
Polo‑like kináza 1 jako cíl protinádorové terapie
Polo‑like Kinase 1 as a Target for Anti‑tumor Therapy
Individual proteins from polo-like kinase (Plk) family fulfil different but critical functions in regulating cell cycle and coordinate cell response to DNA damage. The most studied one from this five member family is Plk1. It is a serine/ threonine kinase that plays a pivotal role in many aspects of mitosis and its deregulation is common in various tumor types where the elevated level is mostly associated with worse prognosis. From the therapeutical point of view, intertwined relationship between Plk1 and p53 protein is very interesting and will be discussed. Not only for these reasons, Plk1 has become an attractive target for anti‑tumor drug development. The most promising seems to be ATP binding site inhibitor Volasertib (BI 6727) which provided a survival benefit for patients with acute myeloid leukemia and is now tested in phase III clinical trial. A new generation of Plk1 inhibitors that target the second druggable domain of Plk1, the polo- box domain, is currently being tested preclinically and are believed to improve Plk1 specificity.
Key words:
polo like kinase 1 – tumor suppressor protein p53 – ATP competitive inhibitors – polo- box domain – drug evaluation studies
This study was supported by the European Regional Development Fund and the State Budget of the Czech Republic (RECAMO, CZ.1.05/2.1.00/03.0101), MEYS – NPS I – LO1413, MH CZ – DRO (MMCI, 00209805) and BBMRI_CZ (LM2010004).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
9. 4. 2015
Accepted:
19. 6. 2015
Authors:
I. Procházková; B. Vojtěšek
Authors‘ workplace:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
Published in:
Klin Onkol 2015; 28(Supplementum 2): 32-39
doi:
https://doi.org/10.14735/amko20152S32
Overview
Jednotlivé proteiny z rodiny polo-like kináz (Plk) plní rozdílné, avšak kritické funkce při regulaci buněčného cyklu a koordinují buněčnou odpověď na poškození DNA. Nejvíce prostudovaným z pěti členů této rodiny je protein Plk1. Jedná se o serin/ treonin kinázu, která hraje klíčovou roli v mnoha fázích mitózy, a s její deregulací se setkáváme u různých typů nádorů, kde je její zvýšená hladina většinou asociována s horší prognózou. Z pohledu léčby je také zajímavý vztah Plk1 a proteinu p53. Nejen z těchto důvodů se Plk1 stala jedním z atraktivních cílů pro vývoj protinádorových léčiv. Nejnadějněji se nyní jeví inhibitor ATP vazebné oblasti Plk1 volasertib (BI 6727), který v doposud provedených klinických studiích prodloužil přežití pacientů s akutní myeloidní leukemií a nyní je testován v klinické studii fáze III. Ve fázi preklinického testování se nachází také několik inhibitorů polo‑ box domény (druhého možného místa inhibice polo‑like kináz), které by měly zajistit větší specifitu vůči Plk1.
Klíčová slova:
polo like kináza 1 – nádorový supresor p53 – ATP kompetitivní inhibitory – polo box doména – klinické studie
Rodina proteinů polo-like kináz
Proteiny z rodiny polo‑like kináz (Plk), jež čítají celkem pět členů, jsou významné regulátory mitózy, meiózy i cytokineze, zkrátka procesu buněčného dělení. Všechny tyto serin/ treonin kinázy obsahují zakonzervovanou kinázovou katalytickou doménu na N‑ konci a jednu či více C‑ koncových „polo‑ box“ domén, jež se uplatňují při vazbě substrátu. Sekvenční podobnost jednotlivých členů vůči Plk1 je 38 % (Plk2, Plk3), resp. 26 % (Plk4, Plk5). Nejvíce prostudovaným členem rodiny je Plk1, a proto jejím fyziologickým funkcím budeme věnovat zvláštní prostor níže a úvodem stručně představíme ostatní členy rodiny Plk.
Plk2 je, na rozdíl od Plk1, které můžeme přisuzovat spíše roli onkoproteinu, považována za nádorový supresor. Některé studie naznačují vliv na inhibici růstu nádorů v souvislosti s tím, že aktivace Plk2 je nutná pro mitózu [1], konkrétně pro duplikaci centrozomu [2]. Plk2 má také potenciální roli v signalizaci v kontrolním bodě replikace skrze interakce s checkpoint kinázou I i II a proteinem p53. Bylo také ukázáno, že Plk2 deficientní lidské nádorové buňky jsou citlivé na replikační stres [3]. Identifikace jasné role Plk2 jako nádorového supresoru je komplikována zajímavou skutečností, že ztráta exprese Plk2 vede ke zvýšené hladině Plk3, která je sama o sobě také považována za nádorový supresor [4].
Hladina Plk3 zůstává v průběhu buněčného cyklu poměrně stálá, ale její kinázová aktivita vrcholí v pozdní S fázi a ve fázi G2 [5]. Obecně můžeme říci, že její role je protichůdná k pro‑proliferační roli Plk1. Role Plk3 jako nádorového supresoru spočívá mimo jiné ve fosforylaci serinu 20 proteinu p53, což brání jeho interakci s ubikvitin ligázou MDM2, čímž dochází ke stabilizaci p53 a k zabránění jeho degradace [6]. Dalším substrátem Plk3 je také nádorový supresor PTEN, jehož fosforylace též vede k jeho stabilizaci [7]. Srovnání profilů genové exprese Plk3 ukazuje na jeho sníženou hladinu v nádorech [8] a naopak jeho overexprese v savčích buňkách vede k zástavě růstu a indukci kondenzace chromatinu a apoptóze.
Exprese proteinu Plk4 (označován i jako SAK) vrcholí v průběhu mitózy a jeho funkce je nezbytná pro duplikaci centrozomu. Plk4 opět interaguje s proteiny regulujícími proliferaci, např. p53 [9] a Cdc25C [10]. Co se týče vztahu tohoto proteinu k nádorové transformaci, aberantní exprese byla detekována v nádorech kolorekta [11]. Na buněčných liniích nádoru prsu bylo rovněž ukázáno, že deplece Plk4 snižuje jejich proliferaci ve srovnání s buňkami normální tkáně [12]. Byla také vyvinuta řada inhibitorů Plk4, které ukázaly protinádorový efekt na buněčných a in vivo modelech nádoru prsu [13].
Posledním, nejpozději objeveným členem rodiny polo‑like kináz je Plk5, jejíž role je spíše podobná Plk2 a Plk3 než Plk1 a Plk4, i když obsahuje nefunkční zkrácenou kinázovou doménu [14]. Overexprese Plk5 vede k zástavě buněčného cyklu v G1 fázi a také k apoptóze, podobně jako je tomu u Plk3. Je exprimována hlavně v diferencovaných tkáních, jako např. mozeček, a její exprese je epigeneticky umlčena u glioblastomů [15,16]. Komplexní a podrobnější informace o jednotlivých členech proteinů rodiny Plk lze najít v přehledovém článku De Cárcer et al [15].
Fyziologické funkce Plk1
Plk1 je jeden z klíčových proteinů podílejících se na progresi buněčného cyklu (obr. 1). Buněčný cyklus je série velmi přísně řízených procesů vrcholící rozdělením mateřské buňky na dvě dceřiné. Přechod mezi jednotlivými fázemi buněčného cyklu (G1 – gap 1, S – synthesis, G2 – gap 2 a M – mitosis) je regulován fosforylacemi a ubikvitinacemi proteinů, které jsou řízeny cykliny a cyklin‑dependentními kinázami. Exprese Plk1 je přímo spojena s jednotlivými fázemi cyklu, přičemž hladina proteinu se začíná zvyšovat v S fázi, vrcholí při přechodu G2– M, ustálí se v průběhu mitózy a ostře klesá po ukončení M fáze [17]. Konkrétní zapojení Plk1 do M fáze spočívá ve fosforylaci (aktivaci) komplexu cyklin B1/ Cdk1, který je hlavním regulátorem vstupu buňky do M fáze [18– 21]. Plk1 se také podílí na procesu tvorby centrozomů a skládání mitotického vřeténka fosforylací Ninein‑like proteinu (NLP), což je protein asociovaný v interfázi s centrozomy [22]. Takto fosforylovaný NLP umožňuje buňce přepnout z interfáze do mitózy tím, že NLP disociuje z centrozomu a umožní jeho „dozrání“ [22]. Další substrát fosforylovaný Plk1 je protein p150Glued, který je důležitý pro „destrukci“ jaderného obalu, k níž dochází v průběhu mitózy na konci profáze [23]. Plk1 se také podílí na správném přichycení mikrotubulů kinetochoru fosforylací klíčových proteinů CLIP‑ 170 [24] a Sgt1 [25] – tento proces umožňuje rovnoměrnou segregaci chromozomů. Plk1 též iniciuje uvolnění z mitózy aktivací anafázi podporujícího komplexu, čímž hraje významnou roli při cytokinezi [26].

Funkce Plk1 nejsou omezeny pouze na buněčnou proliferaci, ale tato kináza se uplatňuje i při stresové signalizaci, konkrétně při odpovědi buňky na poškození DNA [27,28]. V nádorových buňkách vede snížení hladiny Plk1 k nahromadění poškozené DNA a následné apoptóze, což ale neplatí u buněk normálních bez přítomnosti replikačního stresu [29].
Jelikož je Plk1 důležitá kináza při mitotickém dělení buněk, není překvapením, že tomuto faktu odpovídá i její distribuce v tkáních. V průběhu embryogeneze je Plk1 exprimována ve vysoce proliferujících tkáních a stejně tak je tomu i u dospělého člověka, kde můžeme Plk1 nalézt ve varlatech, slezině a kostní dřeni, nikoli však v ostatních neproliferujících tkáních, včetně slinivky, srdce, kůže mozku a ledvin [17,30]. U mnoha nádorových onemocnění je ovšem exprese Plk1 zvýšená ve srovnání s fyziologickým stavem, jak o tom bude pojednáno v následující kapitole.
Ve srovnání s Plk1 je tkáňová distribuce Plk2 a Plk3 širší a není tak úzce spojena s vysoce proliferujícími tkáněmi [31]. Jak již bylo uvedeno výše, hladina Plk5 je u proliferujících buněk dokonce snížena. Rozdílná tkáňově specifická exprese Plk je důležitým faktorem pro možnou budoucí klinickou aplikaci vyvíjených inhibitorů Plk1. Podrobnější informace o fyziologických funkcích Plk1 lze nalézt v přehledových článcích zahraničních autorů [32– 34].
Plk1 v nádorové transformaci
Na rozdíl od normálních buněk, kde se Plk1 uplatňuje především v průběhu mitózy, u buněk nádorových dochází k přemístění Plk1 do jádra dříve než při přechodu G2– M, a dokonce zde může být detekován i v G1– S fázi, což svědčí o specifické funkci Plk1 u nádorových buněk v interfázi (období buněčného cyklu mezi dvěma M fázemi). Některé studie naznačují, že se může také uplatňovat v S fázi, kde fosforyluje proteiny nezbytné pro replikaci DNA [35– 38].
Exprese Plk1 souvisí s růstem a progresí rozličných typů nádorů. Její zvýšenou hladinu můžeme najít u nemalobuněčného karcinomu plic, kde nepřímo koreluje s přežitím pacientů a je silným negativním prognostickým faktorem [39]. Stejná je situace u nádorů hlavy a krku [40,41] i jícnu [42]. Zvýšená hladina Plk1 se vyskytuje také u nádorů prsu [43– 45], jater [46], slinivky [47,48], prostaty [49], stejně tak jako u kolorektálního karcinomu [11,50], nádorů endometria [51] a vaječníků [52,53], kde byla prokázána korelace zvyšující se exprese Plk1 s „grade“ či „stage“ nádoru. Na druhou stranu spojitost zvýšené exprese Plk1 a prognózy nebyla prokázána u nádorů prostaty, slinivky a prsu [40,45,47,49].
Asi není třeba nějak zvlášť představovat a vyzdvihovat protein a nádorový supresor p53, který je jednou z centrálních molekul onkogeneze, a ztráta či změna jeho funkce (ať už mutacemi v genu TP53 nebo inaktivací jeho signální dráhy) je přítomna u většiny nádorů [54– 56]. Jako „strážce genomu“ [57] je aktivován obecně buněčným stresem a zasahuje např. do procesů opravy DNA, zastavení buněčného cyklu, apoptózy a senescence. Jeho aktivita a hladina v buňce je regulována celou řadou proteinů a posttranslačními modifikacemi [58,59]. Jednou z cest k úspěšnější léčbě onkologických pacientů by mohlo být obecně obnovení funkce proteinu p53 u nádorových buněk [56,60].
V současnosti existuje mnoho důkazů o tom, že Plk1 a signální dráha p53 se navzájem ovlivňují. Dráha p53 tlumí aktivitu promotoru genu Plk1 hned několika způsoby. Jeden z nich má na svědomí protein p21WAF1, mediátor signalizační dráhy p53, jenž inhibuje expresi Plk1 cílením na represorové elementy v promotoru Plk1 [61]. Dále např. p53 negativně reguluje expresi proteinu FoxM1, což je onkogenní transkripční faktor, který stimuluje expresi Plk1 [61– 64] a je jí samotnou pozitivně regulován prostřednictvím fosforylací [65]. Bylo ukázáno, že protein p53 reguluje expresi Plk1 také přímo, a to tím, že se váže na promotor Plk1 na dvou různých místech (p53 response element 1 a 2) [66].
Jelikož mutovaný p53 není schopen zabránit expresi Plk1, byla publikována studie ukazující na významnou korelaci pozitivního imunohistochemického barvení Plk1 u pacientek s primárními nádory prsu s přítomností nefunkčního mutovaného proteinu p53. Tyto pacientky vykazovaly také významně horší přežití oproti pacientkám pouze s pozitivním barvením Plk1 nebo pouze s mutací p53 [45].
Jak bylo popsáno výše, regulace Plk1 a p53 je vzájemná. To znamená, že Plk1 se nenechá jen pasivně řídit dráhou proteinu p53, nýbrž také negativně reguluje p53, a to jak přímo, tak pomocí nepřímých mechanizmů. U buněčné linie plicního karcinomu H1299 bylo prokázáno, že se Plk1 fyzicky váže na DNA‑ vazebnou doménu proteinu p53, čímž dochází k inhibici funkce tohoto proteinu [67]. Tato negativní regulace by mohla být fundamentálním mechanizmem pro roli Plk1 v onkogenezi. Jelikož se mnoho bodových mutací vyskytuje právě v DNA‑ vazebné doméně p53, bude zajímavé zjistit, jestli některá z těchto mutací brání v interakci s Plk1, a tudíž vede k deregulaci inhibice proteinu p53. Nepřímým mechanizmem inaktivace proteinu p53 pomocí Plk1 je fosforylace proteinů, které jsou odpovědné za degradaci p53. Jedním z nich je protein MDM2, který svojí E3 ubikvitin ligázovou aktivitou právě negativně reguluje aktivitu proteinu p53 [68]. Plk1 fosforyluje MDM2 na serinu 260, čímž dochází ke stimulaci této degradace [69]. Plk1 fosforyluje také topoizomerázu I‑ vázající protein (Topors), jež vykazuje jak ubikvitin, tak SUMO‑ 1 E3 ligázovou aktivitu a váže se rovněž na p53. Fosforylace serinu 718 na proteinu Topors vede k potlačení sumoylace a naopak k posílení ubikvitinace proteinu p53 a jeho následné degradaci [70]. Konečně posledním nepřímým mechanizmem regulace p53 kinázou Plk1, o němž se zmíníme, je i fosforylace serinu 435 proteinu GTSE1 (G2 a S‑ phase‑ expressed 1 protein), která také vede k degradaci p53 [71].
Dříve bylo ukázáno, že u nádorových buněk s nefunkčním proteinem p53 vede snížená hladina Plk1 k větší cytotoxicitě v porovnání s normálními buňkami [29,72– 75]. Ve studii provedené na buněčné linii kolorektálního karcinomu HCT116 TP53– / – vedla inhibice Plk1 k výraznému snížení tvorby nádorů na zvířecích modelech [76]. Obdobný závěr, tedy větší citlivost k inhibici Plk1 u nádorových buněk s defektním proteinem p53 (v porovnání s nemutovaným proteinem p53), lze vyvodit také ze studií dalších inhibitorů Plk1 [72,77]. Skutečnost, že nádorové buňky s nefunkčním proteinem p53 by byly více náchylné k inhibici Plk1 než buňky zdravé, by mohla představovat významné terapeutické okno. Na druhou stranu byly publikovány práce testující různé inhibitory Plk1, které ukazují, že inhibitory Plk1 cílí na všechny rychle proliferující buňky bez rozdílu (rakovinné a normální) [78– 81]. Tento fakt by mohl vysvětlit silné vedlejší účinky pozorované při aplikaci inhibitorů Plk1 v rámci klinických studií [82– 84]. Bylo také ukázáno, že citlivost k inhibici Plk1 se neliší na základě statutu proteinu p53 [85– 88], jinými slovy, že ztráta funkce proteinu p53 není přímo spojena s vyšší citlivostí k inhibici Plk1 [89]. Je jasné, že tato výše uvedená kontroverzní pozorování bude nutné vysvětlit a bude zajímavé sledovat výsledky těchto studií.
Inhibitory Plk1
Jednou z hlavních funkcí Plk1 je regulace tvorby mitotického vřeténka, inhibice Plk1 proto vede k závažným defektům při jeho tvorbě, jejichž výsledkem je zástava mitózy, a k indukci apoptózy [90,91]. Díky této centrální roli v regulaci buněčného cyklu je Plk1 považována za vhodný cíl protinádorové terapie [74,91– 97]. Hluboká kavita ATP‑ vazebné domény kináz je obecným cílem návrhu ATP kompetitivních inhibitorů. Na druhou stranu základní problém při návrhu takových inhibitorů je vysoká strukturní zakonzervovanost těchto ATP‑ vazebných oblastí, což má za následek sníženou specifitu a vyšší pravděpodobnost nežádoucích efektů. Přesto bylo vyvinuto několik inhibitorů Plk1 cílených na ATP‑ vazebnou doménu. Přehled všech inhibitorů Plk1, které uvedeme v textu, je znázorněn v obr. 2.
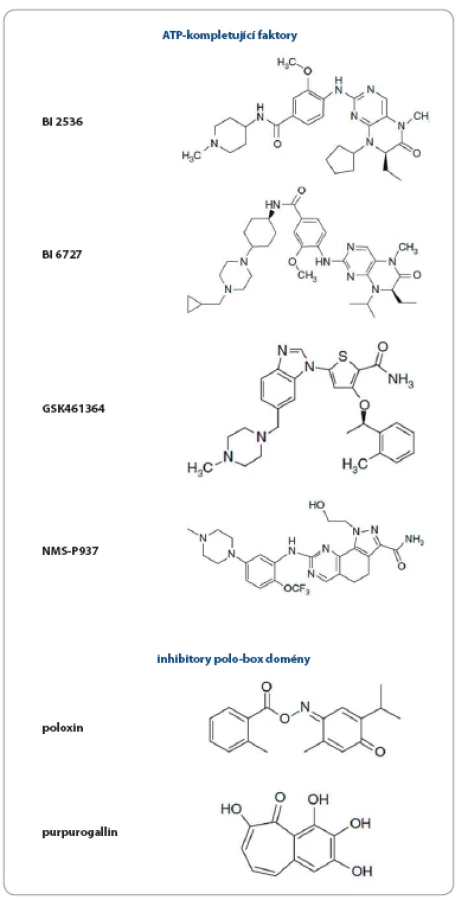
Za takový „prototyp“ je považován inhibitor společnosti Boehringer Ingelheim s názvem BI 2536 (derivát dihydropteridinonu), který posloužil k mnoha studiím osvětlujícím roli Plk1 u nádorových buněk a k objasnění konsekvencí a terapeutického potenciálu inhibice Plk1 [83,98]. Ukázalo se, že způsobuje mitotické abnormality a apoptózu u mnoha buněčných nádorových linií [79,98] a u vysokého procenta buněk dochází k tvorbě monopolárních vřetének. Tento inhibitor prošel klinickými studiemi fáze I a II, ale nebyla u něj prokázána dostatečná protinádorová aktivita [83,99– 101]. Z velké části je to přisuzováno jeho slabým farmakokinetickým vlastnostem, stejně tak jako aktivitě proti Plk3 (která je považována za nádorový supresor) a s tím spojeným nejednoznačným efektem na buněčnou proliferaci.
V další generaci byl proto vyvinut inhibitor BI 6727 – volasertib s jednoznačně vylepšenými farmakokinetickými vlastnostmi [102]. I když velmi silně inhibuje Plk1, stále vykazuje i nezanedbatelnou aktivitu vůči Plk2 a Plk3 (IC50 = 0,87, 5 a 56 nM). BI 6727 je velmi účinný u několika typů nádorů včetně melanomu, rakoviny střeva a hematopoetických malignit. Prošel několika klinickými studiemi fáze I a II [103,104] a u pacientů s akutní myeloidní leukemií probíhá nyní klinická studie fáze III [105].
Inhibitor společnosti Glaxo Smith Kline GSK461364 je derivát thiofenu, který vykazuje více než 1 000krát větší selektivitu pro inhibici Plk1 oproti Plk2 a Plk3. Stejně tak jako BI2536 je účinný u mnoha nádorových buněčných linií, větší citlivost byla navíc prokázána u linií s nefunkčním proteinem p53 [77]. Inhibitor byl testován v I. fázi klinické studie u pacientů se solidními nádory a non‑Hodgkinovým lymfomem [106].
Poslední ATP‑ kompetitivní inhibitor Plk1, o němž se zmíníme, je NMS‑ P937 [107]. Jedná se o sloučeninu s jádrem pyrazolochinazolinu, která indukuje zástavu buněčného cyklu v G2– M fázi a vykazuje antiproliferační aktivitu u více než stovky hematologických a solidních nádorů [108]. Výhodou je orální podání a inhibitor byl testován v klinické studii fáze I u pacientů s pokročilými/ metastatickými solidními tumory [109].
Jak bylo naznačeno výše, většina ATP‑ kompetitivních inhibitorů vykazuje aktivitu také proti Plk2 a Plk3, což není překvapivé vzhledem k vysoké sekvenční homologii povrchu ATP‑ vázající kavity (až 90 %) [110]. Druhou skupinou Plk1 inhibitorů jsou tudíž takové, jež cílí na tzv. polo‑ box doménu (PBD), která se nachází v C‑ koncové části a obsahuje specifickou vazebnou oblast pro interakce se substrátem, zajišťuje danou subcelulární lokalizaci Plk1 (centrozomy, kinetochor, vřeténko, Golgiho aparát atd.) a podílí se také na regulaci samotné kinázové aktivity [111,112]. Blokování PBD domény specifické pro Plk by mělo zaručit větší specifitu takových inhibitorů.
První objevenou nepeptidovou sloučeninou kompetující o vazbu na PBD Plk1 byl poloxin [113]. Jeho základní strukturou je thymochinon, což je biologicky aktivní sloučenina obsažená v oleji z černuchy seté (Nigella sativa, lidově černý kmín), který je znám pro své protizánětlivé, antioxidační a antineoplastické vlastnosti [114,115]. Poloxin indukuje fragmentaci centrozomu, a tím zastavuje mitózu. Dále silně potlačuje proliferaci u nádorových linií, což následně vede k apoptóze [78]. Ačkoli objevení poloxinu byl důležitý posun v návrhu PBD Plk1 inhibitorů a přispěl k pochopení funkcí Plk1, ukázal se nakonec jako nespecifický a málo účinný [116].
Další inhibitor PBD domény byl objeven screeningem knihovny přírodních látek. Purpurogallin je extrahován z hálek dubů a u HeLa buněk zabraňuje přemístění Plk1 k centrozomům, čímž dochází k zastavení mitotického dělení [80]. Je známo, že purpurogallin inhibuje i aktivitu proteinů, které neobsahují PBD, např. tyrozin kináz a HIV‑ 1 integrázy, chemická podstata účinku této látky proto musí být dále zkoumána [117– 119].
Jako zajímavé se dále jeví inhibitory PBD ve formě peptidů, které blokují PBD na principu protein‑proteinových interakcí. Takové peptidy jsou nejčastěji odvozeny ze substrátů Plk1 interagujících s PBD (např. CDC25C nebo PBIP) [120,121].
Závěrem tohoto odstavce je třeba říci, že vývoj inhibitorů Plk1 je v současnosti skutečně rychlý, a není proto v možnostech tohoto sdělení podat vyčerpávající a aktuální výčet informací. Rozsáhlejší přehled lze najít např. v přehledových článcích Klause Strebhardta [122,123].
Závěrem
Není pochyb o tom, že Plk1 hraje významnou roli při buněčném dělení. Její deregulace souvisí s nádorovou transformací buněk, a stala se proto atraktivním cílem pro vývoj protinádorových léčiv. Jelikož ostatní členové rodiny Plk (kromě Plk4) vykonávají spíše tumor supresorové funkce, bude nezbytné zajistit specifitu inhibice vůči Plk1. To, stejně jako zabránění inhibice ostatních kináz, by mohly zaručit nově vyvíjené a testované inhibitory cílící na specifickou polo‑ box doménu. Bude ale důležité zároveň zajistit srovnatelnou účinnost s inhibitory ATP‑ vázající domény, které už byly úspěšně testovány i v klinických studiích. Pro implementaci těchto inhibitorů zase bude nezbytné monitorovat časnou odpověď buněk. Pro tyto účely se jistě stane cennou vyvinutá metoda ELISA, která umožňuje kvantifikaci aktivity Plk1 v malých objemech buněčných lyzátů [124].
I přes značný pokrok v osvětlení funkcí Plk1 v kancerogenezi a efektů provázejících její inhibici zbývá spousta otázek týkajících se především konkrétního mechanizmu účinku inhibitorů, spojitosti statutu proteinu p53 a odpovědi na inhibici Plk1, stejně jako srovnání cytotoxicity inhibitorů Plk1 s ostatními užívanými anti‑tubulinovými léčivy, jež budou muset být v budoucnu zodpovězeny.
Práce byla podpořena Evropským fondem pro regionální rozvoj a státním rozpočtem České republiky (RECAMO, CZ.1.05/2.1.00/03.0101), projektem MŠMT – NPU I – LO1413, MZ ČR – RVO (MOÚ, 00209805) a BBMRI_CZ (LM2010004).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
RNDr. Bořivoj Vojtěšek, DrSc.
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: vojtesek@mou.cz
Obdrženo: 9. 4. 2015
Přijato: 19. 6. 2015
Sources
1. Burns TF, Fei P, Scata KA et al. Silencing of the novel p53 target gene Snk/ Plk2 leads to mitotic catastrophe in paclitaxel (taxol)- exposed cells. Mol Cell Biol 2003; 23(16): 5556– 5571.
2. Warnke S, Kemmler S, Hames RS et al. Polo‑like kinase‑ 2 is required for centriole duplication in mammalian cells. Curr Biol CB 2004; 14(13): 1200– 1207.
3. Matthew EM, Yen TJ, Dicker DT et al. Replication stress, defective S‑ phase checkpoint and increased death in Plk2- deficient human cancer cells. Cell Cycle Georget Tex 2007; 6(20): 2571– 2578.
4. Syed N, Smith P, Sullivan A et al. Transcriptional silencing of polo‑like kinase 2 (SNK/ PLK2) is a frequent event in B‑ cell malignancies. Blood 2006; 107(1): 250– 256.
5. Wang Q, Xie S, Chen J et al. Cell cycle arrest and apoptosis induced by human polo‑like kinase 3 is mediated through perturbation of microtubule integrity. Mol Cell Biol 2002; 22(10): 3450– 3459.
6. Xie S, Wu H, Wang Q et al. Plk3 functionally links DNA damage to cell cycle arrest and apoptosis at least in part via the p53 pathway. J Biol Chem 2001; 276(46): 43305– 43312.
7. Xu D, Yao Y, Jiang X et al. Regulation of PTEN stability and activity by Plk3. J Biol Chem 2010; 285(51): 39935– 39942. doi: 10.1074/ jbc.M110.166462.
8. Winkles JA, Alberts GF. Differential regulation of polo‑like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues. Oncogene 2005; 24(2): 260– 266.
9. Swallow CJ, Ko MA, Siddiqui NU et al. Sak/ Plk4 and mitotic fidelity. Oncogene 2005; 24(2): 306– 312.
10. Bonni S, Ganuelas ML, Petrinac S et al. Human Plk4 phosphorylates Cdc25C. Cell Cycle Georget Tex 2008; 7(4): 545– 547.
11. Macmillan JC, Hudson JW, Bull S et al. Comparative expression of the mitotic regulators SAK and PLK in colorectal cancer. Ann Surg Oncol 2001; 8(9): 729– 740.
12. Mason J, Wei S, Luo X et al. Inhibition of polo‑like kinase 4 as an anti‑cancer strategy. Cancer Res 2011; 71 (Suppl 8): abstr. LB‑ 215.
13. Laufer R, Forrest B, Li S‑ W et al. The discovery of PLK4 inhibitors: (E)- 3- ((1H‑ Indazol‑ 6- yl)methylene)indolin‑2- ones as novel antiproliferative agents. J Med Chem 2013; 56(15): 6069– 6087. doi: 10.1021/ jm400380m.
14. Andrysik Z, Bernstein WZ, Deng L et al. The novel mouse polo‑like kinase 5 responds to DNA damage and localizes in the nucleolus. Nucleic Acids Res 2010; 38(9): 2931– 2943. doi: 10.1093/ nar/ gkq011.
15. De Cárcer G, Manning G, Malumbres M. From Plk1 to Plk5: functional evolution of polo‑like kinases. Cell Cycle Georget Tex 2011; 10(14): 2255– 2262.
16. de Cárcer G, Escobar B, Higuero AM et al. Plk5, a polo box domain‑only protein with specific roles in neuron differentiation and glioblastoma suppression. Mol Cell Biol 2011; 31(6): 1225– 1239. doi: 10.1128/ MCB.00607‑ 10.
17. Golsteyn RM, Schultz SJ, Bartek J et al. Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J Cell Sci 1994; 107(6): 1509– 1517.
18. Toyoshima‑ Morimoto F, Taniguchi E, Shinya N et al. Polo‑like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature 2001; 410(6825): 215– 220.
19. Kumagai A, Dunphy WG. Purification and molecular cloning of Plx1, a Cdc25- regulatory kinase from Xenopus egg extracts. Science 1996; 273(5280): 1377– 1380.
20. Abrieu A, Brassac T, Galas S et al. The Polo‑like kinase Plk1 is a component of the MPF amplification loop at the G2/ M‑ phase transition of the cell cycle in Xenopus eggs. J Cell Sci 1998; 111(12): 1751– 1757.
21. Toyoshima‑ Morimoto F, Taniguchi E, Nishida E. Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep 2002; 3(4): 341– 348.
22. Casenghi M, Meraldi P, Weinhart U et al. Polo‑like kinase 1 regulates Nlp, a centrosome protein involved in microtubule nucleation. Dev Cell 2003; 5(1): 113– 125.
23. Li H, Liu XS, Yang X et al. Polo‑like kinase 1 phosphorylation of p150Glued facilitates nuclear envelope breakdown during prophase. Proc Natl Acad Sci U S A 2010; 107(33): 14633– 14638. doi: 10.1073/ pnas.1006615107.
24. Li H, Liu XS, Yang X et al. Phosphorylation of CLIP‑ 170 by Plk1 and CK2 promotes timely formation of kinetochore‑microtubule attachments. EMBO J 2010; 29(17): 2953– 2965. doi: 10.1038/ emboj.2010.174.
25. Liu XS, Song B, Tang J et al. Plk1 phosphorylates Sgt1 at the kinetochores to promote timely kinetochore‑microtubule attachment. Mol Cell Biol 2012; 32(19): 4053– 4067. doi: 10.1128/ MCB.00516‑ 12.
26. Hansen DV, Loktev AV, Ban KH et al. Plk1 regulates activation of the anaphase promoting complex by phosphorylating and triggering SCFbetaTrCP‑ dependent destruction of the APC Inhibitor Emi1. Mol Biol Cell 2004; 15(12): 5623– 5634.
27. Yoo HY, Kumagai A, Shevchenko A et al. Adaptation of a DNA replication checkpoint response depends upon inactivation of claspin by the polo‑like kinase. Cell 2004; 117(5): 575– 588.
28. Mamely I, van Vugt MA, Smits VA et al. Polo‑like kinase‑ 1 controls proteasome‑ dependent degradation of Claspin during checkpoint recovery. Curr Biol CB 2006; 16(19): 1950– 1955.
29. Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol 2006; 26(6): 2093– 2108.
30. Clay FJ, McEwen SJ, Bertoncello I et al. Identification and cloning of a protein kinase‑ encoding mouse gene, Plk, related to the polo gene of drosophila. Proc Natl Acad Sci U S A 1993; 90(11): 4882– 4886.
31. Yim H, Erikson RL. Plk1‑ targeted therapies in TP53- or RAS‑ mutated cancer. Mutat Res Rev Mutat Res 2014; 761: 31– 39. doi: 10.1016/ j.mrrev.2014.02.005.
32. Archambault V, Lépine G, Kachaner D. Understanding the polo kinase machine. Oncogene. In press 2015. doi: 10.1038/ onc.2014.451.
33. Hyun SY, Hwang HI, Jang YJ. Polo‑like kinase‑ 1 in DNA damage response. BMB Rep 2014; 47(5): 249– 255.
34. Song B, Liu XS, Liu X. Polo‑like kinase 1 (Plk1): an Unexpected Player in DNA Replication. Cell Div 2012; 7: 3. doi: 10.1186/ 1747‑ 1028‑ 7‑ 3.
35. Yim H, Erikson RL. Polo‑like kinase 1 depletion induces DNA damage in early S prior to caspase activation. Mol Cell Biol 2009; 29(10): 2609– 2621. doi: 10.1128/ MCB.01277‑ 08.
36. Li H, Wang Y, Liu X. Plk1‑ dependent phosphorylation regulates functions of DNA topoisomerase II alpha in cell cycle progression. J Biol Chem 2008; 283(10): 6209– 6221. doi: 10.1074/ jbc.M709007200.
37. Shen M, Cai Y, Yang Y et al. Centrosomal protein FOR20 is essential for S‑ phase progression by recruiting Plk1 to centrosomes. Cell Res 2013; 23(11): 1284– 1295. doi: 10.1038/ cr.2013.127.
38. Tsvetkov L, Stern DF. Interaction of chromatin‑associated Plk1 and Mcm7. J Biol Chem 2005; 280(12): 11943– 11947.
39. Wolf G, Elez R, Doermer A et al. Prognostic significance of polo‑like kinase (PLK) expression in non‑small cell lung cancer. Oncogene 1997; 14(5): 543– 549.
40. Knecht R, Oberhauser C, Strebhardt K. PLK (polo‑like kinase), a new prognostic marker for oropharyngeal carcinomas. Int J Cancer J Int Cancer 2000; 89(6): 535– 536.
41. Knecht R, Elez R, Oechler M et al. Prognostic significance of polo‑like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res 1999; 59(12): 2794– 2797.
42. Tokumitsu Y, Mori M, Tanaka S et al. Prognostic significance of polo‑like kinase expression in esophageal carcinoma. Int J Oncol 1999; 15(4): 687– 692.
43. Weichert W, Kristiansen G, Winzer KJ et al. Polo‑like kinase isoforms in breast cancer: expression patterns and prognostic implications. Virchows Arch 2005; 446(4): 442– 450.
44. Wolf G, Hildenbrand R, Schwar C et al. Polo‑like kinase: a novel marker of proliferation: correlation with estrogen‑ receptor expression in human breast cancer. Pathol Res Pract 2000; 196(11): 753– 759.
45. King SI, Purdie CA, Bray SE et al. Immunohistochemical detection of polo‑like kinase‑ 1 (PLK1) in primary breast cancer is associated with TP53 mutation and poor clinical outcome. Breast Cancer Res BCR 2012; 14(2): R40.
46. Pellegrino R, Calvisi DF, Ladu S et al. Oncogenic and tumor suppressive roles of polo‑like kinases in human hepatocellular carcinoma. Hepatol Baltim Md 2010; 51(3): 857– 868. doi: 10.1002/ hep.23467.
47. Weichert W, Schmidt M, Jacob J et al. Overexpression of polo‑like kinase 1 is a common and early event in pancreatic cancer. Pancreatol 2005; 5(2– 3): 259– 265.
48. Gray PJ Jr, Bearss DJ, Han H et al. Identification of human polo‑like kinase 1 as a potential therapeutic target in pancreatic cancer. Mol Cancer Ther 2004; 3(5): 641– 646.
49. Weichert W, Schmidt M, Gekeler V et al. Polo‑like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate 2004; 60(3): 240– 245.
50. Weichert W, Kristiansen G, Schmidt M et al. Polo‑like kinase 1 expression is a prognostic factor in human colon cancer. World J Gastroenterol 2005; 11(36): 5644– 5650.
51. Takai N, Miyazaki T, Fujisawa K et al. Polo‑like kinase (PLK) expression in endometrial carcinoma. Cancer Lett 2001; 169(1): 41– 49.
52. Takai N, Miyazaki T, Fujisawa K et al. Expression of polo‑like kinase in ovarian cancer is associated with histological grade and clinical stage. Cancer Lett 2001; 164(1): 41– 49.
53. Weichert W, Denkert C, Schmidt M et al. Polo‑like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br J Cancer 2004; 90(4): 815– 821.
54. Nigro JM, Baker SJ, Preisinger AC et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989; 342(6250): 705– 708.
55. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408(6810): 307– 310.
56. Brown CJ, Lain S, Verma CS et al. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 2009; 9(12): 862– 873. doi: 10.1038/ nrc2763.
57. Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358(6381): 15– 16.
58. Junttila MR, Evan GI. p53 – a Jack of all trades but master of none. Nat Rev Cancer 2009; 9(11): 821– 829. doi: 10.1038/ nrc2728.
59. Bode AM, Dong Z. Post‑translational modification of p53 in tumorigenesis. Nat Rev Cancer 2004; 4(10): 793– 805.
60. Cheok CF, Verma CS, Baselga J et al. Translating p53 into the clinic. Nat Rev Clin Oncol 2011; 8(1): 25– 37. doi: 10.1038/ nrclinonc.2010.174.
61. Zhu H, Chang BD, Uchiumi T et al. Identification of promoter elements responsible for transcriptional inhibition of polo‑like kinase 1 and topoisomerase II alpha genes by p21(WAF1/ CIP1/ SDI1). Cell Cycle 2002; 1(1): 59– 66.
62. Pandit B, Halasi M, Gartel AL. p53 negatively regulates expression of FoxM1. Cell Cycle 2009; 8(20): 3425– 3427.
63. Barsotti AM, Prives C. Pro‑proliferative FoxM1 is a target of p53- mediated repression. Oncogene 2009; 28(48): 4295– 4305. doi: 10.1038/ onc.2009.282.
64. Wang IC, Chen YJ, Hughes D et al. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2- Cks1) ubiquitin ligase. Mol Cell Biol 2005; 25(24): 10875– 10894.
65. Fu Z, Malureanu L, Huang J et al. Plk1‑ dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol 2008; 10(9): 1076– 1082. doi: 10.1038/ ncb1767.
66. McKenzie L, King S, Marcar L et al. p53- dependent repression of polo‑like kinase‑ 1 (PLK1). Cell Cycle 2010; 9(20): 4200– 4212.
67. Ando K, Ozaki T, Yamamoto H et al. Polo‑like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J Biol Chem 2004; 279(24): 25549– 25561.
68. Kruse JP, Gu W. Modes of p53 regulation. Cell 2009; 137(4): 609– 622. doi: 10.1016/ j.cell.2009.04.050.
69. Dias SS, Hogan C, Ochocka AM et al. Polo‑like kinase‑ 1 phosphorylates MDM2 at Ser260 and stimulates MDM2- mediated p53 turnover. FEBS Lett 2009; 583(22): 3543– 3548. doi: 10.1016/ j.febslet.2009.09.057.
70. Yang X, Li H, Zhou Z et al. Plk1‑ mediated phosphorylation of topors regulates p53 stability. J Biol Chem 2009; 284(28): 18588– 18592. doi: 10.1074/ jbc.C109.001560.
71. Liu XS, Li H, Song B et al. Polo‑like kinase 1 phosphorylation of G2 and S‑ phase‑ expressed 1 protein is essential for p53 inactivation during G2 checkpoint recovery. EMBO Rep 2010; 11(8): 626– 632. doi: 10.1038/ embor.2010.90.
72. Guan R, Tapang P, Leverson JD et al. Small interfering RNA‑ mediated polo‑like kinase 1 depletion preferentially reduces the survival of p53- defective, oncogenic transformed cells and inhibits tumor growth in animals. Cancer Res 2005; 65(7): 2698– 2704.
73. Lansing TJ, McConnell RT, Duckett DR et al. In vitro biological activity of a novel small‑molecule inhibitor of polo‑like kinase 1. Mol Cancer Ther 2007; 6(2): 450– 459.
74. Spänkuch‑ Schmitt B, Bereiter‑ Hahn J, Kaufmann M et al. Effect of RNA silencing of polo‑like kinase‑ 1 (PLK1) on apoptosis and spindle formation in human cancer cells. J Natl Cancer Inst 2002; 94(24): 1863– 1877.
75. Raab M, Kappel S, Krämer A et al. Toxicity modelling of Plk1‑ targeted therapies in genetically engineered mice and cultured primary mammalian cells. Nat Commun 2011; 2: 395. doi: 10.1038/ ncomms1395.
76. Sur S, Pagliarini R, Bunz F et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A 2009; 106(10): 3964– 3969. doi: 10.1073/ pnas.0813333106.
77. Degenhardt Y, Greshock J, Laquerre S et al. Sensitivity of cancer cells to Plk1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability. Mol Cancer Ther 2010; 9(7): 2079– 2089. doi: 10.1158/ 1535‑ 7163.MCT‑ 10‑ 0095.
78. Yuan J, Sanhaji M, Krämer A et al. Polo‑ box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am J Pathol 2011; 179(4): 2091– 2099. doi: 10.1016/ j.ajpath.2011.06.031.
79. Steegmaier M, Hoffmann M, Baum A et al. BI 2536, a potent and selective inhibitor of polo‑like kinase 1, inhibits tumor growth in vivo. Curr Biol 2007; 17(4): 316– 322.
80. Watanabe N, Sekine T, Takagi M et al. Deficiency in chromosome congression by the inhibition of Plk1 polo box domain‑dependent recognition. J Biol Chem 2009; 284(4): 2344– 2353. doi: 10.1074/ jbc.M805308200.
81. Lu B, Mahmud H, Maass AH et al. The Plk1 inhibitor BI 2536 temporarily arrests primary cardiac fibroblasts in mitosis and generates aneuploidy in vitro. PloS One 2010; 5(9): e12963. doi: 10.1371/ journal.pone.0012963.
82. Mross K, Dittrich C, Aulitzky WE et al. A randomised phase II trial of the Polo‑like kinase inhibitor BI 2536 in chemo‑ naïve patients with unresectable exocrine adenocarcinoma of the pancreas – a study within the Central European Society Anticancer Drug Research (CESAR) collaborative network. Br J Cancer 2012; 107(2): 280– 286. doi: 10.1038/ bjc.2012.257.
83. Sebastian M, Reck M, Waller CF et al. The efficacy and safety of BI 2536, a novel Plk‑ 1 inhibitor, in patients with stage IIIB/ IV non‑small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open‑ label, randomized phase II clinical trial. J Thorac Oncol 2010; 5(7): 1060– 1067. doi: 10.1097/ JTO.0b013e3181d95dd4.
84. Frost A, Mross K, Steinbild S et al. Phase i study of the Plk1 inhibitor BI 2536 administered intravenously on three consecutive days in advanced solid tumours. Curr Oncol 2012; 19(1): e28– e35. doi: 10.3747/ co.19.866.
85. Sanhaji M, Kreis NN, Zimmer B et al. p53 is not directly relevant to the response of polo‑like kinase 1 inhibitors. Cell Cycle 2012; 11(3): 543– 553. doi: 10.4161/ cc.11.3.19076.
86. Sanhaji M, Louwen F, Zimmer B et al. Polo‑like kinase 1inhibitors, mitotic stress and the tumor suppressor p53. Cell Cycle 2013; 12(9): 1340– 1351. doi: 10.4161/ cc.24573.
87. Khan SH, Wahl GM. p53 and pRb prevent rereplication in response to microtubule inhibitors by mediating a reversible G1 arrest. Cancer Res 1998; 58(3): 396– 401.
88. Stewart ZA, Mays D, Pietenpol JA. Defective G1- S cell cycle checkpoint function sensitizes cells to microtubule inhibitor‑induced apoptosis. Cancer Res 1999; 59(15): 3831– 3837.
89. Louwen F, Yuan J. Battle of the eternal rivals: restoring functional p53 and inhibiting polo‑like kinase 1 as cancer therapy. Oncotarget 2013; 4(7): 958– 971.
90. Kappel S, Matthess Y, Kaufmann M et al. Silencing of mammalian genes by tetracycline‑ inducible shRNA expression. Nat Protoc 2007; 2(12): 3257– 3269.
91. Matthess Y, Kappel S, Spänkuch B et al. Conditional inhibition of cancer cell proliferation by tetracycline‑ responsive, H1 promoter‑driven silencing of PLK1. Oncogene 2005; 24(18): 2973– 2980.
92. Lane HA, Nigg EA. Antibody microinjection reveals an essential role for human polo‑like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol 1996; 135(6 Pt 2): 1701– 1713.
93. Cogswell JP, Brown CE, Bisi JE et al. Dominant‑ negative polo‑like kinase 1 induces mitotic catastrophe independent of cdc25C function. Cell Growth Differ 2000; 11(12): 615– 623.
94. Spänkuch‑ Schmitt B, Wolf G, Solbach C et al. Downregulation of human polo‑like kinase activity by antisense oligonucleotides induces growth inhibition in cancer cells. Oncogene 2002; 21(20): 3162– 3171.
95. Yuan J, Krämer A, Eckerdt F et al. Efficient internalization of the polo‑ box of polo‑like kinase 1 fused to an Antennapedia peptide results in inhibition of cancer cell proliferation. Cancer Res 2002; 62(15): 4186– 4190.
96. Spänkuch B, Matthess Y, Knecht R et al. Cancer inhibition in nude mice after systemic application of U6 promoter‑driven short hairpin RNAs against PLK1. J Natl Cancer Inst 2004; 96(11): 862– 872.
97. Kappel S, Matthess Y, Zimmer B et al. Tumor inhibition by genomically integrated inducible RNAi‑ cassettes. Nucleic Acids Res 2006; 34(16): 4527– 4536.
98. Lénárt P, Petronczki M, Steegmaier M et al. The small‑molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo‑like kinase 1. Curr Biol 2007; 17(4): 304– 315.
99. Hofheinz RD, Al‑ Batran SE, Hochhaus A et al. An open‑ label, phase I study of the polo‑like kinase‑ 1 inhibitor, BI 2536, in patients with advanced solid tumors. Clin Cancer 2010; 16(18): 4666– 4674. doi: 10.1158/ 1078‑ 0432.CCR‑ 10‑ 0318.
100. Mross K, Frost A, Steinbild S et al. Phase I dose escalation and pharmacokinetic study of BI 2536, a novel polo‑like kinase 1 inhibitor, in patients with advanced solid tumors. J Clin Oncol 2008; 26(34): 5511– 5517. doi: 10.1200/ JCO.2008.16.1547.
101. Schöffski P, Blay JY, De Greve J et al. Multicentric parallel phase II trial of the polo‑like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network Of Core Institutes (NOCI). Eur J Cancer 2010; 46(12): 2206– 2215. doi: 10.1016/ j.ejca.2010.03.039.
102. Rudolph D, Steegmaier M, Hoffmann M et al. BI 6727, a polo‑like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res Off J Am Assoc Cancer Res 2009; 15(9): 3094– 3102. doi: 10.1158/ 1078‑ 0432.CCR‑ 08‑ 2445.
103. Maertens J, Lubbert M, Fiedler W et al. Phase I/ II study of volasertib (BI 6727), an intravenous polo‑like kinase (Plk) inhibitor, in patients with acute myeloid leukemia (AML): results from the randomized phase II part for volasertib in combination with low‑dose cytarabine (LDAC) versus LDAC monotherapy in patients with previously untreated AML ineligible for intensive treatment. Blood (ASH Annu Meet Abstr) 2012; 120(21): abstr. 411.
104. Stadler WM, Vaughn DJ, Sonpavde G et al. An open‑ label, single‑arm, phase 2 trial of the polo‑like kinase inhibitor volasertib (BI 6727) in patients with locally advanced or metastatic urothelial cancer. Cancer 2014; 120(7): 976– 982. doi: 10.1002/ cncr.28519.
105. ClinicalTrials.gov [homepage on the Internet]. A service of the U.S. National Institutes of Health; c2000– 15 [updated 2015 March 8; cited 2015 April 15]. Available from: https:/ / clinicaltrials.gov/ ct2/ show/ study/ NCT01721876?term=NCT01721876 &rank=1.
106. Olmos D, Barker D, Sharma R et al. Phase I study of GSK461364, a specific and competitive polo‑like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin Cancer Res 2011; 17(10): 3420– 3430. doi: 10.1158/ 1078‑ 0432.CCR‑ 10‑ 2946.
107. Beria I, Bossi RT, Brasca MG et al. NMS‑ P937, a 4,5- dihydro‑1H‑ pyrazolo[4,3- h]quinazoline derivative as potent and selective polo‑like kinase 1 inhibitor. Bioorg Med Chem Lett 2011; 21(10): 2969– 2974. doi: 10.1016/ j.bmcl.2011.03.054.
108. Valsasina B, Beria I, Alli C et al. NMS‑ P937, an orally available, specific small‑molecule polo‑like kinase 1 inhibitor with antitumor activity in solid and hematologic malignancies. Mol Cancer Ther 2012; 11(4): 1006– 1016. doi: 10.1158/ 1535‑ 7163.MCT‑ 11‑ 0765.
109. ClinicalTrials.gov [homepage on the Internet]. A service of the U.S. National Institutes of Health; c2000– 15 [updated 2012 September 6; cited 2015 April 15]. Available from: https:/ / clinicaltrials.gov/ ct2/ show/ NCT01014429?term=NMS‑ 1286937&rank=1.
110. Johnson EF, Stewart KD, Woods KW et al. Pharmacological and functional comparison of the polo‑like kinase family: insight into inhibitor and substrate specificity. Biochemistry 2007; 46(33): 9551– 9563.
111. Elia AE, Cantley LC, Yaffe MB. Proteomic screen finds pSer/ pThr‑binding domain localizing Plk1 to mitotic substrates. Science 2003; 299(5610): 1228– 1231.
112. Elia AEH, Rellos P, Haire LF et al. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo‑ box domain. Cell 2003; 115(1): 83– 95.
113. Reindl W, Yuan J, Krämer A et al. Inhibition of polo‑like kinase 1 by blocking polo‑ box domain‑dependent protein‑protein interactions. Chem Biol 2008; 15(5): 459– 466. doi: 10.1016/ j.chembiol.2008.03.013.
114. Gali‑ Muhtasib H, Roessner A, Schneider‑ Stock R. Thymoquinone: a promising anti‑cancer drug from natural sources. Int J Biochem Cell Biol 2006; 38(8): 1249– 1253.
115. Kaseb AO, Chinnakannu K, Chen D et al. Androgen receptor and E2F‑ 1 targeted thymoquinone therapy for hormone‑ refractory prostate cancer. Cancer Res 2007; 67(16): 7782– 7788.
116. Craig SN, Wyatt MD, McInnes C. Current assess-ment of polo‑like kinases as anti‑tumor drug targets. Expert Opin Drug Discov 2014; 9(7): 773– 789. doi: 10.1517/ 17460441.2014.918100.
117. Abou‑ Karam M, Shier WT. Inhibition of oncogene product enzyme activity as an approach to cancer chemoprevention. Tyrosine‑ specific protein kinase inhibition by purpurogallin from Quercus sp. nutgall. Phytother Res PTR 1999; 13(4): 337– 340.
118. Farnet CM, Wang B, Hansen M et al. Human immunodeficiency virus type 1 cDNA integration: new aromatic hydroxylated inhibitors and studies of the inhibition mechanism. Antimicrob Agents Chemother 1998; 42(9): 2245– 2253.
119. Inamori Y, Muro C, Sajima E et al. Biological activity of purpurogallin. Biosci Biotechnol Biochem 1997; 61(5): 890– 892.
120. Yun SM, Moulaei T, Lim D et al. Structural and functional analyses of minimal phosphopeptides targeting the polo‑ box domain of polo‑like kinase 1. Nat Struct Mol Biol 2009; 16(8): 876– 882. doi: 10.1038/ nsmb.1628.
121. McInnes C, Estes K, Baxter M et al. Targeting subcellular localization through the polo‑ box domain: non‑ATP competitive inhibitors recapitulate a PLK1 phenotype. Mol Cancer Ther 2012; 11(8): 1683– 1692. doi: 10.1158/ 1535‑ 7163.MCT‑ 12‑ 0006‑ T.
122. Strebhardt K, Becker S, Matthess Y. Thoughts on the current assessment of polo‑like kinase inhibitor drug discovery. Expert Opin Drug Discov 2015; 10(1): 1– 8. doi: 10.1517/ 17460441.2015.962510.
123. Strebhardt K. Multifaceted polo‑like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 2010; 9(8): 643– 660. doi: 10.1038/ nrd3184.
124. Park JE, Li L, Park J et al. Direct quantification of polo‑like kinase 1 activity in cells and tissues using a highly sensitive and specific ELISA assay. Proc Natl Acad Sci U S A 2009; 106(6): 1725– 1730. doi: 10.1073/pnas.0812135106.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2015 Issue Supplementum 2
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
- Obstacle Called Vasospasm: Which Solution Is Most Effective in Microsurgery and How to Pharmacologically Assist It?
Most read in this issue
- Adenovírusové vektory v génovej terapii
- Nrf2 – dve tváre regulátora antioxidačného systému
- Rekombinantní protilátky a jejich využití v protinádorové terapii
- Co může přinést studium oligomerizace proteinů v procesu onkogeneze?