
Hodnocení variant nejasného významu v genu BRCA2
Evaluation of Variants of Unknown Significance in the BRCA2 gene
Background:
Endogenous processes and exogenous agents cause constant DNA damage. DNA double-strand breaks are among the most serious types of damage. They are mainly repaired by homologous recombination, where the BRCA2 protein plays a dominant role. Heterozygous germline BRCA2 mutations predispose to breast, ovarian, pancreatic and other types of cancer. The presence of a pathogenic mutation in patients or their family members warrants close surveillance and prophylactic surgery. Apart from clearly pathogenic mutations, variants leading only to a single amino acid substitution are often identified. Since the influence of these variants on cancer risk is unknown, they represent a major clinical problem.
Aims:
The aim of this paper is to summarize the current possibilities of predicting pathogenicity of BRCA2 variants. In some cases, genetic methods are able to classify variants with high probability; however, their use is often limited by low frequency of the variants or inaccessibility of samples for mRNA isolation or DNA from family members. Alternatively, functional assays performed in various cellular models may be employed. Multiple functional tests and cellular models are presented and characterized, including their advantages and limitations. A new model of human syngeneic cell lines developed by the authors is presented, in which one BRCA2 allele is deleted and the variant is introduced into the other allele by homologous recombination. This model has the potential to evaluate function of variants without some of the unwanted effects of the other models. Currently, this model is being applied to variants identified in patients with hereditary cancer predisposition in the Masaryk Memorial Cancer Institute.
Conclusion:
Functional assays in cellular models including a new model of syngeneic cell lines described by the authors have a great potential in evaluating clinical importance of unclassified variants in the BRCA2 gene, especially in cases where genetic tests are not applicable.
Key words:
BRCA2 gene – variants of unknown significance – missense mutation – functional assays
The study was supported by:
grant Iga Ns/10536-3. Genetical testing was supported by
the European Regional Development Fund and by the Czech republic´s national budget (OP VaVpI - RECaMO, CZ.1.05/2.1.00/03.0101).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
25. 1. 2012
Accepted:
21. 5. 2012
Authors:
M. Heczková 1; E. Macháčková 2; M. Jirsa 1; J. Špičák 3; L. Foretová 2; T. Hucl 3
Authors‘ workplace:
Centrum experimentální medicíny, Institut klinické a experimentální medicíny, Praha
1; Oddělení genetiky a epidemiologie nádorů, Masarykův onkologický ústav Brno
2; Klinika hepatogastroenterologie, Institut klinické a experimentální medicíny, Praha
3
Published in:
Klin Onkol 2012; 25(Supplementum): 87-95
Overview
Východiska:
Vlivem vnějších a vnitřních faktorů dochází v buňce neustále k poškozování DNA. Mezi nejzávažnější poškození patří tvorba dvouřetězcových zlomů. Jejich bezchybná oprava se uskutečňuje především mechanizmem homologní rekombinace, v němž jednu z klíčových úloh hraje protein BRCA2. Vrozené mutace v genu BRCA2 jsou příčinou vzniku nádorů prsu, ovarií, pankreatu či jiných orgánů. Přítomnost patogenní mutace v BRCA2 u pacientů či jejich rodinných příslušníků je důvodem k jejich dispenzarizaci s cílem včas zachytit nádorové onemocnění a je indikací k profylaktickým chirurgickým výkonům. Vedle zjevně patogenních mutací jsou v genu BRCA2často zjišťovány unikátní bodové varianty vedoucí pouze k záměně jedné aminokyseliny, u kterých je obtížné určit klinický význam. Vzhledem k možnému riziku vzniku nádorového onemocnění u nositelů těchto variant je jejich nejednoznačný význam závažným medicínským problémem.
Cíl:
Cílem tohoto článku je podat přehled o současných možnostech hodnocení patogenity variant v genu BRCA2. Genetické metody jsou v některých případech schopny patogenitu variant s vysokou pravděpodobností predikovat, jejich provedení je však často limitováno nízkou frekvencí varianty či nedostupností vzorků pro izolaci mRNA nebo vzorků DNA od rodinných příslušníků. Alternativou jsou v takových případech metody funkčního hodnocení variant prováděné v různých buněčných modelech. Jednotlivé funkční metody a buněčné modely jsou podrobně charakterizovány včetně jejich výhod a limitací. Dále je představen autory vyvinutý lidský nádorový syngenní buněčný model, ve kterém je jedna alela BRCA2 genu nefunkční a do druhé je homologní rekombinací vnesena studovaná varianta. Tento model má potenciál hodnotit funkci variant s minimem nežádoucích vlivů jiných modelů. V současné době je tento model prakticky zkoušen u variant zjištěných u pacientů s dědičnou nádorovou predispozicí v Masarykově onkologickém ústavu.
Závěr:
Funkční testy v buněčných modelech včetně autory vyvinutého nového modelu syngenních buněčných linií představují velký potenciál k určení patogenity variant s nejasným klinickým významem v BRCA2 genu, především tam, kde jsou genetické metody neproveditelné.
Klíčová slova:
BRCA2 gen – varianty s nejasným významem – missense mutace – funkční testy
Úvod
Působením nejrůznějších vnitřních a zevních faktorů je neustále poškozována DNA. Mají-li buňky přežít, musí být schopny toto poškození v genetickém materiálu opravit. V průběhu evoluce proto vyvinuly řadu různých mechanizmů opravy DNA. Pro každý typ poškození existuje jeden či několik specifických způsobů opravy [1].
Jedním z nejzávažnějších typů poškození DNA jsou dvouřetězcové zlomy (Double-Strand Breaks – DSB) vznikající především působením ultrafialového záření, gama záření, volných radikálů a chemických látek. Zlomy jsou v S či G2 fázi buněčného cyklu, kdy je DNA zdvojená, a sesterská chromatida tak poskytuje matrici, opraveny pomocí homologní rekombinace (HR). Nehomologní způsob opravy (Non-Homologous End Joining – NHEJ) převažuje v G1 fázi buněčného cyklu a představuje přímé spojení konců DNA, při kterém však může dojít ke ztrátě části DNA. Důležitým účastníkem opravy cestou homologní rekombinace je protein BRCA2 [1].
Gen BRCA2 se skládá z 27 exonů a je lokalizovaný na chromozomu 13q12-13. Funkce genu není na biochemické úrovni zcela definovaná, avšak díky své roli v homologní rekombinaci je gen řazen do skupiny genů udržujících neporušený genom. Protein BRCA2 se váže na rekombinázu RAD51, umožní transport RAD51 do jádra na místo poškození, a reguluje tak formování RAD51 nukleoproteinového filamenta, které je nezbytné pro opravu DNA homologní rekombinací [2]. Buňky s absencí proteinu BRCA2 tak mají sníženou schopnost opravy dvouřetězcových zlomů a vykazují zvýšenou citlivost k poškození DNA. Protein BRCA2 ve své struktuře obsahuje osm vazebných míst pro RAD51 lokalizovaných v místě vysoce konzervovaných BRC repetic a na C-konci proteinu. Mezi nimi leží domény zodpovědné za vazbu na DNA. Na C-konci se nachází signální sekvence pro lokalizaci proteinu v jádře [1–3] (obr. 1).
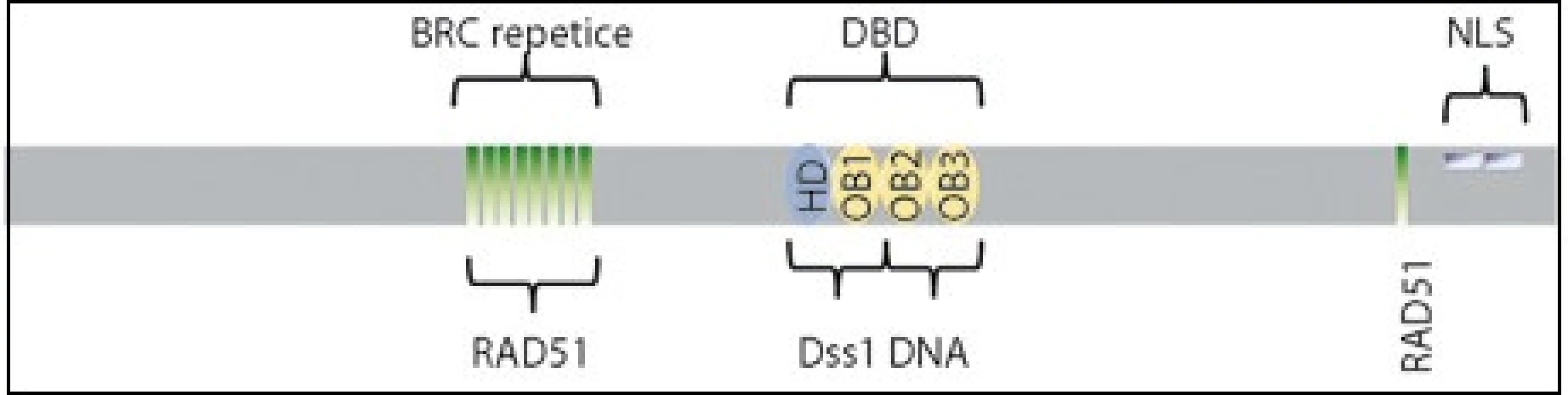
Nádorové onemocnění vzniká jako postupný proces změn (mutací) v genomu. Během času buňky akumulují mutace v onkogenech, tumor supresorových genech či v genech udržujících neporušený genom. Vrozené, či získané alterace v těchto genech pak vedou ke vzniku nádorů.
Germinální bialelické mutace v BRCA2 způsobují vzácný typ Fanconiho anémie (FANCD1 skupina) [4]. Germinální monoalelické (heterozygotní) mutace pak představují druhou nejčastější příčinu familiárního karcinomu prsu a ovarií [5,6]. Kumulativní riziko pro vznik nádoru prsu u nositelů takové mutace do věku 70 let je až 84 % a pro karcinom ovaria 27 % [7]. Heterozygotní BRCA2 mutace taktéž predisponují jedince ke vzniku karcinomu pankreatu – Hahn nalezl mutace v BRCA2 až u 20 % pacientů s familiárním karcinomem pankreatu [8]. Nositelé mutací v BRCA2 mají také zvýšené riziko vzniku nádoru žlučníku, žlučových cest, žaludku a melanomu [6]. Vlastní nádory vznikají většinou v důsledku somatické ztráty (delece) druhé alely.
Pacienti s vysokým rizikem dědičné formy nádorového onemocnění (např. pacienti s familiárním výskytem karcinomu prsu/ovaria, s bilaterálním nádorem prsu ve věku pod 50 let, pacienti s unilaterálním nádorem mladší 35 let, muži s nádorem prsu či specifické histologické podtypy nádorů) jsou indikováni k vyšetření klinickým genetikem a doporučeni k vyšetření přítomnosti mutace v BRCA2 genu [6,9].
Za zjevně patogenní mutace považujeme takové, které znemožňují syntézu funkčního proteinu (nonsense a frame-shift mutace a mutace vyvolávající abnormální sestřih mRNA). Vedle jednoznačně patogenních mutací jsou v řadě genů včetně BRCA2 zjišťovány mutace, které nevedou ke změně čtecího rámce. Nejčastěji se jedná o missense mutace vedoucí k záměně jedné aminokyseliny či o in-frame delece a inzerce, u kterých nedochází ke změně čtecího rámce a důsledkem je delece, či inzerce několika málo aminokyselin. Jejich význam pro funkci proteinu a pro riziko vzniku nádorového onemocnění nelze jednoznačně určit, nazývají se tak mutace s nejasným významem (označované jako UV – Unclassified Variants nebo VUS – Variants of Unknown Significance) [3,10,11]. V případě BRCA2 genu bylo doposud popsáno více než 1 100 různých variant (Breast Cancer Information Core, http://research.nhgri.nih.gov/bic/), které tak představují až 40 % všech zjištěných sekvenčních alterací [7].
Vzhledem k tomu, že v některých souborech jsou takové varianty nalézány až u 13 % žen podstupujících genetické testování, jedná se o významný klinický problém [7]. V současné době stále není možné u většiny těchto variant pacientům říct, zda se jedná o mutace představující riziko vzniku nádorového onemocnění. Zhodnocení patogenity varianty je přitom pro pacienta a jeho rodinu zásadní, neboť nosičství patogenní varianty znamená zvýšené riziko onemocnění u rodinných příslušníků, zatímco u těch členů rodiny, kteří danou patogenní variantu nezdědí, je zvýšené riziko vyloučeno [6,9].
V současné době existuje řada metod, které si dávají za cíl určení patogenity variant. Tyto metody lze rozdělit na přímé, které využívají statistického zpracování genetických a epidemiologických dat, a metody nepřímé, které se odvíjejí od různých vlastností genu či proteinu. Výsledek přímých metod je vyjadřován většinou jako pravděpodobnostní poměr (Likelihood Ratio, LR) asociace varianty s chorobou. Souhrn přímých a nepřímých metod je uveden v tab. 1.
![Souhrn používaných testů k hodnocení patogenity <em>BRCA2</em> variant [10].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/c25a2f3d535ad7626a4ede29a00cb444.png)
Přímé metody
Segregace s chorobou
V případě postižení více členů rodiny a dostupnosti jejich genetického materiálu k vyšetření lze na patogenitu mutace usuzovat na základě segregace dané mutace s chorobou [12]. Jedná se o nejpřímější genetický důkaz souvislosti varianty se vznikem nádorového onemocnění. K průkazu významné pravděpodobnosti je však nutné získat DNA od co největšího množství rodinných členů, což bývá obtížné až nemožné. Velmi vzácně je tak možné kategorizovat varianty na základě samotné segregace [10].
Vyšetření kontrolní populace
Vyšetření kontrolní populace může pomoci odhalit neutrální polymorfizmy, které se v populaci vyskytují často (více než v 1 % případů) a nemají vliv na sledované onemocnění. Např četný výskyt missense záměny p.N372H u 183 ze 476 kontrol nesvědčí pro její možné patogenní působení [7]. Problémem je však nízká frekvence většiny variant (méně než 1 na 1 000). Vedle toho je řada variant specifická pro určitý geografický region, což může komplikovat získání dostatečného množství kontrol [10].
Osobní, rodinná anamnéza a patologická charakteristika nádoru
Předpokládá se, že varianta, která je patogenní, se bude vyskytovat v případech se silnou rodinnou zátěží podobnou takové, jakou známe u patogenních nonsense mutací. Mezi zjišťovaná data patří věk v době diagnózy, počet postižených členů rodiny a jejich věk, přítomnost oboustranného karcinomu či postižení muže v rodině [13].
Nádory nositelů mutací v BRCA2 mohou vykazovat jisté patologicko-anatomické či molekulárně biologické charakteristiky [14]. Za specifický znak je považována tubulární struktura [15]. Jiným parametrem by mohla být ztráta heterozygotnosti (Loss of Heterozygosity – LOH) v místě genu BRCA2, která je nalézána v 80 % nádorů s patogenní nonsense mutací [15]. Naopak ztráta alely nesoucí missense variantu mluví proti její patogenitě [15].
Společná přítomnost s jinou patogenní mutací
Bialelické mutace v BRCA2 vedou k embryonální letalitě či Fanconiho anémii [4,16]. Proto je nepravděpodobné, aby se varianta, která je funkčně významná, vyskytovala společně v pozici trans s jinou patogenní mutací u pacientů, kteří nemají Fanconiho anémii. Stejně tak se dvě patogenní mutace nevyskytují na stejné alele. Z tohoto lze usuzovat, že varianta, která se vyskytuje společně s jinou patogenní mutací, je neutrální (nepatogenní) nebo případně pouze nízko riziková alela. Např. dříve zmiňovaná varianta p.N372H byla zjištěna v přítomnosti mnoha různých patogenních mutací, což ji opět charakterizuje jako neutrální [7]. Absence jiné patogenní mutace ve druhé alele však neklasifikuje variantu jako patogenní [7,10].
Nepřímé metody
Fyzikálně-chemické rozdíly proteinů a konzervace mezi organizmy
Pomocí speciálně vyvinutých modelů, jako např. Granthamova modelu chemických rozdílů, lze usuzovat na fyzikálně-chemické rozdíly mezi wild-type proteinem a proteinem se záměnou aminokyseliny [17]. Na rozdíl mezi patogenní a neutrální mutací lze usuzovat také z míry mezidruhové konzervace u proteinových homologů v místě sledované varianty [18]. Porovnáním sekvencí genu jiných živočichů (např. myši, slepice, kočky, psa či ryby) nebo i nižších organizmů (např. octomilky nebo kvasinky) lze pozorovat různou míru konzervace napříč živočišnými druhy. Vysoká míra konzervace napoví o významnosti aminokyseliny. Mutace v částech genu, které nejsou přítomny u nižších živočichů, může ukazovat na jejich nevýznamnost, nelze však vyloučit, že gen získal v průběhu evoluce nové funkce, za které jsou zodpovědné tyto nové části genu [7]. In silico analýzu toho typu lze dnes provést pomocí speciálních programů, jako např. Align-GVGD [19]; případný vliv varianty na sestřih mRNA lze predikovat např. pomocí programu Max-EntScan [20].
Kombinace metod
Důležitým aspektem klasifikace variant je integrace výsledků jednotlivých metod. Každá metoda generuje různé výsledky, jejichž vztah není vždy jednoznačně určen. Proto se ukázalo jako vhodné výsledky jednotlivých testů kombinovat. Kombinace více testů vyloučí nadměrnou závislost na jednom typu testu, a představuje tedy také určitý kontrolní mechanizmus. Takový multifaktorový algoritmus integrace jednotlivých hodnocení popsal např. Goldgar, který pomocí multifaktoriální analýzy posuzoval segregaci sledované varianty s onemocněním, přítomnost jiné patogenní mutace, stupeň mezidruhové konzervace a závažnost změn fyzikálně-chemických vlastností missense varianty na strukturu proteinu [21]. Jednotlivé výsledky jsou pak zpracovány statistickými metodami, např. podle Bayesova modelu. Původní pravděpodobnost (stanovená na základě in silico analýzy) je modifikována pravděpodobnostmi z jednotlivých genetických testů ve výslednou konečnou pravděpodobnost patogenity [10].
K ještě většímu posílení výpovědní hodnoty slouží společné sdílení výsledků, např. formou databáze [22]. Ex-UV databáze sdružuje všechny publikované varianty hodnocené pomocí Bayesova modelu. Výstupním hodnocením je rozdělení variant podle jednotného IARC klasifikačního systému [23].
Funkční testy
Významným omezujícím faktorem genetických testů je dostupnost dostatečného množství rodinných příslušníků a kontrolních vzorků ke kompletnímu genetickému vyšetření. V řadě případů proto takové vyšetření není možné v dostatečném rozsahu provést. Jedinou metodou klasifikace variant v BRCA2 jsou pak nepřímé funkční metody. Jedná se o in vitro postupy, při kterých se zjišťuje vliv konkrétní odchylky od referenční sekvence na funkci proteinu. Jejich souhrn je uveden v tab. 2.

Imunofluorescence RAD51
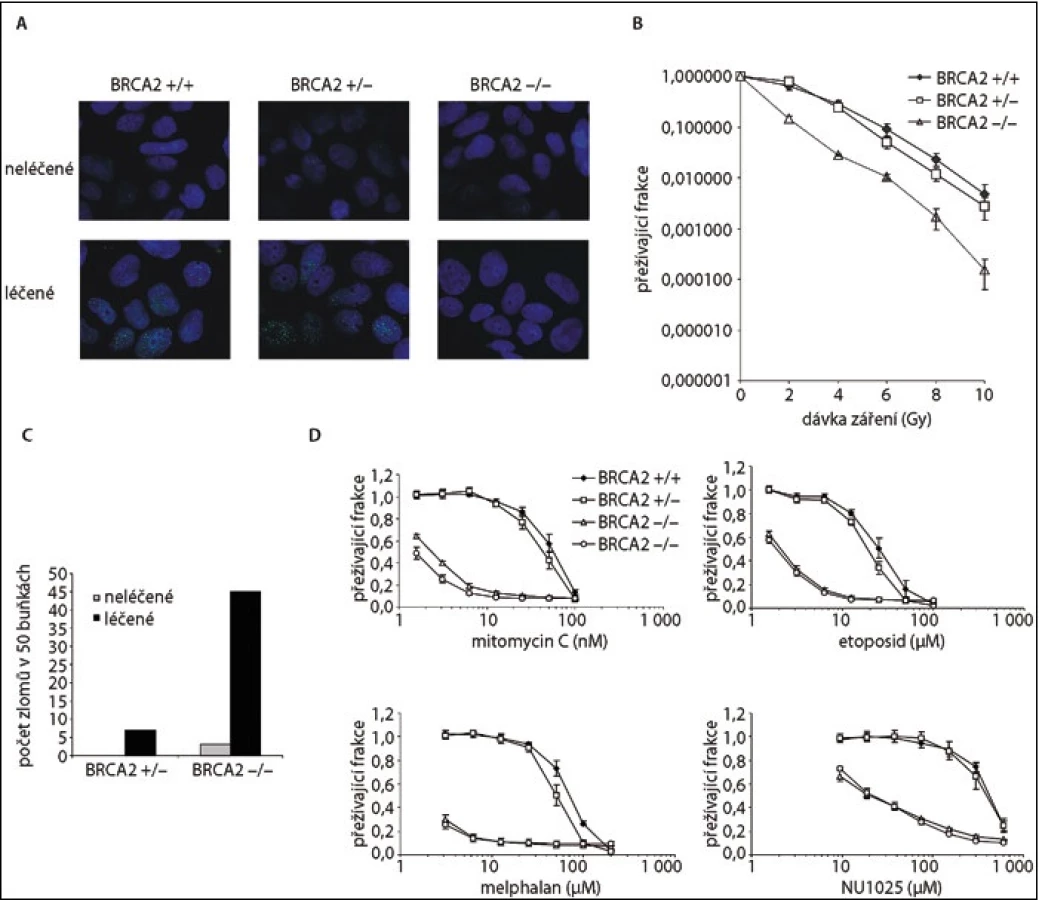
Role BRCA2 v opravě DNA spočívá v jeho vazbě na protein RAD51 a regulaci vzniku nukleárního filamenta, které je nezbytné pro homologní rekombinaci. Imunofluorescenční detekce fokusů RAD51 proteinu v jádře v reakci na poškození DNA je tak nepřímou známkou BRCA2 dependentní homologní rekombinace [2,3]. Buňky rostoucí na krycím sklíčku jsou vystaveny působení mitomycinu C či iradiaci (poškození DNA). V krátkém časovém intervalu je provedeno imunofluorescenční vyšetření a kvantifikace buněk s přítomnými fokusy RAD 51 v jádře [3] (obr. 2).
Homologní rekombinace
Pomocí stabilní transfekce je do genomu buněk integrován GFP (Green Fluorescent Protein) konstrukt. V něm jsou dva za sebou uspořádané GFP geny, každý obsahující jinou inaktivující mutaci. Inaktivace v prvním z nich je zároveň štěpícím místem pro enzym I-SceI. Po transfekci plazmidem produkujícím enzym I-SceI je enzymem vytvořen dvouřetězcový zlom. Exprese aktivního GFP je závislá na opravě mutovaného GFP genu homologní rekombinací po vzniku dvouřetězcového zlomu. GFP vykazuje zelenou fluorescenci a detekujeme ho po expozici ultrafialovému světlu ve fluorescenčním mikroskopu. V původní práci byly provedeny experimenty s buňkami CAPAN1 a s myšími embryonálními buněčnými liniemi [24]. Později byla metoda aplikována na vybrané varianty BRCA2u linie VC8 [7].
Citlivost k agens poškozující DNA
Poškození DNA ve formě meziřetězcových křížových vazeb či zlomů dvojvlákna DNA nemůže být BRCA2-deficitními buňkami opraveno a vede k jejich zániku [3]. Mezi nejčastěji používaná agens patří alkylační látky jako mitomycin C, melfalan nebo cisplatina, případně inhibitory topoizomeráz, např. etoposid. Obdobného účinku je možné dosáhnout i vystavením buněk iradiaci [3]. Buňky v exponenciální fázi růstu jsou vystaveny působení látky poškozující DNA. Jejich proliferace je hodnocena po dobu několika dnů. Absence funkčního BRCA2 vede k zániku prakticky všech buněk, buňky s funkčním BRCA2 umírají pouze při užití vysokých dávek [3,7,25] (obr. 2).
Amplifikace centrozomů
Centrozomy (dělicí tělíska) jsou organely zajišťující vhodné uspořádání mikrobulů k buněčnému dělení. BRCA2-deficitní buňky v buněčných kulturách i v samotných lidských nádorech vykazují amplifikaci centrozomů. Tento fenotyp je nezávislý na expozici buněk látkám poškozujícím DNA a naznačuje funkci BRCA2 v buněčném dělení [4,26,27]. Přesný mechanizmus jeho vzniku však není znám, dříve předpokládaná role BRCA2 v cytokinezi nebyla recentně potvrzena [28]. Amplifikaci centrozomů můžeme tedy zatím hodnotit pouze jako marker deficitu BRCA2. Míra amplifikace se určuje detekcí centrozomů imunofluorescenčním barvením protilátky proti centrinu [29].
Chromozomová nestabilita
BRCA2-deficitní buňky vykazují chromozomovou nestabilitu, která je ještě zvýrazněna po expozici látkám poškozujícím DNA. Jimi vyvolané chromozomální zlomy slouží jako diagnostický test Fanconiho anémie včetně skupiny FANCD1 způsobené mutacemi v BRCA2 [1,3]. Zlomy a radiální chromozomy byly nalezeny ve zvýšené míře u BRCA2-deficitních buněk [3] (obr. 2). Např. u mutace p.Y3308X ukázal karyotyp 68 takových změn ve srovnání s 10 změnami u kontrol [25].
Nitrobuněčná lokalizace
Nukleární lokalizační systém (NLS) se nachází na samém konci genu BRCA2 a většina mutací, které způsobují předčasnou terminaci translace, je tak lokalizována před ním. V experimentech Wu et al byl wild-type BRCA2 protein lokalizován především v jádře ve srovnání s patogenní mutací c.6174delT, u které je mutovaný protein lokalizován v cytoplazmě. Ektopická exprese mutovaného proteinu v cytoplazmě byla rovněž pozorována u mutací c.8395G>C a c.8204G>A, jejichž předpokládaným důsledkem jsou aminokyselinové záměny p.D2723H a p.R2659K. Avšak v případě nepatogenní varianty byl p.T2515I protein lokalizován překvapivě v cytoplazmě i jádře [7]. Metoda se provádí pomocí detekce fluorescenčně značeného proteinu připojeného k exogenně transientně exprimovaným variantám BRCA2.
Buněčné modely
Při interpretaci funkčních testů je nutné brát ohled na buněčný model, který byl k danému testu použit. Buněčné modely ke studiu funkce genu BRCA2 jsou vzácné a obtížně získatelné. Hlavním důvodem je esenciální funkce genu pro udržení neporušeného genomu, a tedy života buňky. Indukovaná ztráta genu je tak často pro buňky letální. Na rozdíl od jiných genů existuje jen jedna buněčná nádorová linie s přirozeně se vyskytující mutací v BRCA2: linie karcinomu pankreatu CAPAN1 (kromě od FANCD1 pacientů odvozených linií leukemických buněk FA-AML1 a SB1690, fibroblastů EUFA423 a lymfoblastů HSC62) [4,30].
Pro funkční hodnocení variant lze použít BRCA2-deficitních či proficitních buněčných linií. V obou případech jsou testy závislé na ektopické expresi konstruktů obsahující BRCA2 s mutací (plazmid, arteficiální chromozom), které jsou transientně či stabilně transfekované do buněk. V prvním případě sledujeme, zda je varianta po transfekci mutovaného konstruktu schopna opravit deficitní fenotyp parenterálních buněk. Ve druhém případě jsou použity proficitní buňky, jejichž wild-type fenotyp je srovnáván s fenotypem mutovanými konstrukty transfekovaných buněk. V tomto případě se předpokládá dominantně-negativní efekt patogenních mutací. Vzhledem k délce sekvence BRCA2 se v některých případech používají dokonce pouze parciální sekvence BRCA2 genu.
Proficitní buňky
K funkčnímu testování variant BRCA2 byly použity buňky HeLa (karcinom děložního čípku) či buňky HEK 293T (transformované embryonální renální buňky) [7]. Wu a později Farrugia použili buňky HEK 293T k testování amplifikace centrozomů. Po transientní transfekci mutovaných konstruktů ukazoval vzestup procentuelního zastoupení buněk s amplifikací centrozomů (více než 4) na funkční významnost varianty. K potvrzení spolehlivosti testu však autoři přece jen použili také deficitní buňky VC8, kde byla naopak patrná perzistence vysokého procenta buněk s amplifikací centrozomů po transfekci konstruktů nesoucích patogenní mutace [7,29].
Nedávno byla provedena studie zkoumající vliv overexprese 15 variant genu BRCA2 transfekovaných do HeLaG1 buněk. Tato buněčná linie umožňuje měřit spontánní homologní rekombinaci mezi dvěma odlišně mutovanými geny pro rezistenci na hygromycin (HygR), kdy frekvence rekombinace je stanovena jako počet rezistentních klonů. Autoři hodnotili vzestup spontánní homologní rekombinace jako známku patogenity [31].
Deficitní buňky
CAPAN1 je jediná deficitní přirozeně se vyskytující solidní nádorová buněčná linie. Její buňky rostou pomalu a jsou špatně transfekabilní. Jedinou isogenní kontrolou jsou buňky CAPAN1 s exogenní overexpresí BRCA2. Tyto buňky však rostou ještě pomaleji, což může být způsobeno nefyziologickými hladinami exprese BRCA2 [16,32].
Jinou možností je vytvoření buněčných linií s indukovaným defektem BRCA2 a pomocí plasmidu nebo umělého chromozomu docílit ektopické tvorby mutovaného nebo wild-type proteinu. Nejznámějším příkladem je buněčná linie VC8 odvozená z křeččích rakovinných buněk V79 náhodnou chemickou mutagenezí. Tyto buňky nesou v 15. a 16. exonu genu XRCC11 (homolog BRCA2) mutaci předčasně ukončující syntézu proteinu [26]. Docílení stabilní exprese ektopického BRCA2 je možné, může být však obtížné. Wu et al tuto linii pro funkční testy BRCA2 variant použili jako první: testovali devět neznámých variant společně s jedním polymorfizmem a jednou patogenní mutací. K funkčnímu hodnocení modelu použili test homologní rekombinace, citlivost vůči mitomycinu C a amplifikaci centrozomů. Sledovali rovněž nitrobuněčnou lokalizaci proteinů. V kombinaci s integrovaným multifaktoriálním pravděpodobnostním poměrem pak identifikovali dvě mutace s patogenním fenotypem, pět variant s neutrálním fenotypem a dvě varianty, jejichž fenotyp nebyl jednoznačný [7]. Celkem 22 missense mutací testovali stejní autoři pomocí dvou funkčních testů, testu homologní rekombinace a amplifikace centrozomů. U 13 variant prokázali ztrátu funkce v alespoň jednom testu, u dvou prokázali aberantní sestřih a u sedmi variant byla funkce zachována. Důležitá byla vysoká korelace výsledků funkčních testů s genetickými pravděpodobnostními modely [29].
Další použitou buněčnou linií jsou myší embryonální kmenové buňky. V těchto buňkách je jedna alela BRCA2 genu inaktivována v místě exonu 11, druhou lze inaktivovat po expresi Cre rekombinázy. Tato situace vede k úmrtí všech buněk. Dodání exogenního lidského BRCA2 formou bakteriálního arteficiálního chromozomu však dokáže odumření zabránit. Na patogenitu variant se tak usuzuje dle jejich možnosti zabránit letalitě buněk s nepřítomným endogenním BRCA2. V případě, že varianta vede k přežití určitého množství klonů (hypomorfní fenotyp), jsou tyto klony podrobeny funkčním testům (homologní rekombinace, přežití po expozici látek poškozující DNA, karyotyp, imunofluorescence RAD51). Tímto buněčným systémem bylo otestováno celkem 17 variant [25].
Problémy buněčných modelů
Používané buněčné modely jsou zatíženy řadou problémů. Využívají často jiné než lidské buňky. Hodnotíme tak jedinečné a citlivé funkce esenciálního proteinu v prostředí jiného živočišného druhu. Funkce, které se za normálních okolností odehrávají v nádorových buňkách, dokonce hodnotíme v nenádorových buňkách. Pomalý růst, obtížná transfekabilita, omezená doba přežívání či dokonce letalita BRCA2-deficitních buněk komplikují použití současných linií. Dalším problémem prakticky u všech modelů je nutnost exogenního dodání BRCA2 ve formě plasmidů či arteficielních chromozomů. Míru exprese pak nelze kontrolovat a může být zcela nefyziologická. Dokonce je často pouze transientní. Experimenty vy-užívající overexpresi jsou nesmírně citlivé a vyžadují mimořádné kontroly. Jiným problémem je užití pouze parciálních proteinů v některých studiích. Takové použití nelze považovat za dostatečné a může vést k chybným závěrům [3,25,32].
Vlastní model
Ve snaze vyhnout se některým nevýhodám doposud dostupných buněčných modelů jsme vytvořili vlastní model BRCA2 deficience. V nádorové buněčné linii DLD1 jsme pomocí homologní rekombinace vytvořili deleci části exonu 11 v hemizygotním a dále i homozygotním stavu. Tyto BRCA2∆ex11/∆ex11 linie jsou i s homozygotní delecí v místě exonu 11 životaschopné a mají, na rozdíl od svých hemizygotních parenterálních buněk, typický fenotyp BRCA2 deficience [3].
V případě použití hemizygotních buněk s jednou deletovanou alelou BRCA2∆ex11/wt lze vnesením mutace do druhé alely hodnotit její funkční význam. Takový model nejblíže napodobuje stav v samotném nádoru. Iniciálně jsme vytvořili knihovnu sedmi mutací ( BRCA2∆ex11/mut) a hodnotili jejich funkční význam. K hodnocení funkce jsme použili dvě metody, imunofluorescenci RAD51 a přežití buněk po expozici mitomycinu C [3] (obr. 2).
Vedle hodnocení variant lze tento model použít i k hodnocení funkce jednotlivých proteinových reziduí. Nedávné studie s použitím parciálních proteinů ukázaly závislost buněk na fosforylaci serinu cyklin-dependentními kinázami v místě 3 291 [33]. Po náhradě serinu v této pozici glutamátem (imituje konstitutivní fosforylaci) a alaninem (znemožňuje fosforylaci) jsme potvrdili, že fosforylace v tomto místě inhibuje vazbu proteinu RAD51, nicméně tato fosforylace nebyla pro buňku nezbytná [3].
Po získání iniciálních výsledků jsme se rozhodli aplikovat naši techniku v klinické praxi. Ve spolupráci s laboratoří Oddělení genetiky a epidemiologie nádorů Masarykova onkologického ústavu jsme zahájili funkční testování variant nejasného významu nalezených v reálných rizikových rodinách vyšetřených v této laboratoři. V našich experimentech provádíme funkční vyšetření všech missense mutací nalezených v exonu 18. Dosavadní výsledky ukazují na dobrou klinickou využitelnost našeho modelu k testování variant (doposud nepublikované výsledky).
Interpretace výsledků
Provedení funkčního testu vyžaduje jeho správnou interpretaci. Funkční testy by měly být vždy provedeny s dostatečnými kontrolami, k jejichž známému fenotypu je testovaná varianta porovnána, optimální je použítí missense varianty s patogenním fenotypem. Buněčné modely spoléhající na overexpresi BRCA2 genu nelze považovat za optimální.
Vždy by měl být proveden více než jeden test, neboť výsledky dvou různých funkčních testů mohou být odlišné. Missense varianty p.R3025W a p.R2784W byly testovány pomocí amplifikace centrozomů a testu homologní rekombinace. Ani jedna z variant nevedla k indukci amplifikace centrozomů, obě však ukázaly redukovanou aktivitu homologní rekombinace [29]. Přestože amplifikace centrozomů je konstantním nálezem u BRCA2-deficitních buněk, mechanizmus jejího vzniku není znám. Dříve předpokládaný podíl BRCA2 při cytokinezi byl recentně zpochybněn [28]. Senzitivita testu amplifikace centrozomů může být nízká, nelze však také vyloučit různé nezávislé funkce genu BRCA2 testované těmito dvěma metodami, z nichž jedna je zachována a druhá ztracena.
Funkční testy provádíme za předpokladu, že jejich abnormální nález znamená, že varianta ovlivňuje u pacienta vznik nádorového onemocnění. Na rozdíl od genetických testů, jako např. testu segregace varianty s chorobou, nelze v současné době u většiny variant tento vztah na základě patologického funkčního testu jednoznačně potvrdit. Výhodou v určení tohoto vztahu je kombinace genetických a funkčních testů. Tato kombinace umožňuje určení senzitivity a specificity funkčních testů [29]. Pouze silná konkordance mezi výsledky genetických a funkčních testů může náš předpoklad definitivně potvrdit. Tato validace však může být provedena pouze u variant, u kterých jsou k dispozici i genetická data.
Vzhledem k náročnosti všech používaných metod a nízké četnosti jednotlivých variant jsou velkým příslibem aktivity typu ENIGMA. Jedná se o mezinárodní inciativu sdružující výzkumné týmy za účelem sdílení dat a optimalizace určování významu variant v BRCA1 a BRCA2 genu [34].
Závěr
Výsledkem genetického vyšetření BRCA2 genu je často nalezení genetické varianty, která nevede k zástavě tvorby proteinu, ale pouze k záměně jedné z aminokyselin. Genetické studie mají omezenou účinnost v určení patogenity těchto variant a často nejsou pro nedostatek potřebných údajů proveditelné. Funkční testy dokáží odlišit známé patogenní varianty, jejich limitací je zatím nedostatečná validace vztahu mezi výsledkem funkčního testu a rizikem vzniku nádorového onemocnění. Představují však velký potenciál pro klinickou praxi.
Práce byla podpořena grantem IGA NS/10536-3. Genetické testování na MOÚ podpořeno Evropským fondem pro regionální rozvoj a státním rozpočtem České republiky (OP VaVpI - RECAMO, CZ.1.05/2.1.00/03.0101).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Tomáš Hucl, Ph.D.
Klinika hepatogastroenterologie
IKEM PRAHA
Vídeňská 9
140 21 Praha 4
e-mail: tohu@ikem.cz
Obdrženo: 25. 1. 2012
Přijato: 21. 5. 2012
Sources
1. Hucl T, Gallmeier E. DNA repair: exploiting the Fanconi anemia pathway as a potential therapeutic target. Physiol Res 2011; 60(3): 453–465.
2. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002; 108(2): 171–182.
3. Hucl T, Rago C, Gallmeier E et al. A syngeneic variance library for functional annotation of human variation: application to BRCA2. Cancer Res 2008; 68(13): 5023–5030.
4. Howlett NG, Taniguchi T, Olson S et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002; 297(5581): 606–609.
5. Ford D, Easton DF, Stratton M et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62(3): 676–689.
6. Foretova L, Petrakova K, Palacova M et al. Genetic testing and prevention of hereditary cancer at the MMCI-over 10 years of experience. Klin Onkol 2010; 23(6): 388–400.
7. Wu K, Hinson SR, Ohashi A et al. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res 2005; 65(2): 417–426.
8. Hahn SA, Greenhalf B, Ellis I et al. BRCA2 germline mutations in familial pancreatic carcinoma. J Natl Cancer Inst 2003; 95(3): 214–221.
9. Foretova L, Navratilova M, Machackova E. Limitations of genetic testing in oncology. Klin Onkol 2009; 22 (Suppl): S65–S68.
10. Goldgar DE, Easton DF, Byrnes GB et al. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum Mutat 2008; 29(11): 1265–1272.
11. Frank TS, Deffenbaugh AM, Reid JE et al. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol 2002; 20(6): 1480–1490.
12. Thompson D, Easton DF, Goldgar DE. A full-likelihood method for the evaluation of causality of sequence variants from family data. Am J Hum Genet 2003; 73(3): 652–655.
13. Easton DF, Deffenbaugh AM, Pruss D et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet 2007; 81(5): 873–883.
14. Hofstra RM, Spurdle AB, Eccles D et al. Tumor characteristics as an analytic tool for classifying genetic variants of uncertain clinical significance. Hum Mutat 2008; 29(11): 1292–1303.
15. Chenevix-Trench G, Healey S, Lakhani S et al. Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res 2006; 66(4): 2019–2027.
16. Gallmeier E, Hucl T, Calhoun ES et al. Gene-specific selection against experimental fanconi anemia gene inactivation in human cancer. Cancer Biol Ther 2007; 6(5): 654–660.
17. Grantham R. Amino acid difference formula to help explain protein evolution. Science 1974; 185(4154): 862–864.
18. Jukes TH, King JL. Deleterious mutations and neutral substitutions. Nature 1971; 231(5298): 114–115.
19. Tavtigian SV, Byrnes GB, Goldgar DE et al. Classification of rare missense substitutions, using risk surfaces, with genetic - and molecular-epidemiology applications. Hum Mutat 2008; 29(11): 1342–1354.
20. Yeo G, Hoon S, Venkatesh B et al. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc Natl Acad Sci U S A 2004; 101(44): 15700–15705.
21. Goldgar DE, Easton DF, Deffenbaugh AM et al. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet 2004; 75(4): 535–544.
22. Vallée MP, Francy TC, Judkins MK et al. Classification of missense substitutions in the BRCA genes: a database dedicated to Ex-UVs. Hum Mutat 2012; 33(1): 22–28.
23. Plon SE, Eccles DM, Easton D et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 2008; 29(11): 1282–1291.
24. Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell 2001; 7(2): 263–272.
25. Kuznetsov SG, Liu P, Sharan SK. Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. Nat Med 2008; 14(8): 875–881.
26. Kraakman-van der Zwet M, Overkamp WJ, van Lange RE et al. BRCA2 (XRCC11) deficiency results in radioresistant DNA synthesis and a higher frequency of spontaneous deletions. Mol Cell Biol 2002; 22(2): 669–679.
27. Tutt A, Gabriel A, Bertwistle D et al. Absence of BRCA2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr Biol 1999; 9(19): 1107–1110.
28. Lekomtsev S, Guizetti J, Pozniakovsky A et al. Evidence that the tumor-suppressor protein BRCA2 does not regulate cytokinesis in human cells. J Cell Sci 2010; 123 (Pt 9): 1395–1400.
29. Farrugia DJ, Agarwal MK, Pankratz VS et al. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res 2008; 68(9): 3523–3531.
30. Gallmeier E, Kern SE. Absence of specific cell killing of the BRCA2-deficient human cancer cell line CAPAN1 by poly(ADP-ribose) polymerase inhibition. Cancer Biol Ther 2005; 4(7): 703–706.
31. Balia C, Galli A, Caligo MA. Effect of the overexpression of BRCA2 unclassified missense variants on spontaneous homologous recombination in human cells. Breast Cancer Res Treat 2011; 129(3): 1001–1009.
32. Gallmeier E, Kern SE. Targeting Fanconi anemia/BRCA2 pathway defects in cancer: the significance of preclinical pharmacogenomic models. Clin Cancer Res 2007; 13(1): 4–10.
33. Esashi F, Christ N, Gannon J et al. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005; 434(7033): 598–604.
34. Spurdle AB, Healey S, Devereau A et al. ENIGMA – evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat 2012; 33(1): 2–7.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2012 Issue Supplementum
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Syndrom Birt-Hogg-Dubé
- Klinický význam analýz genů středního rizika pro hodnocení rizika vzniku karcinomu prsu a dalších nádorů v České republice
- Hereditární difuzní karcinom žaludku
- Klinické dysmorfické syndrómy s tumorigenézou