
Není hypertrofie jako hypertrofie aneb Nezapomínejme na amyloidózu
There’s no hypertrophy like hypertrophy, or don’t forget amyloidosis
Amyloidosis is a systemic disease consisting of the depositing of proteinous substance (amyloid) in body tissues. These deposits can cause cardiac problems. A clinical picture of such an impairment can be, in the early stages of the disease, reminiscent of hypertrophic cardiomyopathy. These findings are documented in case history. Due to their fundamentally different prognosis it is important to know the difference between the two diseases. The article further includes a brief summary of the pathophysiology, diagnosis and treatment of amyloidosis, with a focus on cardiac impairment.
Keywords:
amyloidosis – hypertrophic cardiomyopathy – diagnosis – free light chains
Authors:
D. Zemánek; K. Linhartová
Authors‘ workplace:
Kardiologická klinika 2. LF UK a FN Motol Praha
Published in:
Kardiol Rev Int Med 2011, 13(4): 228-232
Overview
Amyloidóza je systémové onemocnění spočívající v ukládání bílkovinné substance – amyloidu ve tkáních. Ukládáním může být postiženo i srdce. Klinický obraz takového postižení potom může v časných stadiích onemocnění připomínat hypertrofickou kardiomyopatii. Tento nález je dokumentován kazuistikou. Vzhledem k zásadně odlišné prognóze obou onemocnění je důležité znát rozdíly mezi nimi. V článku jsou dále stručně shrnuty patofyziologie, diagnostika a také léčba amyloidózy se zaměřením na srdeční postižení.
Klíčová slova:
amyloidóza – hypertrofická kardiomyopatie – diagnostika – volné lehké řetězce
Úvod
Amyloidóza je systémové onemocnění, při kterém dochází k patologickému ukládání proteinové substance nazývané „amyloid“ v tkáních různých orgánů. Jedním z těchto orgánů může být také srdce. Morfologický a klinický obraz srdečního postižení je variabilní. V typickém případě může zpočátku připomínat hypertrofickou kardiomyopatii (HCM) a později se vyvíjet do obrazu restriktivní kardiomyopatie (RCM). Časná diagnóza je velmi důležitá, protože na rozdíl od typické HCM se jedná o onemocnění s velmi špatnou prognózou srovnatelnou s nádorovým onemocněním [1]. Důležitost odlišení amyloidózy od HCM vzhledem k odlišnému klinickému průběhu je demonstrována kazuistikou.
Kazuistika
66letý nemocný byl odeslán k vyšetření na naší ambulanci pro HCM. Pacient byl asi čtyři roky sledován u ambulantního kardiologa pro bikuspidální aortální chlopeň s nevýznamnou regurgitací, lehkou hypertrofii levé komory srdeční (13–14 mm) a hraniční dilataci ascendentní aorty (48 mm). Jinak byl bez významného přidruženého onemocnění. V dubnu 2011 byl hospitalizován pro oboustranné srdeční selhání na interním oddělení ve spádové nemocnici. Pro suspektní změny při elektrokardiografickém vyšetření (EKG) byla provedena koronarografie, která vyloučila zúžení srdečních tepen. Echokardiograficky byla zjištěna hypertrofie srdečních stěn. Byla zahájena léčba diuretiky a pacient byl po zlepšení klinického stavu propuštěn a ambulantně objednán jako suspektní HCM na naše pracoviště.
Při vyšetření na naší ambulanci v květnu 2011 byl pacient stále limitován výraznou dušností III.–IV. stupně funkční klasifikace. Při fyzikálním vyšetření byly pozorovány známky pravostranného (otoky DK do poloviny lýtek, zvýšená náplň krčních žil a hepatomegalie) i levostranného (bilaterálně bazálně chrůpky) srdečního selhání. Kromě těchto známek nebyla pozorována jiná významná patologie, krevní tlak byl 110/80 mmHg. Pacient byl léčen diuretiky (furosemid, verospiron), betablokátorem a warfarinem.
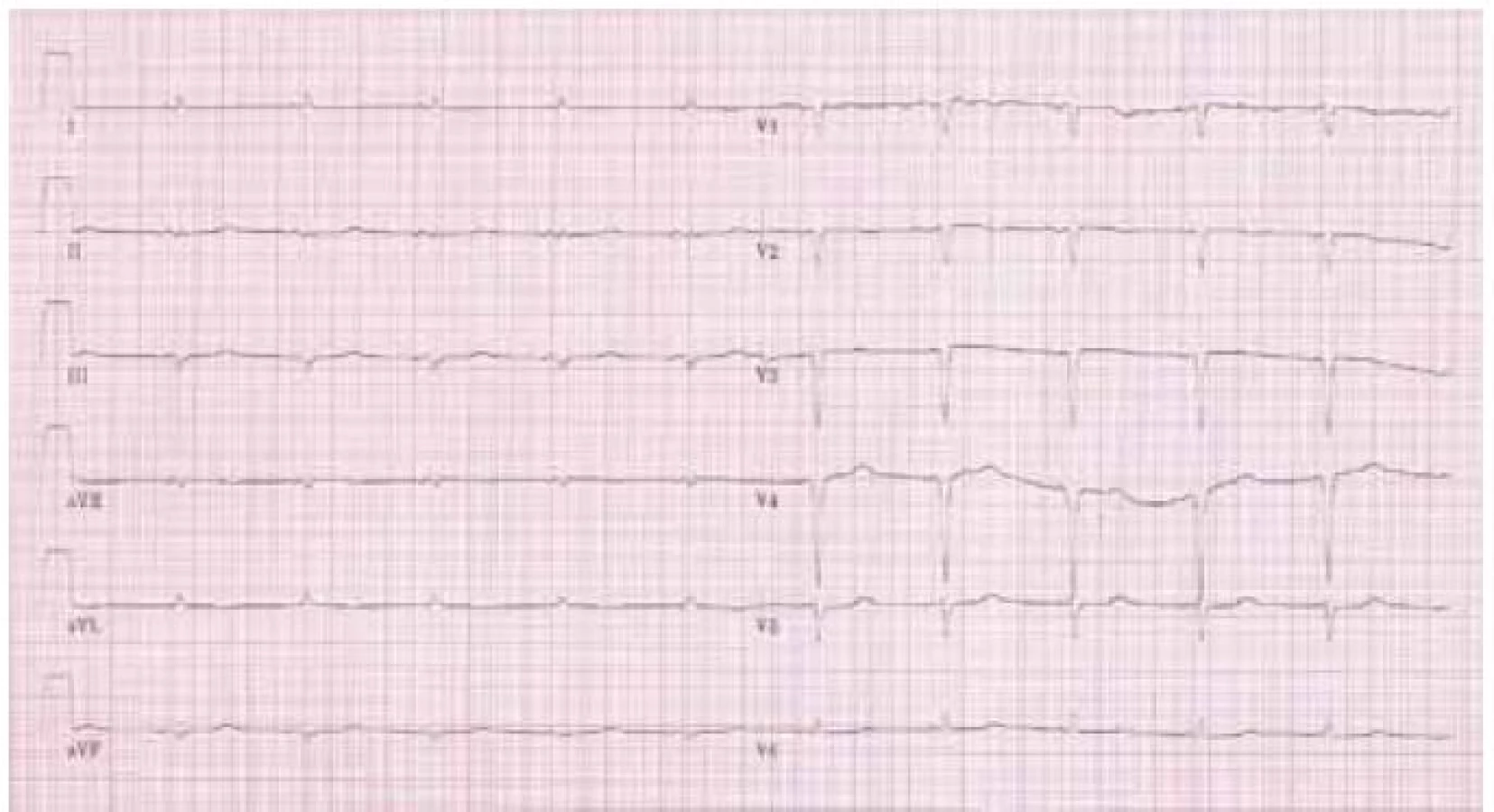
Na klidovém 12svodovém EKG byl přítomen sinusový rytmus s frekvencí komor 85/min, normální převodní intervaly, patologický kmit QS ve svodech III, aVF a V1–V4. Především však byla přítomná nízká „voltáž“ v končetinových svodech (obr. 1).
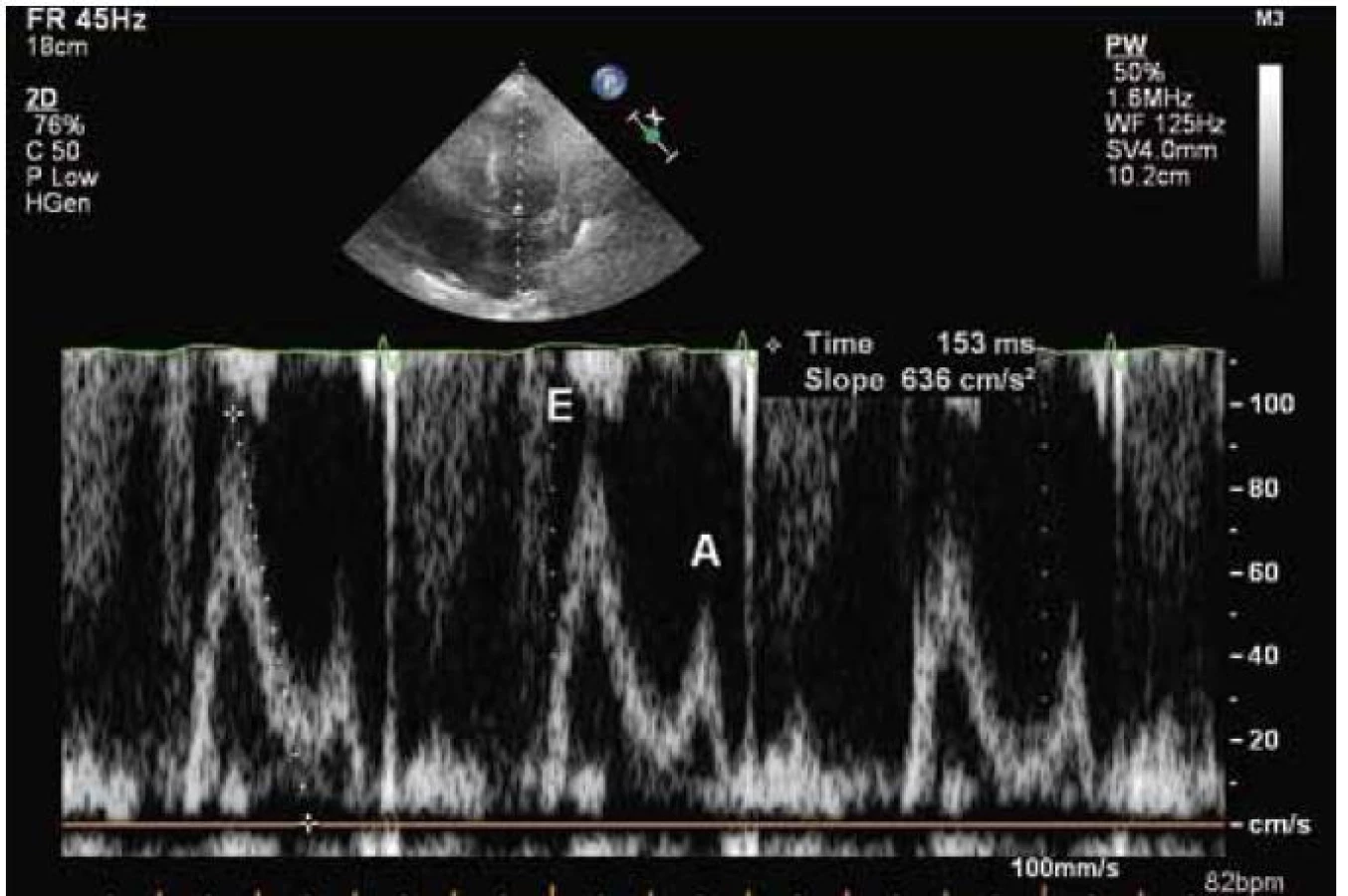
Echokardiograficky byla potvrzena koncentrická hypertrofie levé komory (20 až 22 mm), která rovnoměrně postihovala všechny stěny levé komory (obr. 2). Systolická funkce byla lehce snížená (ejekční frakce byla 45 %) a byla přítomná těžká porucha diastolické funkce (restriktivní typ průtoku na mitrální chlopni – obr. 3) společně s echokardiografickými známkami zvýšeného plnicího tlaku levé komory. Pravá komora byla také hypertrofická s tloušťkou její volné stěny kolem 8 mm. Byl přítomen hemodynamicky nevýznamný perikardiální výpotek.
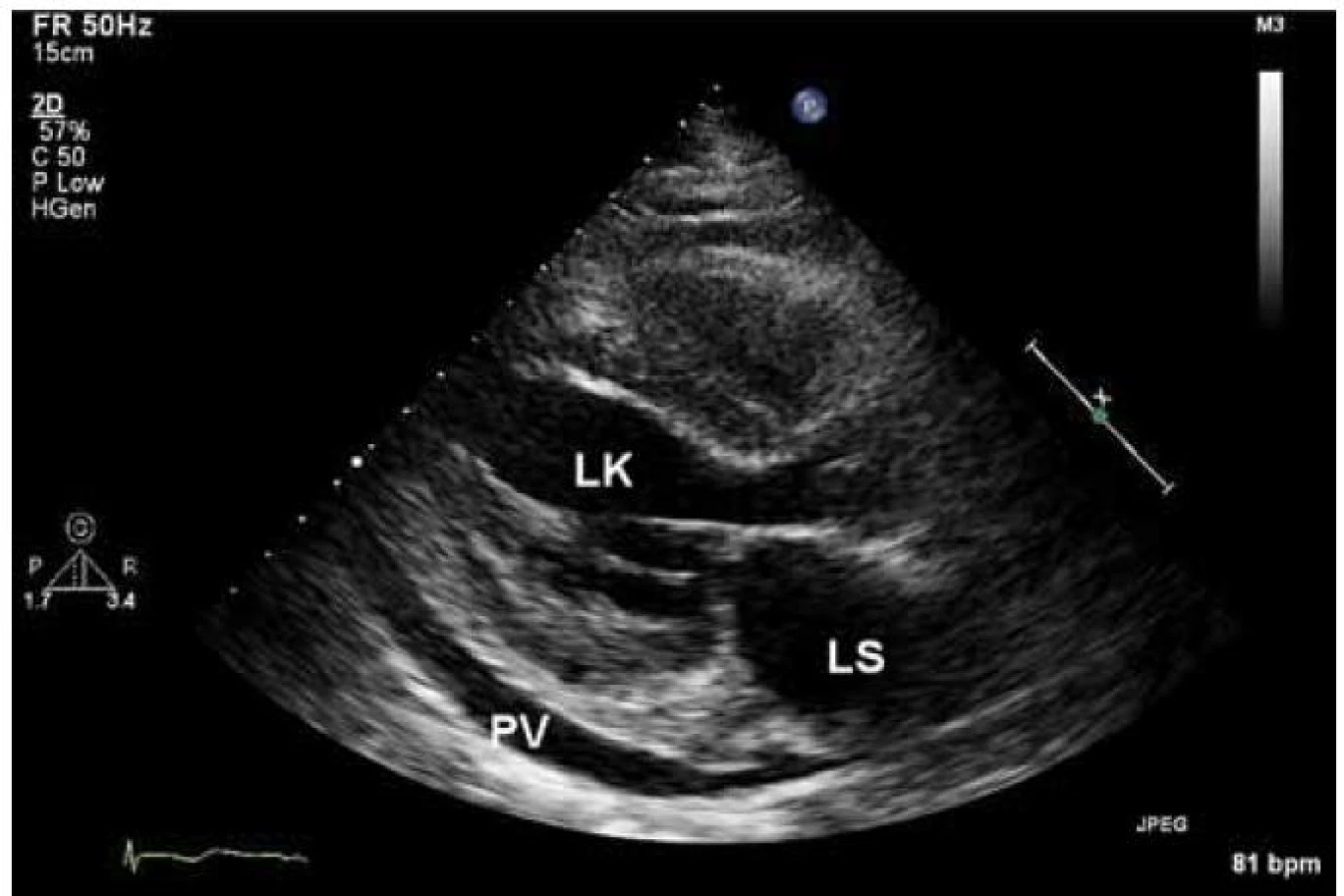
Na základě EKG a echokardiografického vyšetření bylo vysloveno podezření na srdeční amyloidózu a pacient byl odeslán k hospitalizaci. Byl proveden laboratorní odběr ke stanovení hladiny volných lehkých řetězců, při kterém byla zjištěna zvýšená hodnota volných lehkých řetězců typu lambda (942,5 mg/l) a také patologický poměr kappa/lambda (0,01). Na základě těchto hodnot bylo velmi pravděpodobné, že se jedná o AL amyloidózu. Po vysazení warfarinu bylo plánováno provedení trepanobiopsie a biopsie rekta, event. myokardu. Než mohla být plánovaná vyšetření provedena, došlo čtvrtý den od přijetí k srdeční zástavě. Pacient byl sice úspěšně resuscitován a přechodně stabilizován na umělé plicní ventilaci s nutností katecholaminové podpory, ale nakonec zemřel pátý den po přijetí pod obrazem multiorgánového selhání. Pitva prokázala postižení srdce a dalších orgánů AL amyloidózou.
Definice hypertrofické kardiomyopatie a amyloidózy
Ačkoli máme poměrně nová doporučení pro klasifikaci kardiomyopatií, přesto existují v určitých případech pochybnosti, kam určitý nález zařadit. Tyto rozpory se týkají také zařazení amyloidózy. Americká klasifikace správně kritizuje dosavadní morfologické dělení, které nahrazuje zdůrazněním patogenetiky a patofyziologie [2]. Hypertrofická kardiomyopatie je definována jako primární kardiomyopatie na základě mutace sarkomerických proteinů. Amyloidóza je naopak řazena mezi sekundární kardiomyopatie. Problémem je, že přes pokroky v genetice existuje řada pacientů s HCM, u kterých nelze zjistit příčinnou mutaci, a navíc je genetické vyšetření v České republice omezeně dostupné.
Evropská klasifikace je naproti tomu založena na morfologických kritériích. HCM je pak definována na základě zvětšení tloušťky stěny anebo hmotnosti myokardu v nepřítomnosti zevních příčin schopných způsobit tento stav (hypertenze, chlopenní vada) [3]. Přitom není rozhodující, zda je příčinou zesílení srdeční stěny vlastní hypertrofie kardiomyocytů, intracelulární akumulace nebo intersticiální infiltrace. Tato klasifikace je pro běžnou klinickou praxi mnohem použitelnější, protože klasifikuje kardiomyopatie na základě běžně dostupných údajů. Nevýhodou, kterou přiznávají sami autoři, je pak zařazení právě amyloidózy, která mnohdy splňuje kritéria zařazení jako HCM. Podle autorů se jedná o onemocnění na pomezí HCM a RCM, které má některé charakteristické známky (EKG, magnetická rezonance), jež umožňují jeho identifikaci.
Patofyziologie a klasifikace amyloidózy
Amyloidóza je systémové onemocnění, u kterého dochází k extracelulárnímu ukládání proteinové substance „amyloidu“ v tkáních různých orgánů. Amyloid vzniká agregací fibril bílkoviny, u nichž došlo k chybné konformaci. Ty pak mají tendenci k tvorbě velmi stabilních agregátů. Vzniká tak postupně síť vláken, která nepodléhá proteolýze. Na tvorbě a ukládání amyloidu se podílí nejen změna jeho struktury, ale také patologické zvýšení koncentrace amyloidogenního proteinu. K jeho zvýšení dochází například při zvýšené tvorbě monoklonálního imunoglobulinu patologickým klonem kmenových buněk, nebo reaktantu akutní fáze u chronických zánětlivých onemocnění. Negativní působení amyloidu je komplexní a zahrnuje přímou kompresi buněk, zvýšený oxidační stres a také přímou cytotoxicitu proteinových prekurzorů amyloidu. Podle typu proteinové substance dochází také k predilekčnímu postižení některých orgánů.
Ukládání amyloidu v myokardu vede k difuznímu ztlušťování srdečních stěn s jejich následnou zvýšenou tuhostí. To pak vede k poruše především diastolické funkce srdce, která je progresivní a vede až k obrazu restriktivního plnění. Později se přidává také postižení systolické funkce, které je však většinou pouze lehké. Ukládání amyloidu ve stěně srdečních cév pak může vést k ischemii. Obě srdeční síně bývají dilatovány, a to je způsobené jednak diastolickou dysfunkcí, jednak vlastní infiltrací myokardu síní. Infiltrace myokardu zvyšuje riziko supraventrikulárních, ale také komorových arytmií či náhlé srdeční smrti.
Postižení myokardu amyloidem je nejčastěji způsobené ukládáním volných lehkých řetězců u AL amyloidózy. Toto onemocnění postihuje stejně často muže a ženy ve věku nad 50 let. Zhruba u 10–15 % případů je spojeno s mnohočetným myelomem a v tomto případě má horší prognózu. V ostatních případech bývá příčinou tvorby amyloidu monoklonální gamapatie. V následujících kapitolách bude pojednáno především o této formě amyloidózy. Relativně časté a klinicky méně významné je postižení srdce u senilní systémové amyloidózy, kdy je amyloid tvořen normálním transthyretinem, typicky u jedinců starších 70 let. Srdce je téměř vždy postiženo a často se jedná o jediné patrné postižení. Ve srovnání s AL amyloidózou má onemocnění benignější průběh. Hereditární amyloidóza je autozomálně dominantní onemocnění s různou geografickou penetrací. Je nejčastěji způsobená mutací transthyretinu, který je produkován převážně v játrech. Obě pohlaví jsou zastoupena rovnoměrně a onemocnění se může klinicky manifestovat poměrně časně s maximem výskytu v 5. a 6. deceniu. Kromě srdečního postižení je často přítomné neurologické postižení. Izolovaná srdeční amyloidóza je charakterizována ukládáním amyloidu tvořeném z lokálně produkovaného atriálního natriuretického faktoru. Její výskyt se zvyšuje s věkem a může se podílet na vzniku supraventrikulárních arytmií. Systémová amyloidóza (dříve sekundární) je celosvětově nejčastější formou s výskytem především v rozvojovém světě, kde je nejčastěji spojena s chronickými záněty. V rozvinutých zemích pak převažuje jako příčina revmatoidní artritida. Mezi postiženými převažují mírně ženy a k vývoji onemocnění je patrná určitá genetická dispozice. Srdce je u tohoto onemocnění postiženo vzácně a typicky jsou postiženy ledviny [4].
Klinický obraz
Vzhledem k tomu, že je amyloidóza systémové onemocnění, tak se klinický obraz skládá z řady příznaků podle toho, který orgán je postižen. Projevy postižení srdce souvisí s poruchou diastolické funkce a zahrnují příznaky levostranného i pravostranného srdečního selhání. Mezi tyto příznaky patří dušnost, únava, nevýkonnost, nechutenství a periferní otoky. Kromě toho se může vyskytnout ortostatická hypotenze, různé arytmie a angina pectoris. Postižení dalších orgánů se pak může projevovat anémií, tvorbou hematomů, nefrotickým syndromem, periferní neuropatií a selháním ledvin nebo jater.
Při fyzikálním vyšetření se pak kromě známek srdečního selhání (chrůpky, hepatosplenomegalie, zvýšená náplň krčních žil, otoky dolních končetin) a známek postižení výše zmíněných orgánů můžeme setkat s některými specifickými nálezy. Ty zahrnují zvětšení jazyka (makroglosie), periorbitální purpuru a syndrom karpálního tunelu. Tyto známky se však vyskytují pouze u omezené části pacientů, například makroglosie u zhruba 10 % pacientů [5].
Diagnostika
Diagnostický proces u amyloidózy je poměrně komplexní a skládá se z určení rozsahu postižení orgánů amyloidem a stanovení jeho typu. Protože diagnostika a léčba jsou multidisciplinární a jsou vedeny hematologem, zaměříme se v následující části především k popisu diagnostiky srdečního postižení. Znalost srdečního nálezu u amyloidózy je velmi důležitá, protože například u AL amyloidózy se často jedná o první zachycené známky onemocnění.
Elektrokardiografické vyšetření je základní a nejdostupnější vyšetřovací metodou v kardiologii. Jeho důležitost je podpořena faktem, že některé nálezy jsou pro amyloidózu specifické a společně s echokardiografickým obrazem nám často téměř umožní stanovit diagnózu. Typicky nacházíme „sníženou voltáž“ v končetinových svodech (všechny končetinové svody mají amplitudu < 0,5 mV), která je přítomna u 46–71 % případů onemocnění především u AL a hereditární amyloidózy [6]. Kromě toho se můžeme setkat s obrazem „pseudoinfarktu“ přední stěny s patologickými Q kmity, ale tyto změny mohou být lokalizovány prakticky kdekoli. Typický nález na EKG je na obr. 1.
Echokardiografický nález u amyloidózy zahrnuje difuzní ztluštění stěn levé komory a často také pravé komory. U myokardu se může popisovat charakteristická textura „zrnitého skla“. Předpokládá se, že jde o akustický „mismatch“ mezi reflexními depozity amyloidu. Nález je však přítomen jen někdy a jeho přítomnost závisí na vyšetřitelnosti a typu zobrazení [7]. Na rozdíl od levé i pravé síně nejsou levá i pravá komora typicky dilatovány. Často také pozorujeme ztluštění mezisíňové přepážky. Ztluštění také zahrnuje chlopenní struktury a papilární svaly. Může být přítomná lehká regurgitační chlopenní vada, ale významné postižení je vzácné. Při vyšetření se také můžeme setkat s malým perikardiálním výpotkem. Charakteristickým nálezem pro amyloidózu jsou echokardiografické známky diastolické dysfunkce, která v časných fázích může odpovídat poruše relaxace, ale v typických případech se setkáváme s obrazem restriktivního plnění u transmitrálního průtoku, který je nalézán u 21–88 % pacientů [8]. Při tkáňově-dopplerovském zobrazení mitrálního prstence se setkáváme se snížením amplitud měřených vln. Obecně lze říct, že echokardiografický obraz není u amyloidózy specifický a v průběhu onemocnění se mění. V časných stadiích se můžeme setkat s mírnou hypertrofií levé komory s poruchou diastolické funkce, která se dále vyvíjí až do obrazu restrikce. Později můžeme pozorovat také hypertrofii pravé komory. Systolická funkce je zprvu normální, později může být snížená. Také perikardiální výpotek patří do pokročilého stadia onemocnění. Obstrukce ve výtokovém traktu levé komory je na rozdíl od HCM extrémně vzácná. Je důležité, že na základě echokardiografického vyšetření nemůžeme rozlišit jednotlivé typy amyloidózy.
Magnetická rezonance je metodou, která má nově významné místo v diagnostice amyloidózy. Kromě zhodnocení vlastní morfologie srdce se ztluštěním jeho stěn nám umožňuje pomocí vyšetření s kontrastem hodnotit vlastní infiltraci amyloidem. Kontrastní vyšetření s gadoliniem technikou pozdního sycení („late enhancement“) zobrazuje u amyloidózy poměrně typické globální postižení často s maximem subendokardiálně (obr. 4). Senzitivita změn u magnetické rezonance u pacientů s amyloidózou je podle některých autorů velmi vysoká a dosahuje hodnot 80–97 % [9,10].
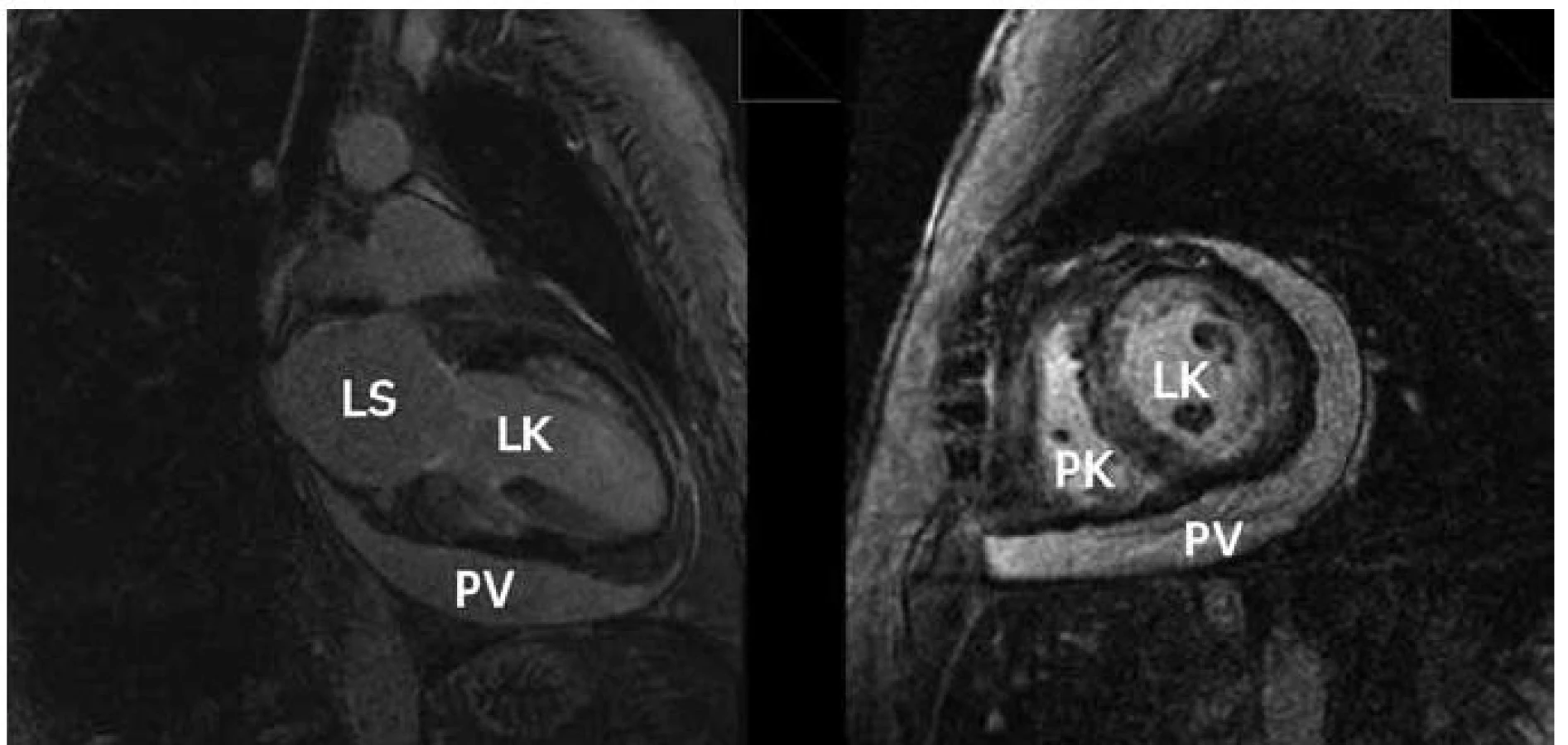
Endomyokardiální biopsie je „zlatým standardem“ v diagnostice srdeční amyloidózy. Mikroskopicky tvoří amyloid amorfní extracelulární depozita, která jsou patrná již při barvením hematoxylinem-eosinem. Barvení Kongo červení je pak specifické a amyloid se barví červeně. Charakteristický je také barevný dvojlom v polarizačním světle. Při průkazu amyloidu je nutné doplnit jeho typizaci, na které závisí další léčba. Protože je endomyokardiální biopsie invazivní výkon, rezervujeme její provedení pro případy určitých diagnostických nejasností. U většiny ostatních stačí typický morfologický obraz srdečního postižení spolu s průkazem amyloidu v biopsii rektální sliznice nebo podkožního tuku.
Laboratorní diagnostika je u pacientů s podezřením na amyloidózu velmi důležitá a spočívá v identifikaci a kvantifikaci patologické bílkoviny. Standardně se provádí vyšetření monoklonálního imunoglobulinu pomocí elektroforézy nebo imunofixace ze séra nebo ještě lépe z moči. Toto vyšetření má dobrou senzitivitu, která dosahuje 71 % pro imunofixaci séra, respektive 84 % u moči [11]. Nicméně i tak není paraprotein v 20 % případů detekován. Proto je dnes preferováno stanovení sérové hladiny volných lehkých řetězců imunoglobulinů typu kappa a lambda, které je 10krát senzitivnější než stanovení pomocí imunofixace. Kromě stanovení vlastní hladiny obou řetězců je důležité také stanovení jejich poměru, protože u jiných imunopatologických stavů sice může stoupnout jejich hladina, ale jejich poměr zůstává zachován. Normální poměr kappa/lambda řetězců je 0,6 (0,26–1,65). Ačkoli abnormalita při tomto vyšetření není nutně podmíněna AL amyloidózou, je jejich normální hodnota prakticky vyloučením tohoto onemocnění. U ostatních laboratorních vyšetření se můžeme setkat s mírnou elevací hladiny troponinu I a T a v důsledku srdečního selhání se zvýšenou hladinou natriuretických peptidů.
Pokud máme podezření na AL amyloidózu, měla by být provedena kostní trepanobiopsie. Nález monoklonálních plazmatických buněk je přítomen u 84 % pacientů s AL amyloidózou při použití imunofluorescenčních technik. Současně může být diagnostikován mnohočetný myelom.
Diferenciální diagnostika
V diferenciální diagnóze je především nutné odlišit amyloidózu od typické HCM. K podezření, že se jedná o amyloidózu, by nás mělo nasměrovat několik skutečností při vyšetření. Na prvním místě obraz „nízké voltáže“ na EKG při současně přítomné hypertrofii levé komory. U echokardiografického vyšetření bychom měli zpozornět, pokud bude přítomen restriktivní mitrální průtok, postižení bude symetrické a myokard bude mít „zrnitou“ texturu. Kromě levé komory bude také postižena pravá komora a mezisíňové septum a bude přítomen malý perikardiální výpotek. U všech těchto nálezů bychom měli provést imunoelektroforézu séra a moči nebo ještě lépe stanovení hladiny volných lehkých řetězců v séru. Jejich zvýšená hladina je velmi suspektní z AL amyloidózy a tito pacienti by pak měli být pokud možno rychle komplexně vyšetřeni včetně histologického průkazu postižení (biopsie). Naopak přítomnost významné obstrukce ve výtokovém traktu svědčí spíše pro HCM. Další možností je provedení magnetické rezonance, která má pro amyloidózu poměrně typický nález. Relativně specifické známky svědčící pro amyloidózu jsou uvedeny v tab. 1.

Léčba
Amyloidóza je systémové onemocnění a srdce je často pouze jedním z orgánů, který je postižen. Proto je diagnostika často multidisciplinární. Vzhledem k častému postižení srdce patří důležité místo kardiologům. Naopak léčba je řízena hematologem a kardiolog má jen pomocnou úlohu v symptomatické léčbě. Ze své povahy se jedná o progresivní onemocnění, u kterého léčba pouze zabrání dalšímu ukládání amyloidu a tím zhoršování klinického stavu. Především u AL amyloidózy je progrese onemocnění velmi rychlá, a tak jsou včasná diagnostika a okamžité zahájení léčby velmi důležité.
Hematologem vedená léčba AL amyloidózy tedy spočívá v zamezení nebo potlačení tvorby amyloidogenního proteinu pomocí kombinované chemoterapie, autologní transplantace kmenových buněk nebo kombinace obou metod. Důležitost včasné diagnostiky onemocnění spočívá v možnosti použití agresivnějších metod léčby spočívajících ve vysokodávkované chemoterapii s následnou autologní transplantací kostní dřeně, která má nejlepší výsledky z hlediska přežití. Problémem je vysoké riziko léčby, pro které je omezena pouze pro časná stadia onemocnění. Riziko se hodnotí na základě parametrů, mezi které patří symptomy srdečního selhání maximálně II. stupně funkční klasifikace, věk do 65 let, hypertrofie levé komory menší než 15 mm. U ostatních pacientů je indikována chemoterapie založená na melfalanu nebo u terminálních stadií léčba kortikoidy. Jsou popsány případy, kdy byla společně s hematologickou léčbou provedena úspěšná transplantace srdce. Důležitost včasné diagnostiky dokládá zlepšení mediánu přežívání pacientů při agresivní léčbě z 6,5 měsíce na 40 měsíců v recentně publikované studii [12].
Léčba ostatních typů amyloidózy je odlišná. U hereditární amyloidózy spočívá v časné transplantaci jater nebo v některých případech v kombinované transplantaci srdce a jater. U senilní a izolované síňové amyloidózy není specifická léčba známa.
Symptomatická léčba srdečních symptomů u amyloidózy zahrnuje restrikci tekutin a opatrnou aplikaci diuretik. Léčba vazodilatancii není dobře snášena pro hypotenze. Léčba digoxinem a blokátory kalciových kanálů je kontraindikována pro jejich vazbu na amyloid, kdy mohou lokálně dosahovat toxických hodnot spojených s poruchami rytmu.
Závěr
Srdeční amyloidóza je závažné onemocnění, které má u neléčených pacientů prognózu srovnatelnou s maligním nádorovým onemocněním. Protože stadium onemocnění, v kterém je zahájena léčba, předurčuje její intenzitu a úspěšnost, je důležité toto onemocnění časně diagnostikovat. Proto by na něj měli kardiologové myslet v rámci diferenciální diagnostiky „tlustého myokardu“ a u pacientů s jinak nevysvětlitelnou hypertrofií levé komory provést stanovení volných lehkých řetězců, které dokáže vyloučit AL amyloidózu.
Doručeno do redakce 4. 10. 2011
Přijato po recenzi 25. 10. 2011
MUDr. David Zemánek
doc. MUDr. Kateřina Linhartová, Ph.D.
Kardiologická klinika 2. LF UK a FN Motol Praha
zejada@seznam.cz
Sources
1. Falk RH. Diagnosis and management of cardiac amyloidoses. Circulataion 2005; 112, 2047–2060.
2. Maron BJ, Towbin JA, Thiene G et al. American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113: 1807–1816.
3. Elliott P, Andersson B, Arbustini E et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008; 29: 270–276.
4. Lachmann HJ, Goodman HJ, Gilbertson JA et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med 2007; 356: 2361–2371.
5. Connors LH, LIM A, Prokaeva T et al. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid 2003; 10: 160–184.
6. Dubrey SW, Cha K, Anderson J et al. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM 1998; 91: 141–157.
7. Linhartová K. Restriktivní kardiomyopatie. In: Veselka J, Linhartová K, Zemánek D (eds). Kardiomyopatie. Praha: Galén 2009: 57–88.
8. Selvanayagam JB, Hawkins PN, Paul B et al. Evaluation and management of the cardiac amyloidosis. J Am Coll Cardiol 2007; 50: 2101–2110.
9. Vogelsberg H, Mahrholdt H, Deluigi CC et al. Cardiovascular magnetic resonance in clinically suspected cardiac amyloidosis: noninvasive imaging compared to endomyocardial biopsy. J Am Coll Cardiol 2008; 51: 1022–1030.
10. Syed IS, Glockner JF, Feng D et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging 2010; 3: 155–164.
11. Gertz MA, Comenzo R, Falk RH et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. Am J Hematol 2005; 79: 319–328.
12. Wechalekar AD, Hawkins PN, Gillmore JD. Perspectives in treatment of AL amyloidosis. Br J Haematol 2008; 140: 365–377.
Labels
Paediatric cardiology Internal medicine Cardiac surgery CardiologyArticle was published in
Cardiology Review

2011 Issue 4
Most read in this issue
- Není hypertrofie jako hypertrofie aneb Nezapomínejme na amyloidózu
- Kalcifikace perikardu
- Nesarkomerické formy hypertrofické kardiomyopatie v dospělosti
- Peripartální kardiomyopatie