortalitou. Hladina KM nad 800 µmol/l znamenala 18násobně vyšší relativní riziko úmrtí ve srovnání s hodnotami pod 400 µmol/l [16].
U pacientů s koronarograficky potvrzenou ICHS hodnota sérové KM v nejvyšším kvartilu představovala pětinásobné riziko úmrtí ve srovnání s nejnižším kvartilem [17]. Také v další studii s koronarograficky ověřenou ICHS hladina KM > 311 µmol/l znamenala v průběhu pěti let sledování 3,5krát vyšší riziko kardiálního úmrtí, výskytu srdečního infarktu, cévní mozkové příhody nebo renálního selhání [18]. Sledování 4 385 osob po dobu osmi let v Rotterdam Study prokázalo, že vysoká hladina KM je silným prediktorem jak srdečního infarktu, tak i cévní mozkové příhody, a to i po zohlednění ostatních KV rizikových faktorů [19]. S významně vyšším rizikem vzniku cévní mozkové příhody byla spojená hladina KM v séru > 295 µmol/l ve studii u 8 tisíc diabetiků, a to i při úpravě na přítomnost ostatních KV rizikových faktorů [20].
Kyselina močová a hypertenze (hyperurikemická hypertenze)
Dostupné experimentální a klinické poznatky podporují názor, že zvýšená hladina KM může vyvolat hypertenzi (obr. 1). První přímý důkaz, že zvýšení KM vede ke zvýšení krevního tlaku, pochází ze zvířecího experimentu. U pokusných krys s hyperurikemií navozenou podáním inhibitoru urikázy došlo po několika týdnech k rozvoji hypertenze, která byla ovlivnitelná podáním inhibitoru xantinoxidázy či urikosurika [21]. Mechanizmem, kterým KM vyvolala hypertenzi, byla renální vazokonstrikce zprostředkovaná snížením endoteliální produkce NO a aktivací RAS [22]. Přetrvávající hyperurikemie u pokusných zvířat vedla k rozvoji mikrovaskulárních renálních změn – arteriolosklerózy stejné jako u esenciální hypertenze, a to i přes kontrolu zvýšeného krevního tlaku diuretiky. Zřejmě jde o přímý a na krevním tlaku nezávislý účinek KM na endoteliální a hladké svalové buňky cévní stěny [23]. Feig se spoluautory [2] uvádí řadu dalších experimentálních prací. V pokusech na tkáňových kulturách (hladké svalové buňky cévní stěny) KM indukovala cévní proliferaci, zánět, oxidativní stres a aktivovala lokální systém renin angiotenzin. Obdobné změny vznikaly u pokusných zvířat s normální hladinou KM během infuze angiotenzinu II nebo blokády syntézy NO. U již přítomných renálních mikrovaskulárních změn dochází k rozvoji sůl-dependentní hypertenze, která trvá i po přerušení infuze angiotenzinu II či přerušení blokády syntézy NO. U hyperurikemických zvířat došlo po vysazení inhibitoru urikázy v době rozvinutého mikrovaskulárního a intersticiálního ledvinného postižení ke zlepšení kontroly krevního tlaku samotnou dietou s nízkým obsahem NaCl [24].

Klinická pozorování hyperurikemie předcházející vznik hypertenze logicky znamenají, že tato odchylka nemůže být následkem hypertenze. Vztah KM k vysokému krevnímu tlaku je zřejmě nejvýznamnější u mladých jedinců s časným začátkem hypertenze [25]. U adolescentů bylo zjištěno, že hyperurikemie je častější u primární hypertenze než u sekundární hypertenze [26]. Intenzita vztahu mezi KM a krevním tlakem u osob s již ustálenou hypertenzí klesá se vzrůstajícím věkem a trváním hypertenze [27]. Jedinci s dlouhotrvající hypertenzí mají již renální mikrovaskulární postižení, které už samo může být primárně zodpovědné za jejich hypertenzi.
Šest velkých epidemiologických studií publikovaných v posledních sedmi letech zjistilo, že hladina KM předpovídá rozvoj hypertenze v následujících letech [28–33]. Normative Aging Study [33] prokázala, že KM je nezávislým prediktorem vzniku hypertenze, a to i při zohlednění dalších faktorů (BMI, obvod pasu, hladiny krevního cukru a tuků, kouření, konzumace alkoholických nápojů).
Kyselina močová a metabolický syndrom
KM byla dlouhodobě považována za obvyklý projev metabolického syndromu (MS) [34]. Průměrná hodnota KM je u MS přibližně o 30–60 µmol/l vyšší než u osob bez MS [35]. Sérová hladina KM stoupá s počtem komponent MS [36–37], a to i po úpravě na věk, pohlaví, hodnotu kreatininové clearance, konzumaci alkoholu a užívání diuretik. Nedávná analýza studie NHANES III [38] prokázala, že prevalence MS stoupá v závislosti na zvyšující se hladině KM. Z jednotlivých komponent MS má nejsilnější korelaci s KM obvod pasu [35]. Příčinou zvýšené koncentrace KM u MS může být zvýšená tvorba nebo snížené vylučování, případně kombinace obou možností. Snížené vylučování KM patrně souvisí se zvýšenou tubulární reabsorpcí sodíku vyvolanou hyperinzulinemií [39]. Stejný mechanizmus poklesu ledvinné exkrece KM byl zjištěn i u obezity a arteriální hypertenze [40], které patří mezi nejčastější projevy MS. Ze zvýšené tvorby KM u MS se obviňuje nadměrný přívod fruktózy stravou [41]. U pacientů s MS a orgánovým postižením by se mohla podílet na nadprodukci KM i lokální ischemie.
Kyselina močová a onemocnění ledvin
Výskyt zvýšených hladin KM v séru byl dlouhodobě spojován s chronickými onemocněními ledvin (Chronic Kidney Disease – CKD), ale bez kauzální role v rozvoji ledvinové dysfunkce [7]. Výjimkou jsou urátová urolitiáza a akutní urátová nefropatie provázející chemoterapeutickou léčbu hemato-onkologických onemocnění. Ve starších studiích bylo pozorováno významné postižení renální funkce u nemocných s dnou (až u 40%). Zásadní úlohu KM oslabovala přítomnost dalších onemocnění a faktorů spojených s dnou a hyperurikemií – hypertenze, diabetu, obezity, užívání alkoholu a nesteroidních antirevmatik.
Experimentální poznatky o úloze kyseliny močové při poškození ledvin
U hyperurikemických krys byl prokázán rozvoj systémové i glomerulární hypertenze, proteinurie, renální dysfunkce, glomerulosklerózy a intersticiální fibrózy [42]. Hyperurikemie navozená u skupiny pokusných zvířat s již existujícím poškozením ledvin vede k výrazné progresi renálních změn ve srovnání se skupinou bez hyperurikemie [43]. To znamená, že hyperurikemie nejen vyvolává poškození ledvin, ale také může vést k progresi již přítomné nefropatie [44–45]. Experimentální mikropunkční studie potvrdily, že zvýšená hladina KM vede ke glomerulární hypertenzi a vazokonstrikci kůry ledvin [46]. K poškození tkáně ledvin přispívá i hyperurikemií podporovaná zvýšená produkce zánětlivých mediátorů [47–48].
Epidemiologické studie u běžné populace
Z výsledků prospektivních populačních studií je zřejmé, že hyperurikemie je nezávislým renálním rizikovým faktorem i po započtení řady běžných a známých rizikových faktorů vývoje CKD. Pětileté sledování 49 413 japonských mužů ve věku 25–60 let prokázalo silný vztah mezi sérovou hladinou KM a vznikem chronického selhání ledvin [49]. Osoby v nejvyšším kvartilu KM (> 508 µmol/l) měly v porovnání s druhým kvartilem (297–386 µmol/l) osminásobně vyšší riziko vzniku renálního selhání. V další populační studii 48 177 osob z Okinawy starších 20 let byla zjištěna prevalence hyperurikemie 31,9% u mužů a 13,6% u žen. Po osmi letech byla u 103 jedinců (53 mužů a 50 žen) zahájena hemodialyzační léčba pro terminální selhání ledvin. Koncentrace KM ≥ 357 µmol/l byla nezávislým prediktorem selhání ledvin, a to významněji u žen [50]. V rámci epidemiologické studie zahrnující 21 475 zdravých dobrovolníků sledovaných sedm let bylo hodnoceno relativní riziko vývoje CKD 3. stadia (pokles glomerulární filtrace na < 60ml/min/1,73 m2). U osob s mírnou hyperurikemií (420–540 μmol/l) bylo zjištěno o 26% vyšší riziko CKD ve srovnání s jedinci s normální KM (< 420 μmol/l). U výrazné hyperurikemie (> 540 μmol/l) zvýšení rizika dosáhlo 63%, a to i po korekci na řadu dalších ukazatelů (vstupní glomerulární filtrace, pohlaví, věk, jednotlivé parametry metabolického syndromu, střední arteriální tlak a užívání antihypertenziv [51]. Vliv hyperurikemie na rozvoj CKD potvrdilo sledování 13 338 pacientů ze studií ARIC a Cardiovascular Health Study [52]. Každý vzestup sérové koncentrace KM o 60 μmol/l byl provázen 7% zvýšením rizika vzniku CKD (pokles alespoň o 15ml/min/1,73 m2 na < 60ml/min/1,73 m2) i po korekci na řadu dalších sledovaných parametrů. Letos publikovaná dosud největší kohortová studie sledovala 25 let u 177 570 pacientů vliv tradičních a nových rizikových faktorů na rozvoj chronického selhání ledvin. Jedinci v nejvyšším kvartilu sérové hladiny KM měli více než dvojnásobně vyšší adjustované riziko vzniku selhání ledvin oproti jedincům v nejnižším kvartilu. Průkaz nového a léčbou ovlivnitelného rizikového faktoru je, spolu s kontrolou hypertenze a farmakologickou blokádou systému renin angiotenzin, příslibem možné nové intervence ke snížení rizika vzniku terminálního selhání ledvin [53]. Je nezbytné provedení randomizované kontrolované studie ke zjištění, zda by snižování sérové hladiny KM bylo účinnou renoprotektivní strategií.
Studie u pacientů s prokázaným chronickým onemocněním ledvin
Při desetiletém sledování osob s biopticky ověřenou chronickou glomerulonefritidou (IgA nefropatií) bylo zjištěno, že hyperurikemie přítomná v době provedení renální biopsie je, spolu s hypertriglyceridemií, hypertenzí a proteinurií, odpovědná za progresivní průběh onemocnění [54]. Přerušení dlouhodobého podávání allopurinolu u nemocných s mírnou hyperurikemií a CKD 3. a 4. stadia vedlo po jednom roce sledování k významnému zhoršení hypertenze, akceleraci poklesu funkce ledvin a zvýšení močové exkrece transformujícího růstového faktoru beta 1. Tyto změny byly ovlivnitelné farmakologickou blokádou systému renin angiotenzin [55]. Naopak prospektivní sedmiletá studie MMKD (Mild to Moderate Kidney Study) u 227 pacientů s nediabetickým renálním onemocněním neprokázala, že KM je nezávislým predikátorem progrese CKD [56].
Předpokládá se, že by hyperurikemie po orgánové transplantaci mohla být potenciálním rizikovým faktorem pro rozvoj postižení ledvinného štěpu a pro vznik renální dysfunkce u transplantací jiných orgánů [57]. Pacienti s hyperurikemií mají větší riziko vzniku kontrastní nefropatie [58].
Léčba hyperurikemie
Současné možnosti ovlivnění zvýšené hladiny KM zahrnují:
- dietní a režimová opatření – omezení konzumace potravin s obsahem purinů (maso a masné produkty – játra, ledvinky, srdce, brzlík, drůbež, mořské ryby, extrakty z kvasnic, hrášek, fazole, špenát) a také alkoholických nápojů;
- cílenou farmakologickou intervenci – převážně podání kompetitivních inhibitorů xantinoxidázy (allopurinol) a spíš výjimečně urikosurik (probenecid, sulfinpyrazon, benzbromaron a benziodaron);
- úpravu přidružené léčby – nízké dávkování tiazidových a kličkových diuretik; při indikaci léků blokujících systém renin angiotenzin aldosteron je výhodné upřednostnit losartan, který jako jediný z antagonistů receptorů angiotenzinu II (AT1 blokátorů) má prokázaný hyperurikemii snižující účinek [59].
Rozhodování, zda léčit nebo neléčit asymptomatickou hyperurikemii, se řídí stupněm KV rizika [60–61]. K farmakologické léčbě hyperurikemie přistupujeme u diabetiků, obézních a hypertenzních pacientů s vysokým KV rizikem na základě předpokladu, že hyperurikemie může být projevem zánětu, ischemie a oxidativního stresu v KV systému. Léčba je nezbytná u pacientů s dnou, urolitiázou, CKD a esenciální hypertenzí provázenou hypertenzní nefrosklerózou [62–64].
U osob s mírnou hyperurikemií při nekomplikované obezitě, hypertenzi a inzulinové rezistenci není indikace farmakologické léčby allopurinolem nebo urikosuriky. Je však žádoucí zavedení dietních a režimových opatření. V případě hypertenze je vhodné upravit složení antihypertenzivní léčby.
Závěr
I přes příznivé výsledky uvedených studií prokazujících kauzální vztah mezi KM a rizikem KV a renálních onemocnění, zůstávají určitá omezení a nejasnosti. Dosud postrádáme podrobnější znalosti všech biologických funkcí KM ve vztahu ke KV onemocněním. Nemáme jednoznačné vysvětlení pro existující rozporuplnost v účincích KM – na jedné straně možnost prozánětlivého působení na cévní endotel i adipocyty a naproti tomu antioxidační efekt KM. Rovněž nevíme, zda je příznivý efekt allopurinolu dán snížením sérové hladiny kyseliny močové nebo potlačením tvorby s xantin oxidázou spojených volných kyslíkových radikálů.
Bude nutné vyčkat výsledků dalších klinických studií k definitivnímu potvrzení, zda bude snížení hyperurikemie přínosné pro prevenci a léčbu kardiovaskulárních a renálních onemocnění.
Doručeno do redakce 8. 7. 2009
Přijato po recenzi 8. 7. 2009
prof. MUDr. Václav Monhart, CSc.
Interní klinika 1. LF UK a ÚVN, Praha
vaclav.monhart@uvn.cz
Sources
1. Alderman M, Aiyer KJV. Uric Acid: Role in Cardiovascular Disease and Effects of Losartan. Curr Med Res Opin 2004; 20 : 369–379.
2. Feig DI, Kang DH, Johnson RJ. Uric Acid and Cardiovascular Risk. N Engl J Med 2008; 359 : 1811–1821.
3. Yusuf S, Hawken S, Ounpuu S et al. INTERHEART Study Investigators. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004; 364 : 937–952.
4. Vasan RS, Sullivan LM, Wilson PW et al. Relative importance of borderline and elevated levels of coronary heart disease risk factors. Ann Intern Med 2005; 142 : 393–402.
5. Wilson PWF. Overview of the risk factors for cardiovascular disease. UpToDate® 2009 (version 17.1). [http://www.uptodate.com/patients/content/topic.do?topicKey=~QGsob5Yuvb].
6. Lippi G, Montagnana M, Franchini M et al. The paradoxical relationship between serum uric acid and cardiovascular disease. Clin Chim Acta 2008; 392 : 1–7.
7. Edwards NL. The role of hyperuricemia and gout in kidney and cardiovascular disease. Cleve Clin J Med 2008; 75 (Suppl 5): S13–S16.
8. Freedman DS, Williamson DF, Gunter EW et al. Relation of aserum uric acid to mortality ischemic heart disease: the NHANES I Epidemiologic Follow‑up Study. Am J Epidemiol 1995; 141 : 637–644.
9. Moriarity JT, Folsom AR, Iribarren C et al. Serum uric acid and risk of coronary heart disease: Atherosclerosis Risk in Communities (ARIC) Study. Ann Epidemiol 2000; 10 : 136–143.
10. Verdecchia P, Schillaci G, Reboldi G et al. Relation between serum uric acid and risk of cardiovascular disease in essential hypertension. The PIUMA study. Hypertension 2000; 36 : 1072–1078.
11. Fang J, Alderman MH. Serum uric acid and cardiovascular mortality: The NHANES I Epidemiologic Follow‑up Study, 1971–1992. National Health and Nutrition Examination Survey. JAMA 2000; 283 : 2404–2410.
12. Culleton BF, Larson MG, Kannel WB et al. Serum uric acid and risk for cardiovascular disease and death: the Framingham Heart Study. Ann Intern Med 1999; 131 : 7–13.
13. Ward HJ. Uric acid as a independent risk factor in the treatment of hypertension. Lancet 1998; 352 : 670–671.
14. Alderman MH, Cohen H, Madhavan S. Distribution and determinants of cardiovascular events during 20 years of successful antihypertensive treatment. J Hypertens 1998; 16 : 761–769.
15. Franse LV, Pahor M, Di Bari M et al. Serum uric acid, diuretic treatment and risk of cardiovascular events in the Systolic Hypertension in the Elderly Program (SHEP). J Hypertens 2000; 18 : 1149–1154.
16. Anker SD, Doehner W, Rauchhaus M et al. Uric acid and survival in chronic heart failure: validation and application in metabolic, functional, and hemodynamic staging. Circulation 2003; 107 : 1991–1997.
17. Bickel C, Rupprecht HJ, Blankenberg S et al. Serum uric acid as an independent predictor of mortality in patients with angiographically proven coronary artery disease. Am J Cardiol 2002; 89 : 12–17.
18. Short RA, Johnson RJ, Tuttle KR. Uric Acid, Microalbuminuria and Cardiovascular Events in High‑Risk Patients. Am J Nephrology 2005; 25 : 36–44.
19. Bos MJ, Koudstaal PJ, Hofman A et al. Uric acid is a risk factor for myocardial infarction and stroke: the Rotterdam Study. Stroke 2006; 37 : 1503–1507.
20. Lehto S, Niskanen L, Rönnemaa T et al. Serum uric acid is a strong predictor of stroke in patients with non‑insulin‑dependent diabetes mellitus. Stroke 1998; 29 : 635–639.
21. Mazzali M, Hughes J, Kim YG et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001; 38 : 1101–1106.
22. Mazzali M, Kanellis J, Han L et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure‑independent mechanism. Am J Physiol Renal Physiol 2002; 82: F991–F997.
23. Feig DI, Nakagawa T, Karumanchi SA et al. Hypothesis: Uric acid, nephron number, and the pathogenesis of essential hypertension. Kidney Int 2004; 66 : 281–287.
24. Watanabe S, Kang DH, Feng L et al. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension 2002; 40 : 355–360.
25. Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on the blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA 2008; 300 : 924–932.
26. Feig DI, Johnson RJ. Hyperuricemia in child-hood primary hypertension. Hypertension 2003; 42 : 247–252.
27. Brand FN, McGee DL, Kannel WB et al. Hyperuricemia as a risk factor of coronary heart disease: the Framingham Study. Am J Epidemiol 1985; 121 : 11–18.
28. Nakanishi N, Okamoto M, Yoshida H et al. Serum uric acid and risk for development of hypertension and impaired fasting glucose or Type II diabetes in Japanese male office workers. Eur J Epidemiol 2003; 18 : 523–530.
29. Alper AB jr, Chen W, Yau L et al. Childhood uric acid predicts adult blood pressure: the Bogalusa Heart Study. Hypertension 2005; 45 : 34–38.
30. Masuo K, Kawaguchi H, Mikami H et al. Serum uric acid and plasma norepinephrine concentrations predict subsequent weight gain and blood pressure elevation. Hypertension 2003; 42 : 474–480.
31. Nagahama K, Inoue T, Iseki K et al. Hyperuricemia as a predictor of hypertension in a screened cohort in Okinawa, Japan. Hypertens Res 2004; 27 : 835–841.
32. Sundström J, Sullivan L, D’Agostino RB et al. Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence. Hypertension 2005; 45 : 28–33.
33. Perlstein TS, Gumieniak O, Williams GH et al. Uric acid and the development of Hypertension: the Normative Aging Study. Hypertension 2006; 48 : 1031–1036.
34. Sarafidis PA, Nilsson PM. The metabolic syndrome: a glance at its history. J Hypertens 2006; 24 : 621–626.
35. Puig JG, Martínez MA, Mora M et al. Serum urate, Metabolic Syndrome, and Cardiovascular Risk Factors: A Population-Based Study. Nucleos Nucleot Nucl Acids 2008; 27 : 620–623.
36. Puig JG, Martínez MA. Hyperuricemia, gout, and the metabolic syndrome: serum urate and the metabolic syndrome. Curr Opin Rheumatol 2008; 20 : 187–191.
37. Hjortnaes J, Algra A, Olijhoek J et al. Serum uric acid levels and risk for vascular diseases in patients with metabolic syndrome. J Rheumatol 2007; 34 : 1882–1887.
38. Choi HK, Ford ES, Li C et al. Prevalence of the metabolic syndrome in patients with gout: the Third National Health and Nutrition Examination Survey. Arthritis Rheum 2007; 57 : 109–115.
39. Strazzullo P, Barbato A, Galletti F et al. Abnormalities of renal sodium handling in the metabolic syndrome: results of the Olivetti Heart Study. J Hypertens 2006; 24 : 1633–1639.
40. Strazzullo P, Puig JG. Uric acid and oxidative stress: relative impact on cardiovascular risk. Nutr Metab Cardiovasc Dis 2007; 17 : 409–414.
41. Nakagawa T, Tuttle K, Short RA et al. Hypothesis: fructose‑induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat Clin Pract Nephrol 2005; 1 : 80–86.
42. Nakagawa T, Mazzali M, Kang DH et al. Hyperuricemia cause glomerular hypertrophy in the rat. Am J Nephrol 2003; 23 : 2–7.
43. Kang DH, Nakagawa T, Feng L et al. A role of uric acid in the progression of renal disease. J Am Soc Nephrol 2002; 13 : 2888–2897.
44. Kang DH, Nakagawa T. Uric acid and chronic renal disease: possible implication of hyperuricemia on progression of renal disease. Semin Nephrol 2005; 25 : 43–49.
45. Nakagawa T, Mazzali M, Kang HD et al. Uric acid: A uremic toxin? Blood Purif 2006; 24 : 67–70.
46. Sánchez-Lozada LG, Tapia E, Avila-Casado C et al. Mild hyperuricemia induces glomerular hypertension in normal rats. Am J Physiol Renal Physiol 2002; 283: F1105–F1110.
47. Kang DH, Park SK, Lee IK et al. Uric acid‑induced C‑reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol 2005; 16 : 3553–3562.
48. Kanellis J, Watanabe S, Li JH et al. Uric acid stimulates monocyte chemoattractant protein‑1 production in vascular smooth Musile cells via mitogen‑activated protein kinase and cyclooxygenase-2. Hypertension 2003; 41 : 1287–1293.
49. Tomita M, Mizuno S, Yamanaka H et al. Does hyperuricemia affect mortality? A prospective cohort study of Japanese male workers. J Epidemiol 2000; 10 : 403–409.
50. Iseki K, Ikemiya Y, Inoue T et al. Significance of hyperuricemia as a risk factor for developing ESRD in a screened cohort. Am J Kidney Dis 2004; 44 : 642–650.
51. Obermayr RP, Temml C, Gutjahr G et al. Elevated uric acid increases the risk for kidney disease. J Am Soc Nephrol 2008; 19 : 2407–2413.
52. Weiner DE, Tighiouart H, Elsayed EF et al. Uric acid and incident kidney disease in the community. J Am Soc Nephrol 2007; 18 : 287–292.
53. Hsu CY, Iribarren C, McCulloch CE et al. Risk factors for end‑stage renal disease: 25-year follow‑up. Arch Intern Med 2009; 169 : 342–350.
54. Syrjänen J, Mustonen J, Pasternack A. Hypertriglyceridaemia and hyperuricaemia are risk factors for progression of IgA nephropathy. Nephrol Dial Transplant 2000; 15 : 34–42.
55. Talaat KM, el-Sheikh AR. The effect of mild hyperuricemia on urinary transforming growth factor beta and the progression of chronic kidney disease. Am J Nephrol 2007; 27 : 435–440.
56. Sturm G, Kollerits B, Neyer U et al. Uric acid as a risk factor for progression of non‑diabetic chronic kidney disease? The Mild to Moderate Kidney Disease (MMKD) Study. Exper Gerontol 2008; 43 : 347–352.
57. Mazzali M. Uric acid and transplantation. Semin Nephrol 2005; 25 : 50–55.
58. Toprak O, Cirit M, Esi E et al. Hyperuricemia as a risk factor for contrast‑induced nephropathy in patients with chronic kidney disease. Catheter Cardiovasc Interv 2006; 67 : 227–235.
59. Ruilope LM. The renal effects of losartan. Recep Cardiovasc Dis 1998; 5 : 1–3.
60. Baker JF, Krishnan E, Chen L et al. Serum uric acid and cardiovascular disease: recent developments, and where do they leave us? Am J Med 2005; 118 : 816–826.
61. Wannamethee SG. Serum uric acid and risk of coronary heart disease. Curr Pharm Des 2005; 11 : 4125–4132.
62. Krishnan E, Baker JF, Furst DE et al. Gout and the risk of acute myocardial infarction. Arthritis Rheum 2006; 54 : 2688–2696.
63. Choi HK, Curhan G. Independent impact of gout on mortality and risk for coronary heart disease. Circulation 2007; 116 : 894–900.
64. Siu YP, Leung KT, Tong MK et al. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis 2006; 47 : 51–59.
Labels
Paediatric cardiology Internal medicine Cardiac surgery CardiologyArticle was published in
Cardiology Review

2009 Issue 3
Most read in this issue
- Holterova monitorace EKG
- Současné názory na roli mírné hyperhomocysteinemie jako rizikového faktoru kardiovaskulárních chorob
- Kyselina močová a kardiorenální riziko
- HRT a kardiovaskulární riziko