
Obrovskobuněčná myokarditida a sarkoidóza srdce – update 2015
Giant cell myocarditis and cardiac sarcoidosis – update 2015
Giant cell myocarditis (GCM) and cardiac sarcoidosis are rare forms of myocarditis resistant to conventional management of heart failure and arrhythmias. GCM usually presents as fulminant myocarditis or subacute progressive non-ischemic heart failure. Advanced atrioventricular blockade and/ or ventricular tachycardias either accompany the aforementioned heart failure syndromes or represent predominant clinical manifestations of GCM. Except for the fulminant clinical course, cardiac sarcoidosis has a clinical presentation similar to GCM. However, its progression is slower, usually across a time interval of several months. The diagnosis of GCM is based mainly on endomyocardial biopsy. In contrast, endomycardial biopsy has a low sensitivity for the diagnosis of cardiac sarcoidosis. In the case of non-diagnostic endomyocardial biopsy, we need histological detection of extracardiac sarcoidosis and signs of cardiac involvement revealed by magnetic resonance imaging or positron emission tomography to diagnose cardiac sarcoidosis. Immunosuppressive treatment is a key component of the management of both diseases. Non-pharmacologic methods of heart failure and arrhythmia management are often necessary.
Keywords:
giant cell myocarditis – cardiac sarcoidosis
Authors:
doc. MUDr. Miloš Kubánek, Ph.D. 1; L. Voska 2
Authors‘ workplace:
Klinika kardiologie, Kardiocentrum, IKEM, Praha
1; Pracoviště klinické a transplantační patologie, Transplantcentrum, IKEM, Praha
2
Published in:
Kardiol Rev Int Med 2015, 17(4): 295-299
Category:
Cardiology Review
Overview
Obrovskobuněčná myokarditida (GCM) a sarkoidóza srdce jsou vzácné formy myokarditidy, jež jsou rezistentní na konvenční léčbu srdečního selhání a arytmií. GCM se manifestuje především jako fulminantní myokarditida nebo subakutně progredující neischemické srdeční selhání. Pokročilá atrioventrikulární blokáda a/ nebo komorové tachykardie doprovázejí předchozí manifestace GCM nebo se vyskytují u GCM izolovaně. Sarkoidóza srdce má kromě fulminantního průběhu podobné klinické projevy jako GCM, progreduje však v řádu měsíců. Diagnostika GCM se opírá především o endomyokardiální biopsii. V případě srdeční sarkoidózy má endomyokardiální biopsie malou senzitivitu, při nediagnostické biopsii vycházíme z histologického průkazu sarkoidózy extrakardiálně a známek srdečního postižení na magnetické rezonanci nebo pozitronové emisní tomografii. U obou onemocnění je přínosná imunosupresivní léčba, kterou je třeba často doplnit dalšími nefarmakologickými léčebnými postupy.
Klíčová slova:
obrovskobuněčná myokarditida – srdeční sarkoidóza
Úvod
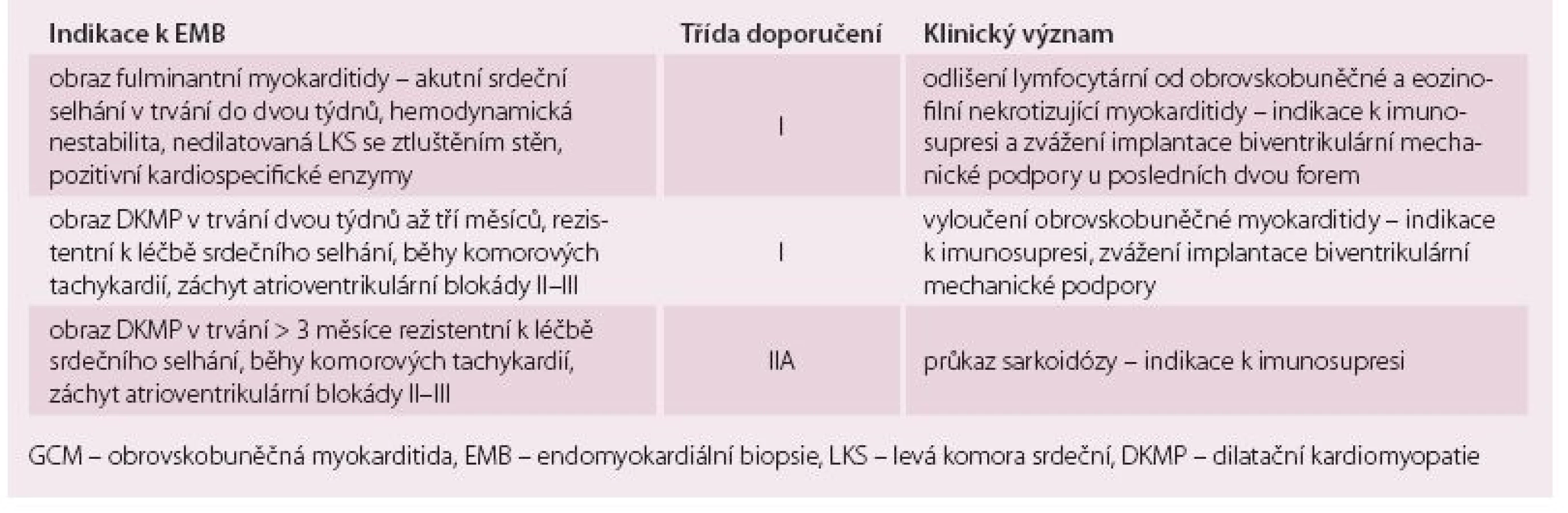
Obrovskobuněčná myokarditida (giant cell myocarditis – GCM) a sarkoidóza srdce jsou prognosticky závažné formy myokarditidy, které jsou rezistentní na konvenční léčbu srdečního selhání a arytmií. Průběh obou onemocnění lze modifikovat imunosupresivní léčbou, proto je důležitá časná diagnostika. Tab. 1 shrnuje klinické situace [1,2], kdy je třeba u pacienta s podezřením na myokarditidu, na GCM a sarkoidózu srdce myslet.
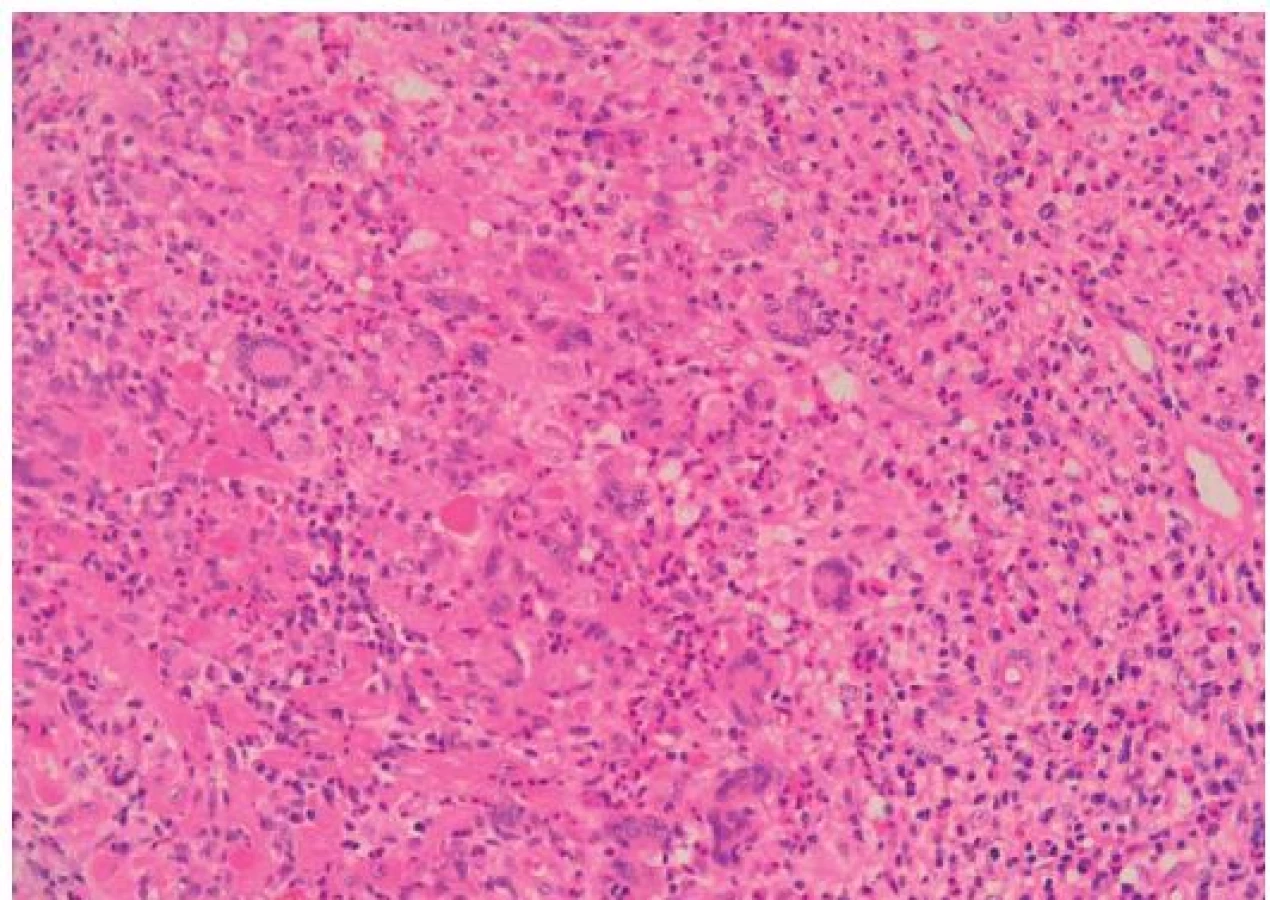
Histopatologie GCM a sarkoidózy myokardu
GCM popsal v roce 1905 Saltykow jako fatální případ myokarditidy charakterizovaný přítomností obrovských mnohojaderných buněk, difuzní zánětlivou infiltrací a nekrózou okolních kardiomyocytů [3,4]. Tato triáda je dodnes základem histopatologické diagnostiky GCM. Zánětlivé infiltráty obsahují především T lymfocyty a četné eozinofily, pokud se tvoří granulomy, jsou nepravidelně tvarované (obr. 1). Naproti tomu pro sarkoidózu myokardu je typická tvorba dobře ohraničených nekazeifikujících granulomů z epiteloidních buněk s přítomností mnohojaderných buněk, které někdy obsahují asteroidní inkluze, tzv. Schaumannova tělíska. Tyto granulomatózní změny jsou provázeny fibrózou, pouze málo aktivním zánětlivým infiltrátem a minimálním výskytem nekrózy kardiomyocytů (obr. 2). Do širší diferenciální diagnostiky granulomatózní myokarditidy patří vyloučení infekční příčiny (syfilis, TBC, mykózy u imunokompromitovaných pacientů), systémových onemocnění (Wegenerova granulomatóza), revmatické karditidy (Aschoffovy uzlíky v intersticiu bez významnější nekrózy myocytů), hypersenzitivní myokarditidy (eozinofilní infiltrát výjimečně provázený ojedinělými obrovskými buňkami), reakce na cizí tělesa (elektrody, kanyly mechanické podpory) a srdečních lymfomů [3–6].
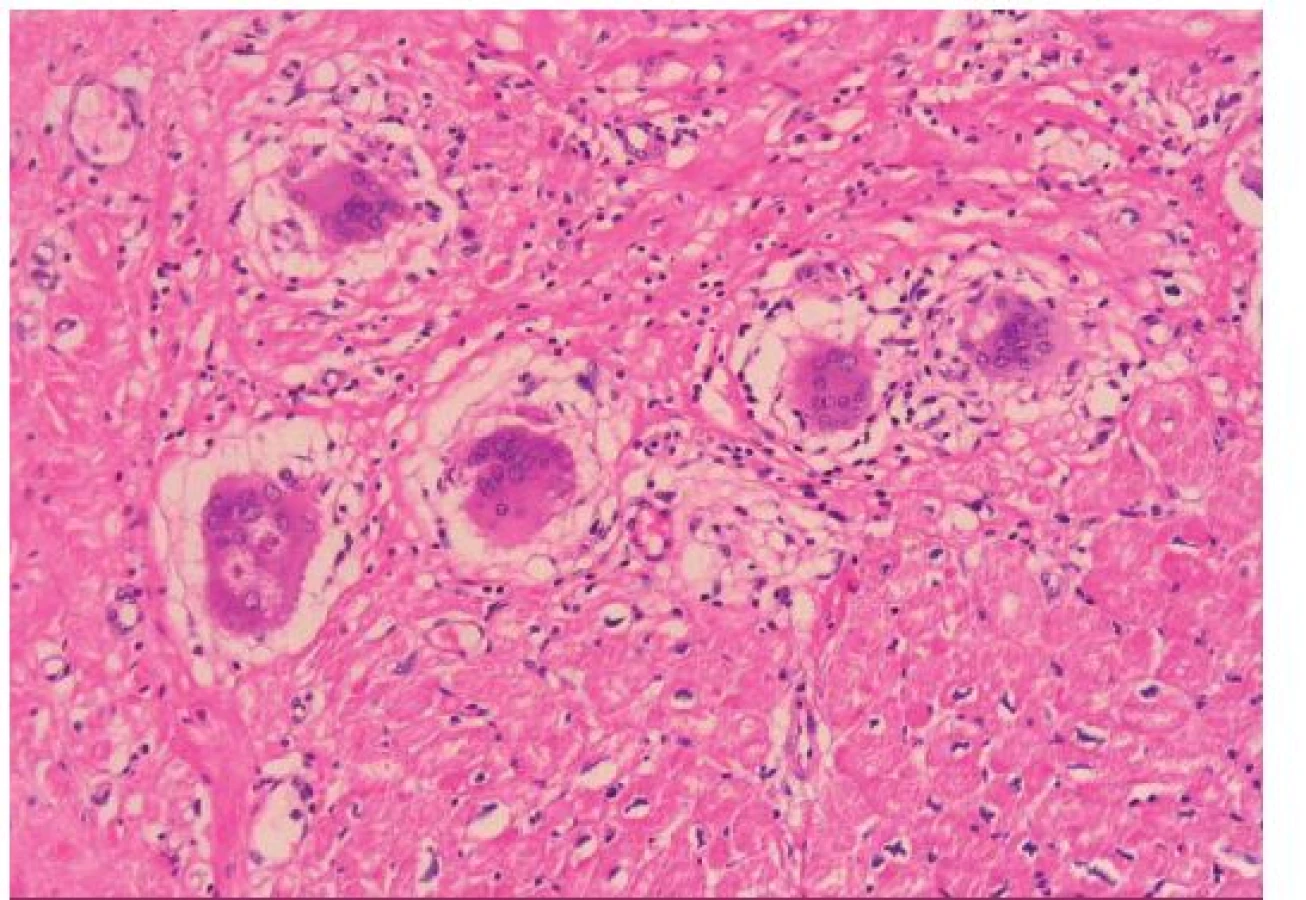
Klinické aspekty GCM
GCM se vyskytuje vzácně, v USA bylo zachyceno u 0,7 % nemocných s provedenou endomyokardiální biopsií (EMB) [5]. Patologové popsali prevalenci 6,6–23,4 případů na 100 000 autopsií [5]. Jedná se o idiopatické onemocnění. Až ve 20 % případů byla dokumentována asociace s autoimunitními chorobami (ulcerózní kolitida, Crohnova nemoc, myozitida kosterních svalů, myastenia gravis, thyroiditida, Takayasuova arteritida aj.). Jindy byla GCM spojena s výskytem nádorů (thymom, bronchogenní karcinom, lymfomy atd.) nebo lékovou alergií (antiepileptika) [3–6]. Autoimunitní etiologii GCM podporují také experimentální data. Obraz GCM lze v experimentu vyvolat imunizací krysy kmene Lewis myozinem [7] nebo přenosem T lymfocytů od postiženého jedince [8].
První klinické popisy onemocnění z 80. let 20. století, vycházely především z nálezů GCM zachycené při autopsii nebo z explantátů srdce při transplantaci [3–5]. Klinický obraz onemocnění upřesnily údaje z multicentrického registru GCM [9] z konce 90. let, kdy již byla běžně dostupná EMB. GCM se projevila v 75 % známkami srdečního selhání, ve 14 % symptomatickými komorovými tachykardiemi a v 5–8 % kompletní atrioventrikulární blokádou [4]. Srdeční selhání probíhalo u části nemocných pod obrazem fulminantní myokarditidy s echokardiografickým nálezem rychle progredující systolické dysfunkce nedilatované levé komory se ztluštěním stěn. Jindy byl rozvoj srdečního selhání subakutní, progrese onemocnění byla patrná v týdnech. Typická byla rezistence na léčbu a častější výskyt maligních arytmií a atrioventrikulární blokády [3–6,9]. Recentní finská práce [10] ukázala, že ve spektru manifestací onemocnění došlo v posledních 20 letech k vývoji. GCM byla v této práci aktivně vyhledávána v rizikových skupinách pacientů. Onemocnění se manifestovalo v 31 % srdečním selháním, v 30 % izolovanou pokročilou atrioventrikulární blokádou, ve 22 % setrvalou komorovou tachykardií, v 13 % bolestmi na hrudi napodobujícími akutní koronární syndrom a ve 4 % oběhovou zástavou při fibrilaci komor [10].
Pro diagnostiku GCM je zásadní EMB. U pacientů z registru GCM z 90. let 20. století s provedenou EMB a dostupným explantátem srdce nebo excizí levé komory při implantaci mechanické srdeční komory byla zjištěna senzitivita EMB pro diagnostiku GCM 82–85 %. Jednalo se však o pokročilé formy onemocnění [11]. Údaje z finského registru [10] prokázaly, že opakování EMB zvýší senzitivitu pro detekci GCM z 68 % při prvním vyšetření na 93 % při rebiopsii. V registru GCM z 90. let byl medián přežívání bez nutnosti srdeční transplantace od manifestace symptomů 5,5 měsíce. U nemocných léčených imunosupresivní léčbou (cyklosporin, kortikoidy a v některých případech azathioprin nebo monoklonální protilátka proti T lymfocytům) byla tato doba významně delší oproti nemocným bez imunosuprese (12,6 měsíce vs. 3 měsíce). Nicméně prognóza onemocnění nebyla příznivá, 70 % nemocných zemřelo nebo podstoupilo transplantaci srdce po roce sledování [4,9]. Jedná se však o retrospektivní práci, kombinovanou imunosupresivní léčbou byly s větší pravděpodobností léčeny lehčí formy onemocnění. Léčba GCM kombinovanou imunosupresí včetně monoklonální protilátky proti T lymfocytům byla testována v jiné práci [12] u devíti pacientů, kde jednoroční přežívání bez nutnosti transplantace dosahovalo 78 %. K podobným výsledkům došli autoři finského registru nemocných s GCM [10], při kombinované imunosupresi přežívalo bez nutnosti srdeční transplantace jeden rok 69 % a pět let 52 %. Tyto výsledky naznačují, že při časné diagnóze a intenzivní léčbě je možno zlepšit prognózu toho onemocnění. Recentně bylo popsáno při léčbě GCM využití novodobé imunosuprese obdobné schématům po transplantaci srdce, konkrétně byl aplikován antithymocytární globulin a kombinace takrolimus, mykofenolát mofetil a kortikoidy [13,14]. V řadě případů je definitivní léčbou srdeční transplantace, často je třeba použít mechanickou srdeční podporu jako most k transplantaci. V registru GCM z 90. let 20. století podstoupilo transplantaci 39 nemocných se srovnatelným pětiletým přežíváním (71 %) jako u jiných diagnóz. V 25 % byla zaznamenána většinou málo symptomatická rekurence GCM v EMB, která byla dobře ovlivnitelná posílením imunosuprese [4,9].
Sarkoidóza srdce
První popis sarkoidózy srdce pochází z roku 1929. Sarkoidóza je systémové onemocnění charakterizované granulomatózním zánětem a možným postižením srdce [15–17]. Histopatologie onemocnění byla popsána na začátku tohoto sdělení. Prevalence sarkoidózy v ČR se pohybuje kolem 60 případů na 100 000 obyvatel. Sarkoidóza myokardu je klinicky manifestní u 3–5 % nemocných se sarkoidózou jiného orgánu, pitevní nálezy u nemocných se sarkoidózou a moderní zobrazovací metody ukazují na podstatně vyšší výskyt srdeční sarkoidózy (až ve 25 %). Obtížně se diagnostikují případy izolované srdeční sarkoidózy bez zjevného extrakardiálního postižení [16–18]. Práce finských autorů ukázala dvacetinásobný vzestup záchytu srdeční sarkoidózy. Tato studie dokumentovala u srdeční sarkoidózy incidenci 5,3 případu a prevalenci 22 případů na milion obyvatel ročně [19]. K postižení srdce dochází především u chronických forem sarkoidózy. Nejčastěji bývají postiženy plíce a lymfatické uzliny, méně často játra, slezina, slinné žlázy, oči, kůže a srdce. Etiologie onemocnění je nejasná. Podle některých hypotéz je granulomatózní proces imunologickou odpovědí na expozici organizmu některými bakteriemi (Mycobacterium sp, Propionibacterium sp.), vlivy životního prostředí nebo profesionální zátěž [6,16–19].
Klinicky se sarkoidóza srdce projevuje srdečním selháním nebo arytmiemi. Srdeční selhání u pacienta se sarkoidózou je nejčastěji způsobeno sarkoidózou myokardu s obrazem dilatační nebo výjimečně restriktivní kardiomyopatie, selháním pravé komory v pokročilých fázích plicního postižení nebo vzácně konstriktivní perikarditidou. Útlak převodního systému granulomatózním procesem v bazální části komorového septa vede k atrioventrikulární blokádě. Difuzní fibrotické změny v myokardu potom vytvářejí substrát pro komorové arytmie. Klinické projevy srdeční sarkoidózy detailně popsal registr finských pacientů z let 1988–2012 [19]. Ze 110 pacientů se srdeční sarkoidózou měly dvě třetiny jedinců izolovanou srdeční formu a jedna třetina srdeční sarkoidózu se současným extrakardiálním postižením. První manifestací byla symptomatická atrioventrikulární blokáda ve 44 %, komorová tachykardie nebo fibrilace komor ve 33 % a srdeční selhání v 18 % případů [19]. V kanadském souboru nemocných ve věku 18–60 let s nevysvětlenou pokročilou atrioventrikulární blokádou byla prokázána pomocí multimodálního vyšetření srdeční sarkoidóza v 34 % případů [20]. V registru komorových tachykardií léčených radiofrekvenční ablací byla srdeční sarkoidóza zachycena v 5 % případů [21]. U pacientů se srdeční sarkoidózou můžeme v EKG také dokumentovat atrioventrikulární blok I. stupně, frekventní komorovou extrasystolii, blok pravého raménka Tawarova nebo obraz jizvy. Na echokardiografii vidíme v časné fázi zánětu lokální ztluštění stěny srdečních komor, později ve fázi jizvení je patrné ztenčení stěny bez vztahu k anatomii koronárního zásobení. Mezi nálezy suspektní ze srdeční sarkoidózy patří aneuryzmata pravé komory nebo ztenčení bazální části komorového septa, jindy vidíme nespecifický obraz globální systolické či diastolické dysfunkce levé komory. Podobně jako echokardiografie je také galiová scintigrafie málo senzitivní metodou [6,15,17]. Více přínosná je magnetická rezonance. Kromě výše zmíněných regionálních poruch kinetiky a změn tloušťky stěny srdečních komor pro srdeční sarkoidózu svědčí pozdní akumulace kontrastní látky s gadoliniem, typicky bývá lokalizována především v bazálních částech komorového septa a boční stěny epikardiálně a intramurálně. Magnetická rezonance (MR) má menší senzitivitu, ale větší specificitu než pozitronové emisní tomografie (PET) s 18fluorodeoxyglukózou (FDG) (75 vs. 87 % a 77 vs. 38 %) [6]. PET využívá pro diagnostiku srdeční sarkoidózy zvýšenou akumulaci 18F-FDG v místech zánětlivé aktivity a výpadky perfuze zobrazené pomocí 82rubidia. Necílená EMB má senzitivitu pro diagnostiku srdeční sarkoidózy pouze 20–25 %. Výtěžnost EMB lze zvýšit odběrem vzorků z postižených míst srdečních komor identifikovaných pomocí neinvazivních metod nebo elektroanatomického mapování [17]. Finská práce ukázala malou senzitivitu laboratorních markerů pro diagnostiku izolované srdeční sarkoidózy (solubilní ACE 15 %, lyzozym 44 %, zvýšená kalciurie 38 %) [19]. Žádná z uvedených metod neumožňuje spolehlivou diagnostiku srdeční sarkoidózy. Vycházíme proto ze skórovacích systémů, nejnověji ze systému WASOG (tab. 2). U všech jedinců s prokázanou extrakardiální sarkoidózou je doporučen screening srdečního postižení pomocí EKG a echokardiografie, někteří autoři doporučují také holterovskou monitoraci EKG. Při abnormálním výsledku těchto vyšetření je třeba doplnit magnetickou rezonanci nebo PET. Na diagnózu sarkoidózy myokardu je třeba pomýšlet také u nemocných s nevysvětlenou pokročilou atrioventrikulární blokádou ve věku 18–60 let nebo nevysvětlenou systolickou dysfunkcí levé komory, zvláště při rezistenci na léčbu srdečního selhání a současném výskytu komorových arytmií a atrioventrikulární blokády vyššího stupně [1,17]. U těchto nemocných pátráme po extrakardiálních projevech sarkoidózy pomocí HRCT hrudníku a/ nebo PET. Pozitronová emisní tomografie umožňuje zacílit biopsii k potvrzení extrakardiální sarkoidózy (např. na nitrohrudní uzliny). K navigaci EMB mohou posloužit magnetická rezonance (MR), PET nebo voltážová mapa [17].

Starší práce uváděly pětileté přežívání nemocných se srdeční sarkoidózou v rozmezí 70–75 %, často ještě nižší, pokud byli do studie zařazeni nemocní s pozdní diagnózou onemocnění (post mortem nebo v explantátu srdce při transplantaci) [22,23]. V současné éře časné diagnostiky a léčby onemocnění bylo ve finské práci pětileté a desetileté přežívání bez potřeby srdeční transplantace 90 % a 83 % [19]. Přítomnost srdečního selhání při manifestaci onemocnění negativně ovlivňovala přežívání bez nutnosti srdeční transplantace, byla spojena s pěti - a desetiletým přežíváním 75 % a 52 %. Izolovaná forma srdeční sarkoidózy byla v porovnání s případy s extrakardiálním postižením asociována s těžším postižením srdce s nižší ejekční frakcí levé komory (LVEF), větším rozsahem pozdní akumulace gadolinia a horší prognózou. Cílový ukazatel složený z mortality, nutnosti srdeční transplantace a výskytu maligních arytmií předpovídaly nízká LVEF a izolovaná forma srdeční sarkoidózy [19].
Kortikoidy jsou základním lékem pro léčbu sarkoidózy srdce, iniciální dávka prednizonu je obvykle 1 mg/ kg a den s postupnou redukcí. Někdy se kombinují s azatioprinem nebo metotrexátem. Indikace k imunosupresivní léčbě vychází z retrospektivních a observačních studií [19,24]. U pacientů s pokročilou atrioventrikulární blokádou lze po léčbě kortikoidy očekávat úpravu atrioventrikulárního převodu skoro v polovině případů, nicméně častá je rekurence blokády [25]. Recentní konsenzus expertů proto doporučuje v těchto případech trvalou stimulaci, vzhledem k riziku maligních komorových arytmií je preferována implantace kardioverteru-defibrilátoru (ICD) [26]. V ostatních případech srdeční sarkoidózy zaléčené imunosupresí a standardní léčbou srdečního selhání je implantace ICD doporučena, pokud jsou splněna kritéria pro primárně nebo sekundárně preventivní implantaci (klinická arytmie a/ nebo LVEF ≤ 35 %), při anamnéze synkopy s pozitivní programovanou stimulací komor, dále u LVEF 36–49 % a/ nebo RVEF < 40 % s pozitivní programovanou stimulací komor [26]. ICD není doporučován u jedinců bez anamnézy synkopy, s normální systolickou funkcí obou komor, bez přítomnosti pozdní akumulace gadolinia na MR, s negativní programovanou stimulací komor a bez indikace k trvalé stimulaci [26]. V literatuře byly popsány příznivé výsledky transplantace srdce u nemocných se srdeční sarkoidózou, které byly srovnatelné s ostatními diagnózami [27]. U nemocných s pokročilým srdečním selháním na podkladě srdeční sarkoidózy bez významnějšího extrakardiálního postižení lze tedy zvážit provedení srdeční transplantace.
Závěr
Vzhledem k obtížné diagnostice, rezistenci na konveční léčbu srdečního selhání a efektivitě imunosupresivní léčby je třeba nemocné s podezřením na GCM a/ nebo sarkoidózu srdce směřovat do komplexních kardiocenter. V rámci vyšetření příčin neischemického srdečního selhání se této problematice intenzivně věnujeme také na naší klinice a rádi se o tyto nemocné postaráme.
Podpořeno výzkumným záměrem Ministerstva zdravotnictví České republiky pro rozvoj výzkumné organizace 00023001 (IKEM, Praha, ČR) – institucionální podpora a dále grantem AZV-MZ 15-27682A.
Doručeno do redakce: 4. 7. 2015
Přijato po recenzi: 22. 10. 2015
MUDr. Miloš Kubánek, Ph.D.
www.ikem.cz
milos.kubanek@ikem.cz
Sources
1. Cooper LT, Baughman KL, Feldman AM et al. The role of endomyocardial biopsy in the management of cardiovascular disease. AHA/ ACCF/ ESC scientific statement. Eur Heart J 2007; 28 : 3076–3093. doi:10.1093/ eurheartj/ ehm456.
2. Caforio AL, Pankuweit S, Arbustini E et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013; 34 : 2636–2648. doi: 10.1093/ eurheartj/ eht210.
3. Cooper LT. Myocarditis. N Engl J Med 2009; 360 : 1526–1528. doi: 10.1056/ NEJMra0800028.
4. Cooper LT. Idiopathic giant cell myocarditis. In: Cooper LT (ed.) Myocarditis. From bench to bedside. Totowa, New Jersey: Humana Press 2003 : 405–420.
5. Bennett MK, Gilotra NA, Harrington C et al. Evaluation of the role of endomyocardial biopsy in 851 patients with unexplained heart failure from 2000–2009 clinical perspective. Circ Heart Fail 2013; 6 : 676–684. doi: 10.1161/ CIRCHEARTFAILURE.112.000087.
6. Blauwet LA, Cooper LT. Idiopathic giant cell myocarditis and cardiac sarcoidosis. Heart Fail Rev 2013; 18 : 733–746. doi: 10.1007/ s10741-012-9358-3.
7. Kodama M, Matsumoto Y, Fujiwara M et al. A novel experimental model of giant cell myocarditis induces in rats by immunization with cardiac myosin fraction. Clin Immunol Immunopathol 1990; 57 : 250–262.
8. Kodama M, Matsumoto Y, Fujiwara M. In vivo lymphocyte-mediated myocardial injuries demostrated by adoptive transfer of experimental autoimmune myocarditis. Circulation 1992; 85 : 1918–1926.
9. Cooper LT jr, Berry GJ, Shabetai R. Multicentre giant cell myocarditis study group investigators. Idiopathic giant-cell myocarditis-natural history and treatment. N Engl J Med 1997; 336 : 1860–1866.
10. Kandolin R, Lehtonen J, Salmenkivi K et al. Diag-nosis, treatment, and outcome of giant-cell myocarditis in the era of combined immunosuppression. Circ Heart Fail 2013; 6 : 15–22. doi: 10.1161/ CIRCHEARTFAILURE.112.969261.
11. Shields RC, Tazelaar HD, Berry GJ et al. The role of right ventricular endomyocardial biopsy for idiopathic giant cell myocarditis. J Card Fail 2002; 8 : 74–78. doi: 10.1054/ jcaf.2002.32196.
12. Cooper LT jr, Hare JM, Tazelaar HD et al. Usefulness of immunosuppression for giant cell myocarditis. Am J Cardiol 2008; 102 : 1535–1539. doi: 10.1016/ j.amjcard.2008.07.041.
13. Grabmaier U, Brenner C, Methe H et al. An alternative immunosuppressive regimen to prolong transplant free survival in a patient with giant cell myocarditis. Int J Cardiol 2013; 168: e27–e28. doi: 10.1016/ j.ijcard.2013.05.078.
14. Chaudhry MA, Correa A, Lee C et al. Modern day management of giant cell myocarditis. Int J Cardiol 2015; 178 : 82–84. doi: 10.1016/ j.ijcard.2014.10.131.
15. Cooper LT. Cardiac sarcoidosis. In: Cooper LT (ed.) Myocarditis. From bench to bedside. Totowa, New Jersey: Humana Press 2003 : 421–436.
16. Dubrey SW, Falk RH. Diagnosis and management of cardiac sarcoidosis. Prog Cardiovasc Dis 2010; 52 : 336–346. doi: 10.1016/ j.pcad.2009.11.010.
17. Kron J, Ellenbogen KA. Cardiac sarcoidosis: Contemporary review. J Cardiovasc Electrophysiol 2015; 26 : 104–109. doi: 10.1111/ jce.12552.
18. Hajšl M, Sedloň P, Černohous M et al. Sarkoidóza srdce jako příčina náhlé srdeční smrti. Cor Vasa 2013; 55: e78–e81. doi: 10.1016/ j.crvasa.2012.08.006.
19. Kandolin R, Lehtonen J, Airaksinen J et al. Cardiac sarcoidosis. Epidemiology, characteristics and outcome over 25 years in a nationwide study. Circulation 2015; 131 : 624–632. doi: 10.1161/ CIRCULATIONAHA.114.011522.
20. Nery PB, Beanlands RS, Nair GM et al. Atrioventricular block as the initial manifestation of cardiac sarcoidosis in middle-aged adults. J Cardiovasc Electrophysiol 2014; 25 : 875–881. doi: 10.1111/ jce.12401.
21. Kumar S, Barbhaiya C, Nagashima K et al. Ventricular tachycardia in cardiac sarcoidosis. Characterisation of ventricular substrate and outcomes of catheter ablation. Circ Arrhythm Electrophysiol 2015; 8 : 87–93. doi: 10.1161/ CIRCEP.114.002145.
22. Okura Y, Dec GW, Hare LM et al. A clinical and histopathologic comparison of cardiac sarcoidosis of sarcoidosis and idiopathic giant cell myocarditis. J Am Coll Cardiol 2003; 41 : 322–329. doi:10.1016/ S0735-1097(02)02715-8.
23. Yazaki Y, Isobe M, Hiroe M et al. Prognostic determinants of long-term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol 2001; 88 : 1006–1010. doi: 10.1016/ S0002-9149(01)01978-6.
24. Chiu CZ, Nakatani S, Zhang G et al. Prevention of left ventricular remodeling by long-term corticosteriod therapy in patients with cardiac sarcoidosis. Am J Cardiol 2005; 95 : 143–146. doi: 10.1016/ j.amjcard.2004.08.083.
25. Sadek MM, Yung D, Birnie DH et al. Corticosteroid therapy for cardiac sarcoidosis: a systematic review. Can J Cardiol 2013; 29 : 1034–1041. doi: 10.1016/ j.cjca.2013.02.004.
26. Birnie DHS, Bobun F, Cooper J et al. HRS expert consensus statement on diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm 2014; 11 : 1305–1323. doi: 10.1016/ j.hrthm.2014.03.043.
27. Zaidi AR, Zaidi A, Vaitkus PT. Outcome of heart transplantation in patients with sarcoid cardiomyopathy. J Heart Lung Transplant 2007; 26 : 714–717. doi: 10.1016/ j.healun.2007.05.006.
Labels
Paediatric cardiology Internal medicine Cardiac surgery CardiologyArticle was published in
Cardiology Review

2015 Issue 4
Most read in this issue
- Perikarditidy
- Myokarditidy a zánětlivé kardiomyopatie
- Srdeční vrozené vady v dospělosti
- Infekční endokarditida – diagnostika a doporučené postupy