
Angioimunoblastický T-lymfom (AITL) jako velmi nepříznivá malignita – zkušenost centra
Angioimmunoblastic T-cell Lymphoma as a Very Poor-Prognosis Malignancy – a Single Centre Experience
Background:
Angioimmunoblastic T-lymphoma (AITL) is a poor prognosis malignancy. Because of relatively rare incidence and lack of publications in Czech, we decided to share our experience. Patients and Methods: Retrospective analysis of newly diagnosed AITL patients treated at our institution between 1/2000–12/2010.
Results:
Twelve patients with median age of 64 (43–82) years were analysed. Two patients over 80 years were treated with corticosteroids. Ten patients were treated with 6 cycles of CHOP-21 chemotherapy resulting in: 2/10 (20%) stable disease, 5/10 (50%) partial remission and 3/10 (30%) complete remission. The median EFS and OS of chemotherapy-treated patients were 8 and 10 months, resp. The EFS and OS were both significantly longer in patients who achieved complete remission within the first line of CHOP or autologous stem cells transplantation therapy: 43 vs 6 (p = 0.0052) and 46 vs 6 months (p = 0.0023), respectively. It was not possible to perform autologous transplantation in 4/7 (57%) patients in need for further reduction of the disease because of poor performance status or early progression of lymphoma and death during salvage chemotherapy.
Conclusion:
AITL is a poor prognosis malignancy with a very high risk of early relapse after CHOP induction chemotherapy. In fit patients, autologous transplantation should be performed immediately after induction chemotherapy; information about availability of stem cells donor, both in the family or any available register, should be found during the induction treatment.
Key words:
lymphoma – chemotherapy – transplantation
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
17. 5. 2011
Accepted:
24. 8. 2011
Authors:
S. Vokurka 1; V. Koza 1; V. Vozobulová 1; P. Jindra 1; K. Steinerová 1; M. Schutzova 1; L. Boudová 2
Authors‘ workplace:
Hematologicko-onkologické oddělení, FN Plzeň
1; Patologicko-anatomický ústav, FN Plzeň
2
Published in:
Klin Onkol 2012; 25(3): 206-211
Category:
Original Articles
Overview
Východiska:
Angioimunoblastický T-lymfom (AITL) je nepříznivá malignita. Pro relativně vzácný výskyt a nedostatek literárních údajů v českém písemnictví jsme zpracovali naše zkušenosti. Soubor a metody: Retrospektivní analýza pacientů s nově diagnostikovaným AITL a léčených na našem pracovišti v 1/2000–12/2010.
Výsledky:
Analyzováno 12 pacientů s mediánem věku 64 (43–82) let. Dva pacienti ve věku nad 80 let byli léčeni kortikoterapií. Deset pacientů bylo léčeno chemoterapií 6× CHOP-21 a bylo dosaženo stavu: stabilní nemoc 2/10 (20 %), parciální remise 5/10 (50 %), kompletní remise 3/10 (30 %). Medián EFS a OS pacientů léčených chemoterapií byl 8 a 10 měsíců. EFS a OS byl signifikantně delší při dosažení stavu CR v rámci 1. linie léčby po 6× CHOP nebo po doplnění autologní transplantace krvetvorných buněk: 43 vs 6 (p = 0,0052) a 46 vs 6 měsíců (p = 0,0023). U 4/7 (57 %) pacientů s potřebou další minimalizace nemoci po indukci nebylo možné realizovat autologní transplantaci pro nepříznivý stav nebo časnou progresi lymfomu a úmrtí na záchrannou chemoterapii.
Závěr:
AITL je nepříznivá malignita s rizikem velmi časné progrese po indukci CHOP-21. Je-li pacient únosný, je třeba doplnit autologní transplantaci krvetvorných buněk obratem po indukci a již v době indukce mít informace o dostupnosti dárce krvetvorných buněk v rodině, eventuálně registrech.
Klíčová slova:
lymfom – chemoterapie – transplantace
Úvod
Angioimunoblastický T-lymfom (AITL) představuje specifickou malignitu definovanou v rámci lymfoidních T-buněčných neoplazií, respektive periferních T-lymfomů aktuální WHO klasifikace [1]. Původně byl tento typ lymfomu označován v 70. letech jako angioimunoblastická nebo imunoblastická lymfadenopatie [2,3], dále jako lymfogranulomatóza X [4], imunoblastický T-lymfom [5] a od roku 1994 v rámci REAL klasifikace již jako angioimunoblastický lymfom [6].
AITL patří mezi relativně vzácné lymfomy. V období 1999–2006 bylo v rámci registru Kooperativní lymfomové skupiny ČR evidováno 3 518 pacientů a z toho 16 (0,5 %) s AITL [7]. V registrech International Lymphoma Study Group (ILSG) pak AITL zastupuje 1,2 % lymfomů [8].
Svým původem je AITL řazen jako malignita vycházející z CD4 pozitivních folikulárních T-helper lymfocytů. Imunofenotyp nádorových buněk charakterizuje pozitivita CD2, CD3, CD5, CD4, CD10, PD1, bcl6 a CXCL13. U 90 % případů bývá prokazována klonalita T-cell receptoru (TCR), nicméně v 10–20 % případů se vyskytuje i klonální přestavba genu pro těžký řetězec imunoglobulinu (IgH) [1]. Imunomorfologický nález mívá v plně vyvinutých případech typický vzhled, v němž dominují výrazně zmnožené a větvené drobné cévy a extrafolikulární proliferace folikulárních dendritických buněk. Nádorové buňky se vyskytují v různém počtu, mají typicky světlou cytoplazmu (tzv. „clear cells“) a různě vyjádřené atypie. Bývá přítomen polymorfní infiltrát tvořený především lymfocyty, eozinofily, plazmatickými buňkami, dendritickými elementy a histiocyty. Součástí bývají i velké B-blasty, často s pozitivitou EBV, které mohou mít vzhled Reedové-Sternbergových buněk. V případech s méně výraznými imunomorfologickými rysy je patologická diagnóza velmi těžká a někdy vyžaduje opakovanou biopsii a klinickopatologické korelace [24].
AITL postihuje většinou starší pacienty s mediánem věku okolo 60 let. V klinickém obrazu dominují převážně pokročilá stadia nemoci doprovázená celkovými příznaky (horečka, hubnutí, noční pocení), téměř vždy generalizovanou lymfadenopatií a často hepato-splenomegalií. Téměř u poloviny případů se objevují doprovodné kožní projevy v podobě exantémů, purpury, svědění nebo urtiky. Symptomatologii mohou doplňovat kloubní obtíže (artritida, artralgie), otoky, ascites. Průběh je většinou akutní a může připomínat systémová nebo infekční onemocnění nebo jiné lymfoproliferace. Transformace v lymfom vysoké agresivity nebývají časté. Laboratorně bývá přítomna většinou anémie, která může být hemolytická s průkazem pozitivity Coombsova testu, dále trombocytopenie, eozinofilie nebo pozitivita autoprotilátek, jako jsou např. revmatoidní faktor a anti-nukleární faktor [9].
V léčbě se uplatňuje především standardní protokol chemoterapie CHOP [9]. Průběh onemocnění je variantní, nicméně ve většině případů je prognóza nepříznivá, s mediánem přežívání méně než 3 roky i v případě zajištění intenzivní léčby a šance na dlouhodobé přežívání je do 30 % [10,11]. Obecně se zdá, že dosažení kompletní remise je zásadním prognostickým faktorem [9].
S ohledem na relativně vzácné postavení AITL a prakticky chybějící literární údaje v českém písemnictví jsme se rozhodli naše zkušenosti s léčbou tohoto lymfomu zpracovat.
Soubor a metodika
Retrospektivní analýza se týká pacientů s nově diagnostikovaným AITL a hospitalizovaných na našem pracovišti v období 1/2000–12/2010. Diagnóza AITL byla stanovena podle kritérií REAL, respektive WHO klasifikace. U všech pacientů bylo zajištěno histologické a imunohistochemické vyšetření uzlin a současně bylo doplněno i molekulárně-genetické vyšetření na přítomnost klonální přestavby receptoru TCR gama. Biopsie byly odečteny zkušenými patology naší fakultní nemocnice – konzultanty Kooperativní lymfomové skupiny [7,21–23].
Chemoterapie
CHOP-21: cyklofosfamid 750 mg/m2 i.v. 1. den, doxorubicin 50 mg/m2 i.v. 1. den, vinkristin 2 mg i.v., prednison 60 mg/m2 p.o. 1.–5. den.
DHAP: cisplatinum 100 mg/m2 i.v. 1. den, cytarabin 4 000 mg/m2 i.v. 2. den, dexamethason 40 mg/den p.o. 1.–4. den.
BEAM s autologní transplantací periferních krvetvorných buněk: BiCNU 300 mg/m2 i.v. jedenkrát denně v den –6, etoposid 100 mg/m2 i.v. dvakrát denně v den –5 až –2, cytosin-arabinosid 200 mg/m2 i.v. dvakrát denně v den –5 až –2, melphalan 140 mg/m2 i.v. v den –1, transplantace autologního štěpu periferních krvetvorných buněk za 24 hod po aplikaci posledního cytostatika přípravy. Leukaferézy štěpu autologních periferních krvetvorných buněk byly realizovány po přípravě s vysokodávkovaným cyklofosfamidem (3 g/m2 i.v. 1. den) s následnou aplikací filgrastimu 10 µg/kg/den do dne sklizně.
FLU/MEL s alogenní transplantací periferních krvetvorných buněk: fludarabin 30 mg/m2 i.v. jedenkrát denně 4 dny, melfalan 140 mg/m2 i.v. jedenkrát denně den před transplantací alogenního štěpu periferních krvetvorných buněk.
Léčebná odpověď
Léčebná odpověď byla hodnocena podle kritérií National Cancer Institute (NCI). Doba celkového přežití (overall survival – OS) byla stanovena jako počet měsíců od stanovení diagnózy do úmrtí. Doba přežití do události (event-free survival – EFS) byla stanovena jako počet měsíců od stanovení diagnózy do první progrese, relapsu nebo úmrtí.
Statistická analýza
Statistické analýzy byly provedeny s využitím programu GraphPad InStat (GraphPad Software). Byly doplněny Kaplan-Maierovy křivky přežití a log rang test, přičemž významnost sledovaných rozdílů byla stanovena na hladině významnosti 5 % (p = 0,05).
Výsledky
Do analýzy bylo zařazeno celkem 12 (1,9 %) pacientů s nově diagnostikovaným AITL ze souboru celkem 631 pacientů s nově diagnostikovaným non-hodgkinským lymfomem (NHL) a hospitalizovaných na našem pracovišti v období 1/2000–12/2010.
Medián věku pacientů byl 64 (43––82) let. Uzlinové postižení bylo přítomno u všech pacientů (100 %), dále se vyskytovala hepatomegalie (50 %), splenomegalie (33 %), ascites a/nebo hydrothorax (25 %) a infiltrace kostní dřeně (25 %), autoimunitní hemolytická anémie (25 %). Klinické „B“ příznaky (noční pocení, horečky, hubnutí) se vyskytovaly u 66 % pacientů. Bližší charakteristiky souboru jsou uvedeny samostatně v tab. 1.
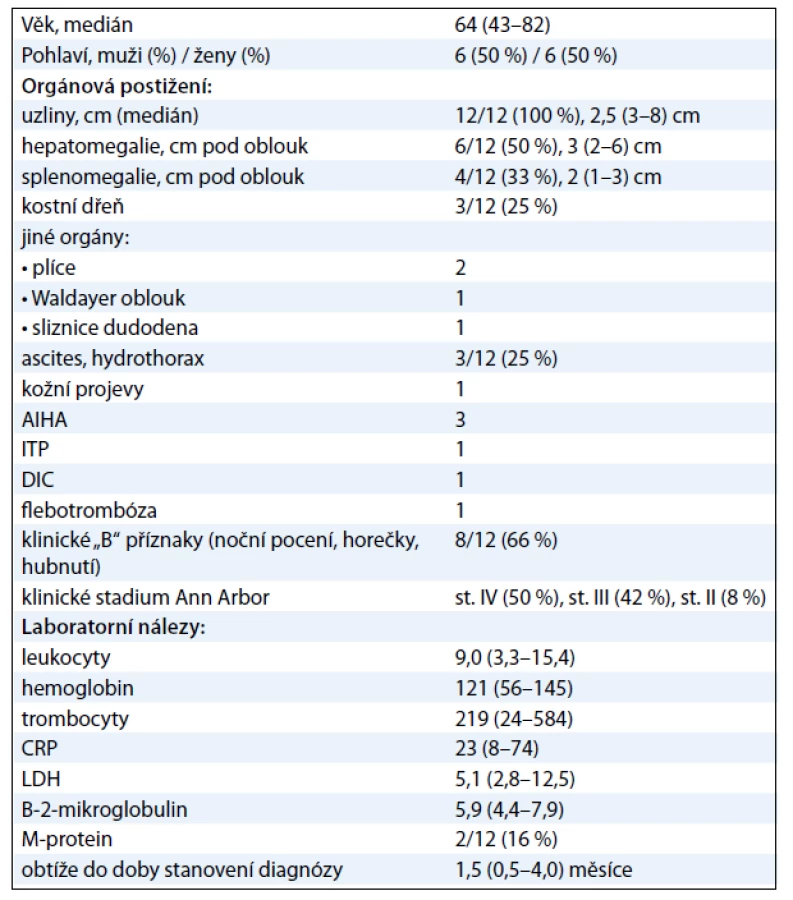
U 2 pacientů s řadou dalších onemocnění a ve věku 81 a 82 let byla zahájena kortikoterapie s prednizonem v úvodní dávce 1 mg/kg/den po dobu 5 dní a dále s redukcí na 10–20 mg/den denně po dobu efektu. Kortikoterapie vedla jen k částečné a přechodné regresi uzlinového postižení nebo klinických „B“ příznaků v trvání do 6 měsíců. Další léčba pacientů pak byla již čistě symptomatická.
Celkem 10 pacientů ve věku 43–72 let podstoupilo indukční chemoterapii se šesti cykly CHOP-21. Po zahájení léčby došlo u všech k ústupu celkových „B“ příznaků a po ukončení indukční série bylo dosaženo následné léčebné odpovědi: stabilní nemoc 2/10 (20 %), parciální remise 5/10 (50 %), kompletní remise 3/10 (30 %).
U všech 3 pacientů v kompletní remisi (CR) po indukci CHOP nebyla dále doplněna žádná další terapie v rámci první linie. U pacientky v původním věku 54 let nadále trvá první kompletní remise 151 měsíců od ukončení indukce. U jednoho pacienta ve věku 64 let byla vysokodávkovaná chemoterapie BEAM s autologní transplantací kontraindikována pro celkově nepříznivý stav s dalšími onemocněními a tento pacient zrelaboval za 3 měsíce po ukončení indukce. U třetího pacienta ve věku 51 let došlo k relapsu za 30 měsíců po ukončení indukce a byla pak doplněna 2. série CHOP, ale již v době přijetí k plánované autologní transplantaci u něj docházelo k novému incipientnímu relapsu AITL, a po autologní transplantaci byla tedy doplněna následně ještě transplantace nepříbuzenská alogenní, přičemž tento pacient zemřel v den +102 po alogenní transplantaci na sepsi s multiorgánovým selháním.
U celkem 5 pacientů v parciální remisi (PR) po indukci CHOP byla vysokodávkovaná chemoterapie BEAM s autologní transplantací doplněna pouze u 2 pacientek s tím, že bylo u obou následně dosaženo stavu CR. U jedné pacientky ve věku 43 let trvala CR 5 měsíců a pacientka zemřela na progresi lymfomu při zdravotní neúnosnosti k intenzivní chemoterapii a druhá pacientka ve věku 67 pak zemřela ve stavu trvající CR za 33 měsíců na sekundární AML. U zbylých 3 pacientů ve věku 53, 63 a 69 let nebyla intenzifikace s autologní transplantací realizována pro floridní a časný relaps lymfomu za 1–2 měsíce po ukončení indukce, přičemž všichni pacienti zemřeli na fulminantní infekční komplikace (2× pneumonie, 1× sepse) prakticky v úvodu zahájené záchranné chemoterapie DHAP.
U 2 pacientů bylo dosaženo pouze stabilizace nemoci (SD), přičemž u pacienta ve věku 72 let byla z důvodu celkového nepříznivého stavu a dalších onemocnění indikována dále již jen symptomatická léčba. U pacientky ve věku 60 let byla obratem doplněna vysokodávkovaná chemoterapie BEAM s autologní transplantací krvetvorných buněk, po které bylo dosaženo stavu PR, a následně pak byla za 1,5 měsíce provedena příbuzenská alogenní transplantace krvetvorných buněk s redukovanou intenzitou přípravy (protokol Flu/Mel), která však již byla realizována v době počínající progrese lymfomu, a pacientka zemřela v den +7 po transplantaci na toxické komplikace a sepsi.
Alogenní transplantace krvetvorných buněk byla tedy provedena u 2 již výše uvedených pacientů. Jinak nebyla provedena a ani plánována s ohledem na vysoký věk a další onemocnění u 4/10 (40 %) chemoterapií léčených pacientů, pro úmrtí během záchranné chemoterapie u 2/10 a pro trvající CR u 2/10 pacientů.
Medián OS chemoterapií léčených pacientů byl 10 měsíců, přičemž přežití bylo statisticky signifikantně delší při dosažení stavu CR, a to buď v rámci 1. linie léčby po 6× CHOP, nebo po doplnění vysokodávkované léčby BEAM s autologní transplantací: 46 vs 6 měsíců (p = 0,0023). Medián EFS byl 8 měsíců, přičemž lepších výsledků bylo statisticky signifikantně dosaženo rovněž u pacientů s dosažením CR v rámci 1. linie léčby: 43 vs 6 měsíců (p = 0,0052), viz grafy 1, 2.


Diskuze
Angioimunoblastický T-lymfom (AITL) je relativně vzácná a obecně prognosticky nepříznivá malignita. V předkládané analýze jsme se snažili shrnout naše poznatky a zkušenosti s léčbou pacientů s tímto onemocněním. Limitací práce je její retrospektivní charakter a současně malý soubor pacientů jednoho pracoviště, což je nutné mít na paměti při hodnocení výsledků. Na druhé straně jde ale o poměrně homogenní skupinu pacientů s jasně definovanou indukční léčbou a s výsledky, které svým charakterem bezpochyby ukazují na rizikovost této malignity. Navíc s výjimkou učebnic je problematika AITL v českém písemnictví prakticky nepublikovaná.
Četnost výskytu AITL 1,9 % v našem souboru pacientů s NHL je srovnatelná s údaji z velkých registrů, kde byla udávána v rozmezí 0,5–1,2 % [7,8]. Lymfom v naší skupině charakteristicky postihoval spíše starší pacienty s mediánem věku 64 (43–82) let, vždy bylo přítomno postižení lymfatických uzlin, často hepatosplenomegalie, a celkově tak dominovala (92 %) klinická stadia III–IV doprovázená u dvou třetin pacientů také celkovými příznaky (noční pocení, horečky, hubnutí). Další doprovodné symptomy nebo poruchy nebyly vzácné a zahrnovaly vedle výskytu hydrothoraxu nebo ascitu především autoimunitní hemolytickou anémii (AIHA), trombocytopenii (ITP), výsev exantému, poruchy koagulace (DIC a flebotrombóza) a výskyt monoklonální gamapatie.
Medián období od prvních příznaků onemocnění (např. povšimnutí si zvětšených uzlin, rozvoje celkových příznaků nebo jiných problémů) byl 1,5 měsíce s maximem do 4 měsíců. Tendence k pozdějšímu diagnostickému závěru byla u pacientů s menším uzlinovým postižením, kdy jiné projevy AITL imponovaly spíše jako možné systémové zánětlivé onemocnění nebo infekce. U 3/12 (25 %) pacientů byla také nutná opakovaná excize a vyšetření uzliny při přetrvávání nebo zhoršování obtíží při původně odečteném nespecifickém histologickém nálezu lymfadenitidy. Finální diagnóza však byla u našich pacientů nakonec vždy potvrzena histologicky, imunohistochemicky a současně byla prokazována molekulárně-geneticky také přítomnost klonální přestavby receptoru TCR gama. Po stanovení diagnózy pak byli pacienti referováni ke zvážení léčby prakticky obratem.
Jako iniciální léčba byla u většiny pacientů (n = 10) zajištěna chemoterapie CHOP-21 a pouze u dvou pacientů ve věku nad 80 let kortikoterapie. Chemoterapie CHOP byla dobře tolerována a rychle vedla k ústupu projevů lymfomu. Bylo dosaženo také 30 % kompletních remisí, což je ve srovnání s efektem u jiných lymfomů málo, ale na druhé straně intenzivnější protokol ESHAP dosahoval ve studiích stavu kompletní remise také jen 33% úspěšnosti [12].
Naši pacienti, kteří dosáhli po CHOP-21 kompletní remise, měli vyšší pravděpodobnost delšího EFS. U ostatních pacientů (n = 7) s dosaženou pouze parciální remisí nebo jen stabilizací nemoci však byla další prognóza krajně nepříznivá, kdy buď s ohledem na celkový stav nebylo možné zajistit další intenzifikaci (1/7), nebo nebylo možné zajistit realizaci odběru periferních krvetvorných buněk pro novou, velmi časnou progresi nemoci (3/7) následovanou většinou pokusem o záchrannou léčbu DHAP a úmrtím. Tedy pouze u 3/7 (43 %) pacientů s potřebou další minimalizace nemoci po indukci bylo možné zajistit a realizovat vysokodávkovanou chemoterapii BEAM s autologní transplantací s tím, že dosažení stavu CR opět zvýšilo pravděpodobnost delšího EFS. Z pohledu absolutních hodnot lze však hodnoty EFS i OS u pacientů s dosažením CR považovat rovněž za prognosticky nepříznivé (hodnota mediánu EFS a OS: 46 a 43 měsíců).
Naše zkušenosti s intenzifikací v podobě vysokodávkované chemoterapie BEAM s autologní transplantací krvetvorných buněk nebo s alogenní transplantací krvetvorných buněk po přípravě Flu/Mel jsou tedy omezené, a to především z důvodu nemožnosti je vůbec realizovat pro nepříznivý stav pacienta, jeho věk či progresi nemoci a úmrtí během záchranné léčby. Lze spekulovat, do jaké míry by případně jiný postup u konkrétního pacienta mohl vést k lepšímu efektu léčby.
V léčbě se jako standardní uplatňuje nadále protokol chemoterapie CHOP, i když částečný efekt mohou mít i některé jiné léky, jako např. fludarabin, interferon alfa, kortikoidy [9]. Využití intenzivnějších protokolů v indukční léčbě, jako jsou např. ESHAP, ACVBP, CHOP, nepříznivou prognózu pacientů zásadněji neovlivnilo [12–15]. Významný vliv na zlepšení přežívání pacientů nebyl pozorován ani při zajištění konsolidace s autologní transplantací krvetvorných buněk [11,16], nicméně z výsledků retrospektivní analýzy EBMT [17] u 146 pacientů přeci jenom vyplývá, že doba přežití bez progrese po 4 letech byla 56 % u pacientů autologně transplantovaných v kompletní remisi a 23 % u pacientů s chemorefrakterní nemocí. Lepší výsledky po autologní transplantaci u selektovaných pacientů v kompletní remisi udávají i Reimer et al [18] s celkovým přežitím 71 % ve 3 letech. Zkušenosti s alogenní transplantací jsou omezené a u mladých pacientů přinesla tato léčba efekt v podobě OS 64 % ve 3 letech [17,19,20].
Na základě našich zkušeností a údajů z dostupné literatury se domníváme, že u pacientů ve věku do 60 let nebo starších v dobré kondici by bylo vhodné při sebemenším podezření na omezený efekt úvodní chemoterapie CHOP-21 zajistit obratem intenzifikaci léčby a zvažovat podání jednoho až dvou cyklů DHAP se současným pokusem o odběr autologních krvetvorných buněk. U pacientů s velmi dobrou léčebnou odpovědí a dosažením kompletní remise po indukci lze uvažovat o možnosti dlouhodobější remise, nicméně se domníváme, že obratem by měl být rovněž doplněn odběr periferních krvetvorných buněk a následně by bez zbytečné prodlevy měla proběhnout konsolidace s vysokodávkovanou chemoterapií BEAM s autologní transplantací. Současně by mělo být vždy se stanovením diagnózy AITL ihned zahájeno vyhledávání dárce krvetvorných buněk v rodině a pátrání po dostupnosti dárců v registrech pro případ potřeby alogenní transplantace při neuspokojivém léčebném průběhu (nedosažení kompletní remise) nebo pro případ relapsu nemoci. I tak je však nutné mít na vědomí, že průběh onemocnění je velmi variabilní a ve většině případů je prognóza krajně nepříznivá s mediánem přežívání méně než 3 roky i v případě zajištění intenzivní léčby
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
MUDr. Samuel Vokurka, Ph.D.
Hematologicko-onkologické oddělení
FN Plzeň
Alej Svobody 80
304 60 Plzeň
e-mail: vokurka@fnplzen.cz
Obdrženo: 17. 5. 2011
Přijato: 24. 8. 2011
Sources
1. Swerdlow SH, Campo E, Harris NL et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon: World Health Organization 2008.
2. Frizzera G, Moran EM, Rappaport H. Angio-immunoblastic lymphadenopathy with dysproteinaemia. Lancet 1974; 1(7866): 1070–1073.
3. Lukes RJ, Tindle BH. Immunoblastic lymphadenopathy. A hyperimmune entity resembling Hodgkin’s disease. N Engl J Med 1975; 292(1): 1–8.
4. Lennert K. Nature, prognosis and nomenclature of angioimmunoblastic (lymphadenopathy, lymphogranulomatosis X or T-zone lymphoma). Dtsch Med Wochenschr 1979; 104(35): 1246–1247.
5. Shimoyama M, Minato K. Clinical, cytological and immunological analysis of T-cell type lymphoid malignancies: a classification of T-cell type lymphoid malignancy (author’s transl). Rinsho Ketsueki 1979; 20(9): 1056–1069.
6. Harris NL, Jaffe ES, Stein H et al. A revised European--American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 1994; 84(5): 1361–1392.
7. Trněný M, Vášová I, Pytlík R et al. Distribuce podtypů non-hodgkinského lymfomu v České republice a jejich přežití. Klin Onkol 2007; 20(5): 341–348.
8. The Non-Hodgkin’s Lymphoma Classification Project. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. Blood 1997; 89(11): 3909–3918.
9. de Leval L, Gisselbrecht C, Gaulard P. Advances in the understanding and management of angioimmunoblastic T-cell lymphoma. Br J Haematol 2010; 148(5): 673–689.
10. Armitage J, Vose J, Weisenburger D. Internatitonal T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol 2008; 26(25): 4124–4130.
11. Mourad N, Mounier N, Brière J et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d‘Etude des Lymphomes de l‘Adulte (GELA) trials. Blood 2008; 111(9): 4463–4470.
12. Delmer A, Fitoussi O, Gaulard P et al. A phase II study of bortezomib in combination with intensified CHOP-like regimen (ACVBP) in patients with previously untreated T-cell lymphoma: Results of the GELA LNH05–1T trial. J Clin Oncol 2009; 27 (Suppl 15): 8554.
13. Escalón MP, Liu NS, Yang Y et al. Prognostic factors and treatment of patients with T-cell non-Hodgkin lymphoma: the M. D. Anderson Cancer Center experience. Cancer 2005; 103(10): 2091–2098.
14. Tilly H, Lepage E, Coiffier B et al. Intensive conventional chemotherapy (ACVBP regimen) compared with standard CHOP for poor-prognosis aggressive non-Hodgkin lymphoma. Blood 2003; 102(13): 4284–4289.
15. Karakas T, Bergmann L, Stutte HJ et al. Peripheral T-cell lymphomas respond well to vincristine, adriamycin, cyclophosphamide, prednisone and etoposide (VACPE) and have a similar outcome as high-grade B-cell lymphomas. Leuk Lymphoma 1996; 24(1–2): 121–129.
16. Mounier N, Gisselbrecht C, Brière J et al. All aggressive lymphoma subtypes do not share similar outcome after front-line autotransplantation: a matched-control analysis by the Groupe d’Etude des Lymphomes de l’Adulte (GELA). Ann Oncol 2004; 15(12): 1790–1797.
17. Kyriakou C, Canals C, Goldstone A et al. High-dose therapy and autologous stem-cell transplantation in angioimmunoblastic lymphoma: complete remission at transplantation is the major determinant of Outcome-Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2008; 26(2): 218–224.
18. Reimer P, Rüdiger T, Geissinger E et al. Autologous stem-cell transplantation as first-line therapy in peripheral T-cell lymphomas: results of a prospective multicenter study. J Clin Oncol 2009; 27(1): 106–113.
19. Le Gouill S, Milpied N, Buzyn A et al. Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 2008; 26(14): 2264–2271.
20. Corradini P, Dodero A, Zallio F et al. Graft-versus-lymphoma effect in relapsed peripheral T-cell non--Hodgkin’s lymphomas after reduced-intensity conditioning followed by allogeneic transplantation of hematopoietic cells. J Clin Oncol 2004; 22(11): 2172–2176.
21. Vášová I, Boudová L, Trněný M et al. Primární testikulární lymfom, klinickopatologická multicentrická retrospektivní studie a data registru Kooperativní lymfomové skupiny (KLS). Klin Onkol 2007; 20(5): 302–306.
22. Boudová L, Kazakov D, Jindra P et al. Primární kožní velkobuněčný anaplastický T-lymfom s koexpresí CD30 a CD56, bohatý na neutrofily a histiocyty. Popis raritního případu s přehledem literatury. Klin Onkol 2007; 20(3): 260–263.
23. Fabian P, Boudová L. Poznámky ke 4. vydání klasifikace lymfomů WHO. Klin Onkol 2009; 22(3): 121–122.
24. Quintanilla-Martinez L, Ott G. Angioimmunoblastic T-cell lymphoma. In: Jaffé ES, Harris NL, Vardiman JW et al (eds). Hematopathology. Philadelphia: Elsevier 2011.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2012 Issue 3
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Angioimunoblastický T-lymfom (AITL) jako velmi nepříznivá malignita – zkušenost centra
- Triple-negativní karcinom prsu: analýza souboru pacientek diagnostikovaných a/nebo léčených v Masarykově onkologickém ústavu v letech 2004 až 2009
- Nové a klinicky využívané onkomarkery karcinómu močového mechúra
- Faktor stimulující kolonie granulocytů (G-CSF) zrychluje hojení vlhké deskvamace kůže vyvolané radiací