
Analýza změn fosfoproteomu nádorové buněčné linie MDA‑ MB‑ 468 v odpovědi na expresi izoforem p63 pomocí hmotnostní spektrometrie
Analysis of Phosphoproteome Changes in MDA ‑ MB ‑ 468 Cancer Cell Line in Response to Expression of p63 Isoforms Using Mass Spectrometry
Compared to normal cells, tumor cells can show different activity of kinases and phosphatases resulting in altered phosphorylation states of proteins affecting their activity within various signaling pathways. The detection of these alterations is essential for development of targeted therapy based on activation/ inhibition of specific signaling pathways. Various methods can be used for detection of protein phosphorylation; however, a comprehensive assessment of phosphoproteome is performed by mass spectrometry. The differences in phosphoproteome were studied using MDA MB 468 cell line (with incorporated genes encoding isoforms of p63) derived from breast carcinoma. Cells with tetracycline‑induced expression of the p63 isoforms were compared to control cells with wild‑type expression. Denatured proteins from cell lysates were digested to peptides, enriched for phosphopeptides and subsequently separated using liquid chromatograph coupled with mass spectrometer Orbitrap Elite. Three different mass spectrometric methods were used for each sample analysis to find the most suitable conditions for the detection of phosphorylated peptides. Then phosphoproteins were identified and quantified. The number of identified phosphoproteins using all chosen mass spectrometric methods was similar; however, each method showed several unique phosphorylated proteins. Our analysis revealed that both p63 isoforms (TAp63α a ∆Np63α) mainly affected phosphorylation of proteins associated with RNA splicing in MDA MB 468 cells.
Key words:
mass spectrometry – phosphoproteins – signaling pathways – p63 isoforms – breast cancer
This study was supported by the European Regional Development Fund and the State Budget of the Czech Republic (RECAMO, CZ.1.05/2.1.00/03.0101), by the project MEYS – NPS I – LO1413, by MH CZ – DRO (MMCI, 002 09805), by IGA NT/14602-3/2013 and BBMRI_CZ (LM2010004).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
9. 4. 2015
Accepted:
20. 7. 2015
Authors:
P. Dvořáková; M. Nekulová; J. Holčáková; B. Vojtěšek; L. Hernychová
Authors‘ workplace:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
Published in:
Klin Onkol 2015; 28(Supplementum 2): 11-19
doi:
https://doi.org/10.14735/amko20152S11
Overview
Nádorové buňky se od buněk normální tkáně liší mimo jiné aktivací různých signálních drah. Tyto změny jsou podmíněny odlišnou aktivitou kináz a fosfatáz způsobujících rozdíly ve fosforylaci proteinů. Detekce těchto změn je zásadní pro cílenou léčbu zaměřenou na inhibici nebo aktivaci konkrétních signálních drah. Existují různé metody, kterými lze fosforylaci proteinů stanovit. Jednou z vhodných metod schopných charakterizovat fosfoproteom je hmotnostní spektrometrie. Tento článek je zaměřen na analýzu změn ve fosfoproteomu buněčné linie MDA MB 468 odvozené od nádoru prsu s vnesenými geny kódujícími izoformy proteinu p63. Buňky s tetracyklinem indukovanou expresí izoforem proteinu p63 byly srovnány s kontrolními buňkami s přirozenou expresí p63. Denaturované proteiny z buněčného lyzátu byly nejprve enzymaticky štěpeny na peptidy a následně obohaceny o fosfopeptidy. Měření probíhalo pomocí kapalinového chromatografu spojeného s hmotnostním spektrometrem Orbitrap Elite. Každý vzorek byl analyzován třemi různými hmotnostně spektrometrickými metodami s cílem najít nejvhodnější podmínky pro detekování fosforylovaných peptidů. Z naměřených dat pak byly identifikovány a kvantifikovány fosfoproteiny. Počet fosfoproteinů identifikovaných jednotlivými hmotnostně spektrometrickými metodami byl srovnatelný, avšak každou metodou bylo získáno několik unikátních fosforylovaných proteinů. Analýzou získaných dat bylo zjištěno, že v buňkách MDA MB 468 obě izoformy proteinu p63 (TAp63α a ∆Np63α) ovlivňují zejména fosforylaci proteinů odpovědných za sestřih RNA.
Klíčová slova:
hmotnostní spektrometrie – fosfoproteiny – signální dráhy – izoformy p63 – rakovina prsu
Úvod
Fosforylace proteinů je jedna z nejrozšířenějších kovalentních posttranslačních modifikací [1] a hraje klíčovou úlohu v regulaci buněčných signálních drah. Proteiny jsou v buňce fosforylovány prostřednictvím proteinových kináz, které katalyzují přenos fosfátové skupiny z adenosintrifosfátu na serin, treonin a tyrozin, k defosforylaci proteinů pak dochází prostřednictvím proteinových fosfatáz. Onkologická onemocnění jsou často spojena s chybnou funkcí proteinových kináz či fosfatáz a místa fosforylací bývají v nádorových buňkách mutována [2]. Fosforylace se rovněž podílí na regulaci proliferace nádorové buňky, signalizaci onkogenních kináz, regulaci transkripce genů a aktivity nádorového supresoru p53 a mnoha dalších proteinů. Kontrola hladiny fosforylace různých proteinů je proto cílem protinádorové terapie, která využívá především různé tyrozinkinázové inhibitory, jako je např. gefitinib, imatinib, sorafenib atd. [3].
K identifikaci a kvantifikaci změn fosforylace proteinů je možné využít různé metody a přístupy. Vhodný je test kinázové aktivity in vitro v přítomnosti substrátu a adenosintrifosfátu. Další možností je inkubování buněk s radioaktivně značeným 32P ortofosfátem s následnou separací získaných proteinů pomocí gelové elektroforézy. Lze také použít metody založené na existenci fosfo specifických protilátek, jako je imunoblot, ELISA (enzyme‑linked immuno sorbent assay) a průtoková cytometrie. Úspěšná detekce fosfoproteinu je však závislá na specifitě a afinitě protilátky a také na její komerční dostupnosti.
Moderní metoda, pomocí které je možné změny ve fosfoproteomu posoudit komplexně, je hmotnostní spektrometrie. Lze ji využít pro identifikaci fosfoproteinů i pro kvantifikaci změn ve fosfoproteomu. K tomu jsou využívány speciální techniky, mezi něž patří metabolické značení aminokyselin aplikované na živé buněčné kultury stable isotope labeling with aminoacids in cell culture – SILAC) [4], nebo izotopické či izobarické značení peptidů [5,6]. Při SILAC jsou během kultivace do nově syntetizovaných proteinů inkorporovány aminokyseliny značené stabilními izotopy lyzinu a argininu, paralelně jako kontrola jsou kultivovány buňky v lehkém médiu s přirozeně se vyskytujícími aminokyselinami (obr. 1).

Z důvodu výskytu nízké koncentrace fosforylovaných proteinů v buňkách je nutné použít metodu, která specificky tyto proteiny zakoncentruje. Existuje celá řada přístupů, kterými lze obohacení provést, a to na úrovni proteinů či peptidů [7]. Obvykle se používají techniky pro obohacování fosfopeptidů, kdy je nejprve směs proteinů proteolyticky štěpena a vzniklé peptidy jsou následně obohaceny o fosforylované formy. Nejčastěji se volí obohacení fosfopeptidů oxidem titaničitým (TiO2) [8] nebo chelatační afinitní chromatografie (immobilized metal ion affinity chromatography – IMAC) [9]. Při TiO2 obohacení se fosforylované peptidy v kyselém prostředí navazují na TiO2 částice přes svou fosfátovou OH ‑ skupinu a po odstranění nefosforylovaných peptidů jsou z částic uvolněny změnou pH. V případě IMAC se záporně nabité fosfopeptidy navazují na ionty kovů chelatované na částice pokryté nitrilotrioctovou nebo iminodioctovou kyselinou.
Detekce fosfopeptidů může být provedena různými typy hmotnostních spektrometrů (mass spectrometer – MS). MS zaznamená efektivní hmotu fosfopeptidu (poměr hmotnosti a náboje, m/ z), jež je navýšena o fosfátový zbytek (98 Da, 80 Da) [10]. Pomocí tandemového hmotnostního spektrometru (MS/ MS) lze kromě identifikace určit i přesnou aminokyselinu, která byla fosforylována. Pro vznik fragmentačních spekter je třeba detekovaný fosfopeptid (prekurzor) izolovat a následně jej fragmentovat na menší ionty (obr. 2). Fragmentace bývá nejčastěji uskutečněna pomocí kolize iontů peptidů s molekulami plynu (collision induced dissociation – CID), při níž fosfopeptidy ztrácí fosfoskupinu (fragment neutrální ztráty) [11]. Měří se tedy sken neutrálních ztrát (neutral loss – NLMS3), kdy je fragmentační spektrum použito pro identifikování fragmentu neutrální ztráty. Tento fragment je izolován a znovu fragmentován [12]. Je také možné využít vícestupňovou aktivaci (multistage activation – MSA), při které jsou prekurzorový ion a ionty neutrálních ztrát fragmentovány dohromady. Dalším vhodným typem je fragmentace, při níž kladně nabitý peptid reaguje s elektrony např. s fluoranthenovým aniontem (electron transfer dissociation – ETD). Nedochází při ní ke ztrátě fosforylací, a tudíž mohou být přímo identifikovány [12].

Před MS je vhodné předřadit kapalinovou chromatografii, díky které dochází ke snížení komplexity vzorku. Směs fosfopeptidů je nejprve zachycena na analytické koloně a poté jsou fosfopeptidy postupně eluovány z kolony přímo do MS.
Naměřená data musí být hodnocena pomocí speciálních programů – fosfoproteomika využívá programy MaxQuant [13] nebo Proteome Discoverer s algoritmem PhosphoRS 3.1 [14] predikující lokalizaci fosforylovaných míst. Spektra získaná MS/ MS analýzou jsou pomocí vyhledávacích algoritmů (Mascot, Sequest) porovnávána s proteinovou databází, při kvantitativních studiích je navíc určena změna fosforylace oproti kontrolnímu vzorku a celkové expresi proteinu. Proteiny vykazující signifikantní kvantitativní změny se mohou za pomoci speciálních programů, např. Ingenuity Pathway Analysis, David, PathVisio, využít ke studiu signálních drah [15].
Analýza fosfoproteomu byla provedena v buněčné linii MDA MB 468 s inducibilní expresí N ‑ koncových izoforem proteinu p63 (TAp63α a ∆Np63α), které se od sebe liší přítomností N koncové transaktivační domény. Linie MDA MB 468 je odvozena od nádoru prsu bazálního typu a vykazuje triple negativní fenotyp, tj. je negativní pro estrogenový a progesteronový receptor a bez zvýšené hladiny Her2. Do této linie byly vneseny geny kódující izoformy proteinu p63, TAp63α nebo ∆Np63α, jejichž exprese je indukovatelná tetracyklinem. Protein p63 má zásadní význam v průběhu ontogeneze epiteliálních struktur včetně mléčné žlázy a jeho hladina je často zvýšená u spinocelulárních karcinomů [16 – 18]. V případě karcinomu prsu byla popsána exprese proteinu p63 u jeho bazálního typu, který je často spojen s triple ‑ negativním fenotypem [19]. Buněčná linie MDA MB 468 je proto vhodným modelem pro studium funkce izoforem p63 u karcinomu prsu. Protein p63 plní funkci transkripčního faktoru a v nádorové buňce je zapojen do mnoha signálních drah, např. EGFR (epidermal growth factor receptor), FGFR (firoblast growth factor receptor), Wnt/ β‑catenin, Notch a dalších [20 – 22]. Předpokládá se tedy, že výrazně ovlivňuje i fosfoproteom nádorové buňky. Izoforma TAp63α plní funkce nádorového supresoru a inhibitoru metastazování [23,24]. Izoforma ∆Np63α byla původně považována pouze za dominantně negativní inhibitor aktivity TAp63 a případně i dalších členů rodiny p53 [25], dnes je však známo, že je také schopna aktivovat transkripci cílových genů [26].
Cílem této práce je nalezení optimální metody pro detekování fosforylovaných proteinů ve výše popsané buněčné linii a zároveň zjistit změny ve fosfoproteomu.
Materiál a metody
V rámci studie byly porovnány fosfoproteiny buněk MDA MB 468 s indukcí a bez indukce exprese izoforem p63 (TAp63α a ∆Np63α) pomocí tetracyklinu. Buněčné lyzáty byly po změření koncentrace smíchány, denaturované proteiny štěpeny a peptidy obohaceny o fosfopeptidy pomocí TiO2. Takto připravené vzorky (celkem tři) byly změřeny třemi různými hmotnostně spektrometrickými metodami s předřazenou kapalinovou chromatografií (LC MS/ MS). Získaná data z každého měření byla zpracována zvlášť pomocí programu Proteome Discoverer (obr. 3).
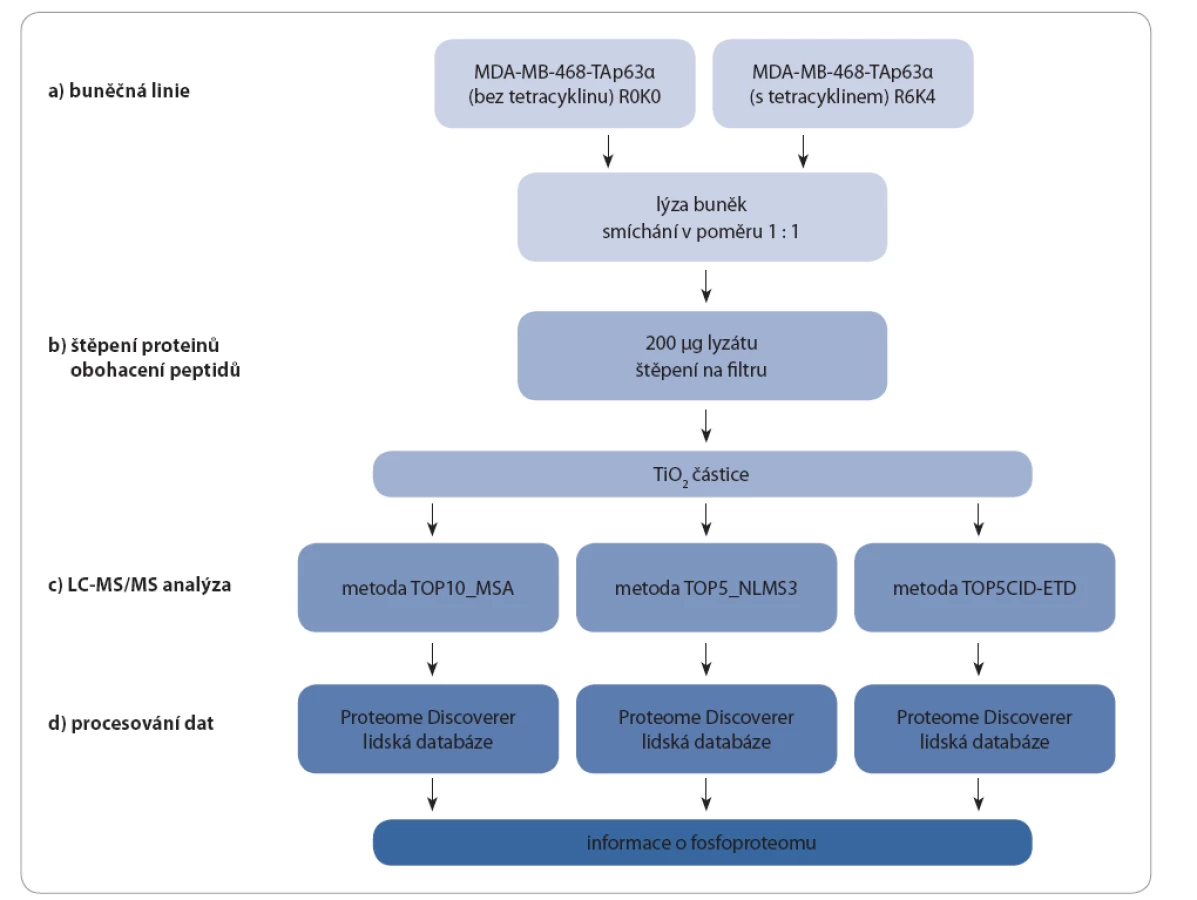
Buněčná linie
Buněčné linie MDA MB 468 - ∆Np63α a MDA MB 468 - TAp63α obsahují stabilně inkorporovaný gen kódující danou izoformu proteinu p63 pod kontrolou cytomegalovirového (CMV) promotoru, jehož exprese je inhibována vazbou tetracyklinového represoru. Přidání tetracyklinu do kultivačního média má za následek zablokování vazby represoru a tím spuštění transkripce příslušného genu.
Jako kontrolní linie byly použity buňky MDA MB 468 - pcDNA6/ TR, které jsou mateřskou linií výše zmíněných dvou linií a exprimují pouze tetracyklinový represor. Buňky byly kultivovány za standardních podmínek v DMEM (Dulbecco’s modified eagle’s medium) obohaceném o 10% fetální bovinní sérum (HyClone, Thermo Scientific, USA). Pro kvantifikaci byla použita metoda SILAC: buňky byly kultivovány v DMEM obsahujícím neznačené aminokyseliny (R0K0) a v médium se značenými aminokyselinami (R6K4)(Dundee Cell Products, UK), s přídavkem 200 mg/ l L prolinu. K buňkám v DMEM R6K4 byl na 24 hod přidán tetracyklin v koncentraci 1 µg/ ml. Buňky (dvě misky o průměru 10 cm) byly lyzovány v pufru obsahujícího 8 M močovinu (Sigma Aldrich, USA), 40 mM NaCl (Lach:ner, ČR), 50 mM tris(hydroxymetyl)aminometan (Bio Rad, USA), 2 mM MgCl2 (Sigma Aldrich, USA) s přídavkem inhibitoru fosfatáz a proteáz. Koncentrace proteinů byla změřena metodou dle Bradfordové. Lyzáty o stejné koncentraci celkového proteinu byly následně smíchány v poměru 1 : 1 (w/ w).
Štěpení proteinů na peptidy a jejich následné obohacení o fosfopeptidy
Na přípravu každého vzorku bylo použito 200 µg celkového denaturovaného proteinu. Štěpení probíhalo na filtru FASP (filter aided sample preparation) [27] za použití trypsinu. Centrifugací byla směs peptidů uvolněna z filtru, odpařena do sucha a poté obohacena o fosfopeptidy pomocí TiO2 částic (GL Sciences, Japan) [28]. Vzorky následně odpařené do sucha byly ihned analyzovány pomocí MS.
LC ‑ MS/ MS metoda
Vzorky byly rozpuštěny ve 20 µl 0,1% kyseliny mravenčí (Sigma Aldrich, USA) a podrobeny LC MS/ MS analýze na kapalinovém chromatografu UltiMate 3000 RSLCnano spojeném on line s MS Orbitrap Elite. Fosfopeptidy byly nejprve odsoleny na předkolonce C18 PepMap 100 (30 µm × 5 mm; velikost částic: 5 µm) a následně byly separovány na analytické koloně Acclaim PepMap RSLC C18, (75 µm × 150 mm; velikost částic: 2 µm). Jako mobilní fáze byly použity 0,1% kyselina mravenčí (A) a 0,08% kyselina mravenčí v 80% acetonitrilu (B). Pro postupnou eluci peptidů z kolony byl zvolen gradient s lineárně se zvyšující organickou frakcí (0 – 4 min 2 % B, 68 min 40 % B,70 – 80 min 98 % B, 81 – 105 min 2 % B). Detekce eluovaných fosfopeptidů, jejichž efektivní hmotnost se nacházela v rozmezí 400 – 2 000, probíhala pomocí MS při rozlišení 120 000 (pro 400 m/ z). Každý vzorek byl měřen třemi rozdílnými metodami – metodou vícestupňové aktivace (CID_MSA), metodou neutrálních ztrát (CID_NLMS3) a metodou kombinující CID a ETD fragmentační techniku (CID_ETD). Nejintenzivnější píky v daném čase byly izolovány a fragmentovány v lineární iontové pasti (10 000 iontů akumulováno po dobu 10 ms s izolačním oknem 2 m/ z; CID: relativní energie 35 %; ETD: aktivační doba 100 ms).
Zpracování dat
Data získaná z LC MS/ MS analýz byla zpracována v programu Proteome Discoverer verze 1.4 s algoritmem PhosphoRS 3.1 a prohledávána proti lidské databázi Uniprot a Swissprot (databáze stažena v červnu 2014) [29]. Byly zvoleny běžně se vyskytující modifikace peptidů a dále fosforylace na serinu, treoninu a tyrozinu. Pro odstranění falešně pozitivních výsledků byly získané peptidy prohledány proti obrácené databázi a pro identifikaci fosfoproteinů byly vybrány pouze peptidy s menší než 1% falešně pozitivní mírou. Jelikož některé proteiny mají vzájemně společné peptidy, jsou tyto proteiny spojovány do proteinových skupin.
Z fosfoproteinů identifikovaných alespoň jednou ze tří metod byly následně vybrány ty, u kterých došlo po působení tetracyklinu ke kvantitativní změně o více než 20 %. Pro odstranění falešně pozitivních výsledků byly z analýzy vyloučeny proteiny, u nichž došlo ke změně exprese i v kontrolní linii MDA MB 468 - pcDNA6/ TR. Hladina těchto proteinů tedy nebyla regulována v závislosti na expresi izoforem p63, ale nespecificky samotným působením tetracyklinu. Takto vybrané proteiny, jejichž exprese souvisí s hladinou izoforem p63, byly použity pro analýzu genové ontologie a signálních drah pomocí veřejně přístupné databáze DAVID Bioinformatics Resources 6.7 [30,31]. Dále byly vybrány proteiny identifikované s vysokou spolehlivostí, tj. všemi třemi metodami, u kterých kvantitativní změna vykazovala stejný trend (nárůst vs. snížení).
Výsledky
V prvním kroku byla zjišťována vhodnost jednotlivých použitých fragmentačních metod a to na základě počtu identifikovaných proteinových skupin v jednotlivých vzorcích. Získaná data jsou uvedena v tab. 1. Počet identifikovaných proteinových skupin jednotlivými metodami byl srovnatelný. Významně více proteinových skupin bylo identifikováno v případě zpracování dat ze všech třech měření dohromady.

V druhém kroku hodnocení dat byly vybrané fosforylované proteiny použity pro analýzu genové ontologie (tab. 2) a mapování signálních drah (tab. 3). Obě izoformy regulovaly fosfoproteiny podílející se na regulaci sestřihu RNA, izoforma TAp63α ovlivňovala buněčný cyklus a apoptózu, izoforma ∆Np63α buněčnou polaritu a organizaci cytoskeletu. Konkrétní příklady proteinů (identifikované všemi třemi metodami) s významným kvantitativním rozdílem jsou uvedeny v tab. 4.



Diskuze
U všech třech analyzovaných vzorků (MDA MB 468 - ∆Np63α; MDA MB 468 - TAp63α; MDA MB 468 - pcDNA6/ TR) byl identifikován srovnatelný počet proteinů. Každou fragmentační metodou (CID_MSA, CID_NLMS3, CID_ETD) byla kromě společných proteinových skupin získána až desetina unikátních (obr. 4). Ze získaných výsledků je patrné, že kombinování fragmentačních metod je pro identifikaci většího množství fosforylovaných proteinů velmi výhodné. Předpokladem k tomuto přístupu je ovšem použití vysokorozlišovacího MS, který nabízí kombinování více typů fragmentací. Na druhou stranu je třeba upozornit na složitou interpretaci získaných dat. V některých případech může být fosforylace a její lokalizace zachycena pouze jednou metodou, zatímco ostatní metody detekují peptid bez fosforylace. Je tedy třeba vybrat strategii, která danému typu experimentu vyhovuje nejlépe.

Z analýzy fosfoproteinů identifikovaných alespoň jednou ze tří metod vyplynulo, že obě izoformy p63 se podílí na regulaci sestřihu RNA. Tento poznatek je ve shodě s výsledky analýzy interaktomu p63, která ukázala, že p63 interaguje s proteiny, jež jsou součástí tzv. spliceozomu [32,33]. Sestřih RNA je zásadní mechanizmus regulace genové exprese, který umožňuje alternativní výběr sestřihových míst, a tím produkci širokého spektra proteinových izoforem [34,35]. Mnoho genů zapojených do regulace buněčného cyklu a apoptózy kóduje proteinové izoformy s odlišnými až antagonistickými funkcemi [36]. Proto je v nádorových buňkách alternativní sestřih často deregulován ve smyslu produkce izoforem s onkogenním účinkem [37]. Nalezení souvislosti exprese izoforem p63 s regulací alternativního sestřihu prostřednictvím změn ve fosforylaci proteinů zapojených do tohoto procesu ukazuje na nový potenciální mechanizmus působení tohoto proteinu v nádorových buňkách.
Závěr
Získání informace o změnách ve fosfoproteomu nádorové buňky je zásadní pro pochopení změn v signálních drahách a může být základem pro vývoj personalizované léčby. Dnes používané techniky jsou schopny zachytit pouze nepatrnou část fosforylovaných proteinů přítomných v daném čase v dané buňce. Metody hmotnostní spektrometrie umožňují v současnosti velmi komplexní pohled na buněčný fosfoproteom, volba konkrétní metody pak závisí na povaze vzorku a požadovaných výsledcích. Jako nejvýhodnější řešení k identifikaci co největšího počtu fosforylovaných proteinů se jeví použití více fragmentačních metod pro měření jednoho vzorku. Tento přístup je spíše než do klinické praxe určen, aspoň prozatím, pouze pro výzkumné účely. Nabízí identifikaci nových proteinů nebo signálních drah aberantně aktivovaných nebo inaktivovaných v nádorových buňkách, které budou v další etapě validovány jako potenciální biomarkery.
Práce byla podpořena Evropským fondem pro regionální rozvoj a státním rozpočtem České republiky (RECAMO, CZ.1.05/2.1.00/03.0101), projektem MŠMT – NPU I – LO1413, MZ ČR – RVO (MOÚ, 00209805), IGA NT/14602-3/2013 a BBMRI_CZ (LM2010004).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Ing. Petra Dvořáková, Ph.D.
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: petra.dvorakova@mou.cz
Obdrženo: 9. 4. 2015
Přijato: 20. 7. 2015
Sources
1. Yates JR, Shabaz M, Heck AJ. Prosphoproteomics. Anal Chem 2014; 86(3): 1313. doi: 10.1021/ ac404019p.
2. Reimand J, Wagih O, Bader GD. The mutational landscape of phosphorylation signaling in cancer. Sci Rep 2013; 3 : 2651. doi: 10.1038/ srep02651.
3. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009; 9(1): 28 – 39. doi: 10.1038/ nrc2559.
4. Oda Y, Huang K, Cross FR et al. Accurate quantitation of protein expression and site ‑ specific phosphorylation. Proc Natl Acad Sci USA 1999; 96(12): 6591 – 6596.
5. Gygi SP, Rist B, Gerber SA et al. Quantitative analysis of complex protein mixtures using isotope ‑ coded affinity tags. Nature Biotechnol 1999; 17(10): 994 – 999.
6. Thompson A, Schafer J, Kuhn K et al. Tandem mass tag: a novel qunatification strategy for comparative analysis of complex protein mixture by MS/ MS. Anal Chem 2003; 75(8): 1895 – 1904.
7. Fíla J, Honys D. Enrichment techniques employed in phosphoproteomics. Amino Acids 2011; 43(3): 1025 – 1047. doi: 10.1007/ s00726 ‑ 011 ‑ 1111 ‑ z.
8. Pinkse MW, Uitto PM, Hilhorst MJ et al. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D ‑ nano LC ‑ ESI ‑ MS/ MS and titanium oxide precolumns. Anal Chem 2004; 76(14): 3935 – 3943.
9. Ficarro SB, McCleland ML, Stukenberg PT et al. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nature Biotechnol 2002; 20(3): 301 – 305.
10. Mann M, Ong S, Grønborg M et al. Analysis of protein phosphorylation using mass spectrometry: deciphering the phosphoproteome. Trends Biotechnol 2002; 20(6): 261 – 268.
11. Hoffert JD, Knepper MA. Taking aim at shotgun phosphoproteomics. Anal Biochem 2008; 375(1): 1 – 10.
12. Boersema PJ, Mohammed S, Heck AJ. Phosphopeptide fragmentation and analysis by mass spectrometry. J Mass Specrom 2009; 44(6): 861 – 878. doi: 10.1002/ jms.1599.
13. MaxQuant.org [homepage on the Internet]. MaxQuant. Max planck institute of biochemistry. Martinsried: Germany; c2015 [cited 2015 March 30]. Available from: http:/ / www.maxquant.org/ .
14. Taus T, Köcher T, Pichler P et al. Universal and confident phosphorylation site localization using phospho RS. J Proteome Res 2011; 10(12): 5354 – 5362. doi: 10.1021/ pr200611n.
15. Pjechová M, Hernychová L, Tomašec P et al. Analýza fosfoproteínov a signálních dráh kvantitatívno ‑ proteomickými metodami. Klin Onkol 2014; 27 (Suppl 1): S116 – S120. doi: 10.14735/ amko20141S116.
16. Yang A, Schweitzer R, Sun D et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999; 398(6729): 714 – 718.
17. Hibi K, Trink B, Patturajan M et al. AIS is an oncogene amplified in squamous cell carcinoma. Proc Natl Acad Sci U S A 2000; 97(10): 5462 – 5467.
18. Nylander K, Vojtesek B, Nenutil R et al. Differential expression of p63 isoforms in normal tissues and neoplastic cells. J Pathol 2002; 198(4): 417 – 427.
19. Leong CO, Vidnovic N, DeYoung MP et al. The p63/ p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest 2007; 117(5): 1370 – 1380.
20. Danilov AV, Neupane D, Nagaraja AS et al. Delta-Np63alpha ‑ mediated induction of epidermal growth factor receptor promotes pancreatic cancer cell growth and chemoresistance. PLoS One 2011; 6(10): e26815. doi: 10.1371/ journal.pone.0026815.
21. Ramsey MR, Wilson C, Ory B et al. FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J Clin Invest 2013; 123(8): 3525 – 3538. doi: 10.1172/ JCI68899.
22. Wu N, Rollin J, Masse I et al. p63 regulates human keratinocyte proliferation via MYC ‑ regulated gene network and differentiation commitment through cell adhesion‑related gene network. J Biol Chem 2012; 287(8): 5627 – 5638. doi: 10.1074/ jbc.M111.328120.
23. Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res 2004; 2(7): 371 – 386.
24. Su X, Chakravarti D, Cho MS et al. TAp63 suppresses metastasis through coordinate regulation of dicer and miRNAs. Nature 2010; 467(7318): 986 – 990. doi: 10.1038/ nature09459.
25. Yang A, Kaghad M, Wang Y et al. p63, a p53 homolog at 3q27 - 29, encodes multiple products with transactivating, death ‑ inducing, and dominant ‑ negative activities. Mol Cell 1998; 2(3): 305 – 316.
26. Ghioni P, Bolognese F, Duijf PH et al. Complex transcriptional effects of p63 isoforms: identification of novel activation and repression domains. Mol Cell Biol 2002; 22(24): 8659 – 8668.
27. Wisniewski JR, Zougman A, Nagaraj N et al. Universal sample preparation method for proteome analysis. Nat Methods 2009; 6(5): 359 – 362. doi: 10.1038/ nmeth.1322.
28. Aryal UK, Ross AR. Enrichment and analysis of phosphopeptides under different experimental conditions using titanium dioxide affinity chromatography and mass spektrometry. Rapid Commun Mass Spectrom 2010; 24(2): 219 – 231. doi: 10.1002/ rcm.4377.
29. UniProt.org [homepage on the Internet]. UniProt, c2002 – 2015 [cited 2015 March 30]. Available from: http:/ / www.uniprot.org/ .
30. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc 2009; 4(1): 44 – 57. doi: 10.1038/ nprot.2008.211.
31. Huang Y, Jeong JS, Okamura J et al. Global tumor protein p53/ p63 interactome. Cell Cycle 2012; 11(12): 2367 – 2379. doi: 10.4161/ cc.20863.
32. Huang da DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37(1): 1 – 13. doi: 10.1093/ nar/ gkn923.
33. Amoresano A, Di Costanzo A, Leo G et al. Identification of deltaNp63alpha protein interactions by mass spectrometry. J Proteome Res 2010; 9(4): 2042 – 2048. doi: 10.1021/ pr9011156.
34. Moore MJ, Wang Q, Kennedy CJ et al. An alternative splicing network links cell ‑ cycle control to apoptosis. Cell 2010; 142(4): 625 – 636. doi: 10.1016/ j.cell.2010.07.019.
35. David CJ, Manley JL. Alternative pre‑mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 2010; 24(21): 2343 – 2364. doi: 10.1101/ gad.1973010.
36. Schwerk C, Schulze ‑ Osthoff K. Regulation of apoptosis by alternative pre‑mRNA splicing. Mol Cell 2005; 19(1): 1 – 13.
37. Liu S, Cheng C. Alternative RNA splicing and cancer. Wiley Interdiscip Rev RNA 2013; 4(5): 547 – 566. doi: 10.1002/ wrna.1178.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2015 Issue Supplementum 2
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Adenovírusové vektory v génovej terapii
- Nrf2 – dve tváre regulátora antioxidačného systému
- Rekombinantní protilátky a jejich využití v protinádorové terapii
- Co může přinést studium oligomerizace proteinů v procesu onkogeneze?