
Fanconiho anémie, komplementační skupina D1 v důsledku bialelické mutace genu BRCA2 – kazuistika
Fanconi Anemia, Complementation Group D1 Caused by Biallelic Mutations of BRCA2 Gene – Case Report
Fanconi anemia is a rare autosomal recessive disorder, clinically and genetically heterogeneous, characterized by typical clinical features, such as short stature, microcephaly, skeletal abnormalities, abnormal skin pigmentations, developmental delay and congenital heart, kidney anomalies etc. Pancytopenia leading to bone marrow failure occurs in the first decade. Patients with Fanconi anemia have a high risk of hematologic malignancies and solid tumors. The diagnosis of Fanconi anemia is based on cytogenetic testing for increased rates of spontaneous chromosomal breakage and increased sensitivity to diepoxybutane or mitomycin C. Fanconi anemia is a heterogeneous disorder, at least 15 complementation groups are described, and 15 genes in which mutations are responsible for all of the 15 Fanconi anemia complementation groups have been identified. Unlike other Fanconi anemia complementation groups, for complementation group D1 (FANCD1), the bone marrow failure is not a typical feature, but early-onset leukemia and specific solid tumors, most often medulloblastoma and Wilms tumor, are typical for this complementation group.
Key words:
Fanconi anemia – complementation group – FANCD1 – BRCA2 gene – leukemia – Wilms tumor – medulloblastoma
This work was supported by grant from Norway NF-CZ11-PDP-3-003-2014, MH ČR – RVO, UH Motol 00064203 and OPPK – CZ-2.16./3.1.00/24022.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
14. 7. 2015
Accepted:
6. 12. 2015
:
A. Puchmajerová 1; K. Švojgr 2; D. Novotná 1; E. Macháčková 3; D. Sumerauer 2; P. Smíšek 2; R. Kodet 4; M. Kynčl 5; A. Křepelová 1; L. Foretová 3
:
Ústav biologie a lékařské genetiky, 2. LF UK a FN v Motole, Praha
1; Klinika dětské hematologie a onkologie 2. LF UK a FN v Motole, Praha
2; Oddělení genetiky a epidemiologie nádorů, Masarykův onkologický ústav, Brno
3; Ústav patologie a molekulární medicíny, 2. LF UK a FN v Motole, Praha
4; Klinika zobrazovacích metod 2. LF UK a FN v Motole, Praha
5
:
Klin Onkol 2016; 29(Supplementum 1): 89-92
:
Case Report
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amko2016S89
Fanconiho anémie je vzácné autozomálně recesivně dědičné onemocnění, klinicky a geneticky heterogenní, charakterizováno typickými klinickými projevy: malý vzrůst, mikrocefalie, skeletální anomálie, abnormální kožní pigmentace, opoždění vývoje, vrozené srdeční vady, vrozené vady ledvin a jiné. V první dekádě života se manifestuje pancytopenií, která vede k selhání kostní dřeně. Pacienti s Fanconiho anémií mají zvýšené riziko hematologických malignit a solidních tumorů. Diagnóza Fanconiho anémie je založena na cytogenetickém vyšetření, které prokazuje zvýšený výskyt spontánních chromozomálních aberací, jejichž počet stoupá po působení diepoxybutanu nebo mitomycinu C. Fanconiho anémie je heterogenní onemocnění, dosud je popsáno 15 komplementačních skupin, každá z nich je způsobena mutacemi některého z 15 kauzálních genů. Pro komplementační skupinu D1 (FANCD1) není typickým projevem, na rozdíl od ostatních skupin, selhávání kostní dřeně, ale časně se manifestující leukemie a specifické solidní tumory, nejčastěji meduloblastom a Wilmsův tumor ledvin.
Klíčová slova:
Fanconiho anémie – komplementační skupina – FANCD1 – gen BRCA2 – leukemie – Wilmsův tumor – meduloblastom
Úvod
Fanconiho anémie (FA) je vzácné, autozomálně recesivní onemocnění, jehož prevalence se uvádí 1 : 350 000. Řadí se mezi syndromy chromozomální instability, pro které je typická porucha reparace DNA. V důsledku toho jsou buňky takového jedince velmi citlivé na působení různých mutagenů, především ionizačního záření a alkylačních činidel. Porucha reparace DNA je nejvíce patrná u buněk, které se rychle dělí, a proto jsou jedinci s FA malého vzrůstu, mají mikrocefalii, dochází u nich k poruchám pohlavního zrání a poruchám imunity. Někdy se objevují i vrozené vady ledvin a srdce, mohou se vyskytnout i skeletální anomálie jako je skolióza a radiální hypo/ aplazie postihující především palce, event. porucha psychomotorického vývoje. V průběhu první dekády se postupně rozvíjí pancytopenie, která velmi často vede až k selhávání kostní dřeně s nutností její transplantace. U 10 – 30 % pacientů s FA se objevují hematologické malignity (myelodysplastický syndrom a akutní myeloidní leukemie) a u 25 – 30 % nehematologické malignity (solidní tumory, především hlavy, krku, kůže, GIT a pohlavních orgánů) [1,2].
Diagnostika FA je několikastupňový proces, který zahrnuje cytogenetické vyšetření, vyšetření buněčné kinetiky, stanovení komplementačních skupin a molekulárně-genetické vyšetření příslušného genu.
Cytogenetické vyšetření vychází z detekce chromozomálních aberací (zlomů, přestaveb, chromatidových výměn) v buněčných-lymfocytárních kulturách, jejichž počet stoupá po přidání diepoxybutanu (DEB) nebo mitomycinu C (MMC) do buněčné kultury [3–5]. Problémy při interpretaci výsledku mohou činit lymfocytární mozaiky, kde mohou být tyto testy falešně negativní. To je přičítáno zpětným mutacím, kompenzačním delecím/ inzercím vedoucím k selekční výhodě lymfocytů bez mutace. Pokud i přes negativní DEB/ MMC test přetrvává vysoké podezření na FA, je vhodné tyto testy provést vyšetřením jiných buněk např. kožních fibroblastů.
Vyšetření buněčné kinetiky využívá zastavení buněk v G2 fázi, k němuž dochází ve vysoké frakci u buněk lymfocytů i fibroblastů pacientů s FA, pokud jsou vystaveny působení (MMC). Průtokovou cytometrií se poté stanovuje percentuální zastoupení buněk v G2 fázi buněčného cyklu.
Následuje zařazení do jedné z 15 komplementačních FA skupin (A, B, C, D1, D2, E, F, G, I, J, L, M, N, O a P), za každou z nich je zodpovědná bialelická mutace příslušného genu. Výjimkou je komplementační skupina FANCB, kde je dědičnost vázaná na X chromozom [1].
Komplementační skupina FANCD1 se od ostatních komplementačních skupin FA liší. Etiologicky jsou za tento typ FA zodpovědné bialelické mutace genu BRCA2. Na rozdíl od výše zmíněných komplementačních skupin u pacientů s FANCD1 nedochází k selhávání kostní dřeně, ale k časnému rozvoji především akutní leukemie, nejčastěji T lymfoblastické leukemie, a solidních tumorů zvláště meduloblastomu a nefroblastomu, ale i dalších nádorů. Kumulativní riziko rozvoje malignity je 97 % do šesti let věku [1,3,6 – 11].
Kazuistika
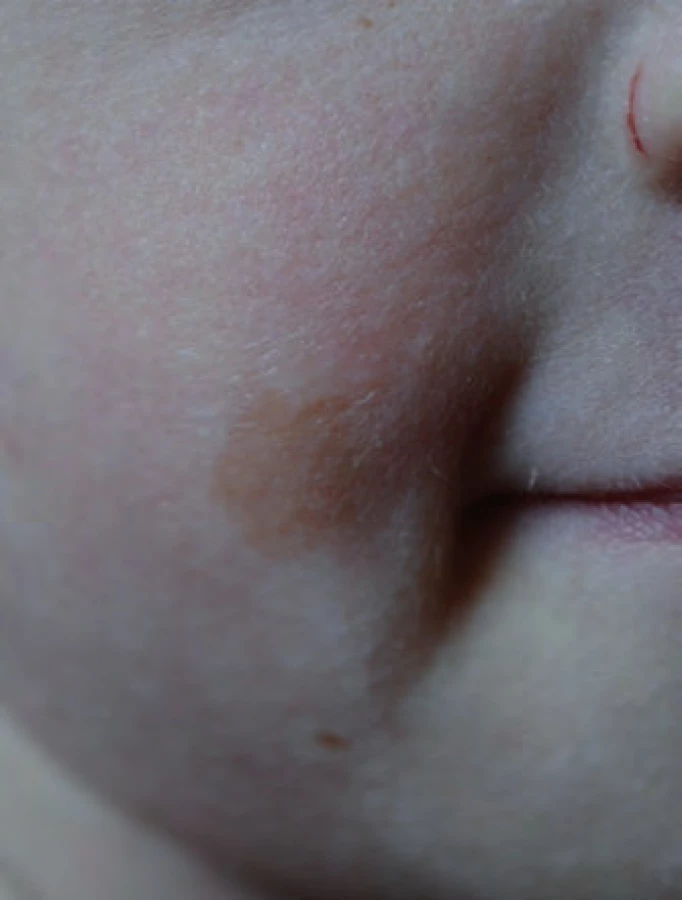
Téměř čtyřletý chlapec byl doporučený ke genetickému vyšetření pro Wilmsův tumor ledviny a pozitivní rodinnou anamnézu nádorových onemocnění [11,12]. Narodil se ze III. gravidity, rizikové pro gemini. V té době měli rodiče jednu zdravou 12letou dceru, syn ze II. gravidity zemřel ve třech letech na akutní sekundární myeloidní leukemii, předtím se od cca 10 měsíců léčil pro kojeneckou akutní T lymfoblastickou leukemii [13]. V roce 2000 byl geneticky vyšetřen, zvažován Nijmegen breakage syndrom (NBS), mutace genu NBS nebyla prokázána, vyšetření zlomů se nezdařilo. U našeho pacienta byl nefroblastom pravé ledviny zjištěn ve věku 2,5 roku, byla provedena iniciální nefrektomie, histologicky intermediální riziko, pro prorůstání nádoru přes pouzdro ledviny II. klinické stadium. Pacient byl následně léčen 27 týdnů trvající chemoterapií vinkristin a aktinomycin D. Dále je chlapec v remisi. Jeho sestra-dvojče byla v době vyšetření zdráva. Matka chlapce se léčila ve 35 letech pro karcinom prsu. Porovnáním fenotypu chlapce, jeho zemřelého staršího bratra a sestry-dvojčete bylo zjištěno, že všichni jsou malého vzrůstu s mikrocefalií a s výskytem café-au-lait makul (obr. 1). Cytogenetickým vyšetřením bylo prokázáno, že chlapec má 35 % spontánních chromozomálních zlomů a přestaveb, po indukci DEB nalezeno 14 % chromozomálních aberací, u jeho sestry bylo prokázáno 26 % spontánních aberací a 13 % aberací po indukci DEB.
Vzhledem ke karcinomu prsu u matky v mladém věku a fenotypu dětí byla zvažována FA typ D1 v důsledku bialelické mutace genu BRCA2. Bylo provedeno molekulárně-genetické vyšetření – přímá sekvenace genu BRCA2 (OEGN, MOÚ Brno). Bylo prokázáno, že chlapec je složený heterozygot pro dvě různé mutace genu BRCA2: c.658_659del2/ p.Val220IlefsX4 a c.9366_9367del2/ p.Ser3123GlnfsX26. Stejný genetický profil byl prokázán i u toho času zdravé sestry-dvojčete s malým vzrůstem, mikrocefalií a vysokou hladinou chromozomálních aberací.
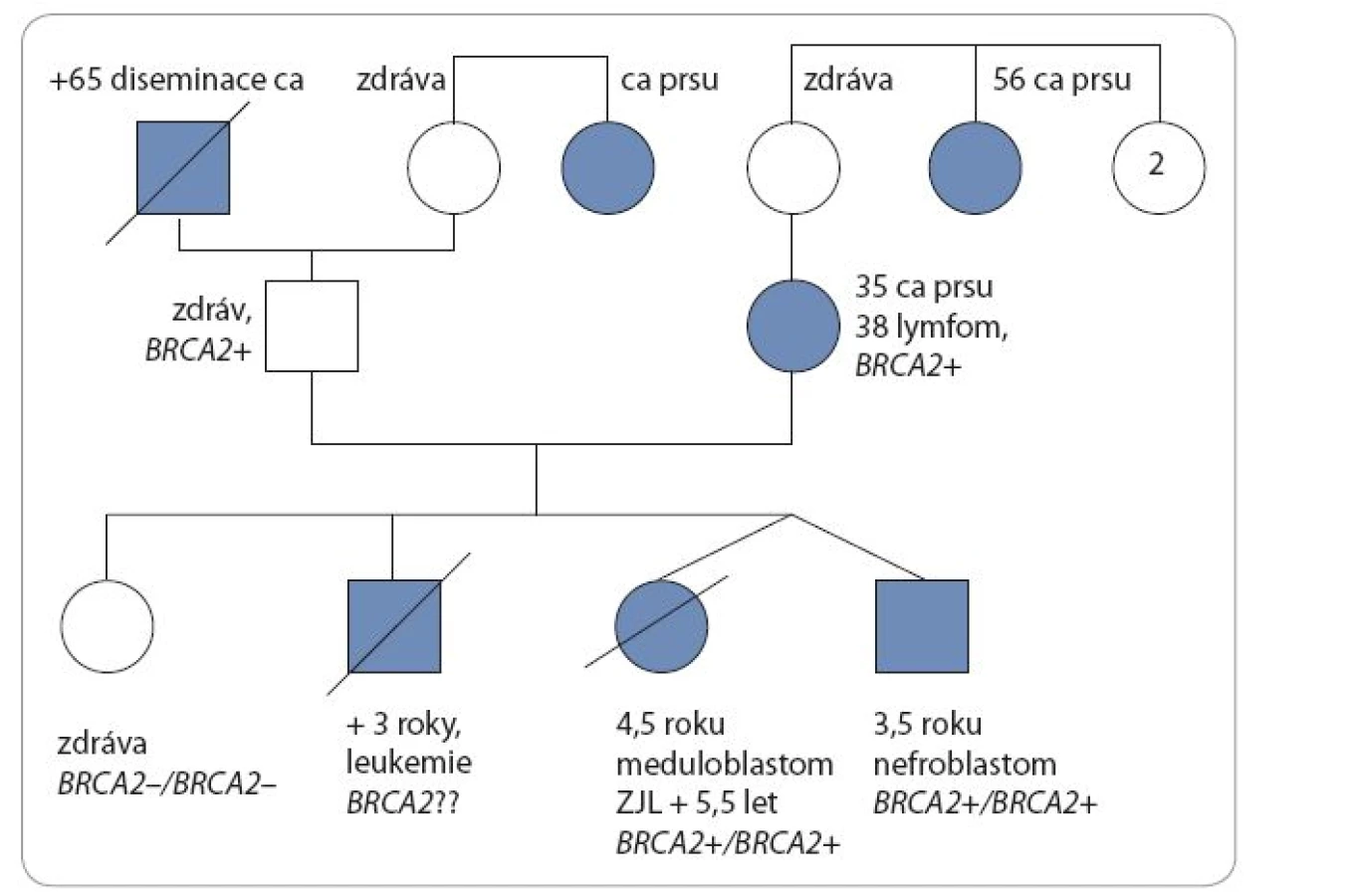
U dětí se znalostí genetické podstaty onemocnění byl zahájen pravidelný screeningový program k časné detekci malignity. Ve čtyřech letech a třech měsících pacient i jeho sestra-dvojče podstoupili vyšetření břicha a retroperitonea ultrazvukem (UZ), aspiraci kostní dřeně a MRI mozku, u obou byl normální nález. Přibližně za tři měsíce po tomto vyšetření se u sestry-dvojčete objevily obtíže s chůzí, poruchy rovnováhy, zvracení a na MRI mozku byla zjištěna v zadní jámě rozsáhlá expanze. Nádor byl neurochirurgy radikálně odstraněn, histologie prokázala meduloblastom, klasický typ, WHO grade IV, β-catenin negativní [14]. Vyšetření v době diagnózy neprokázalo generalizaci onemocnění. Byla zahájena modifikovaná chemoterapie pro pacienty s poruchou reparace DNA – vincristin, doxorubicin, metotrexát (intraventrikulárně do Ommaya rezervoáru). Bezprostředně po chemoterapii se rozvinuly velmi závažné komplikace spojené s těžkou sepsí, slizniční a kožní toxicitou grade IV a několikatýdenní regenerací krevního obrazu. Po zvládnutí těžkého stavu byla za šest týdnů na kontrolním MRI mozku nalezena lokální recidiva meduloblastomu v zadní jámě lební. Nádor byl opět radikálně odstraněn a zahájena paliativní chemoterapie – irinotekan, bevacizumab a intratheálně podávaný lipozomální cytarabin. Chemoterapie byla opakovaně komplikována především infekcemi. Asi šest měsíců po první recidivě se u dívky opět objevily obtíže s chůzí a zvracení, MRI vyšetřením byla nalezena druhá (inoperabilní) recidiva tumoru v zadní jámě lební. Dívka byla dále léčena pouze symptomaticky a během tří týdnů zemřela ve věku pěti let a čtyř měsíců na progresi základního onemocnění. Její bratr-dvojče je v té době v remisi onemocnění. Na obr. 2 je genealogie rodiny.
Diskuze
Rodiny s bialelickou mutací genu BRCA2 byly již několikrát popsány. Byla zjištěna asociace mutace c.658_659del2 (dle HGVS) genu BRCA2 s výskytem nefro-blastomu a meduloblastomu v těchto rodinách [2,6,11,14]. V uvedené rodině se do narození dětí nevyskytovala ve zvýšené míře nádorová onemocnění v mladém věku. Kromě nádoru prsu ve středním věku u maternálních tet každého rodiče nic v rodině nesvědčilo pro možnost nosičství heterozygotní mutace v genu BRCA2. K onemocnění matky karcinomem prsu a následně Hodgkinovým lymfomem došlo až po porodu všech dětí. U bratra, který zemřel na akutní leukemii, bylo vysloveno podezření na syndrom chromozomální instability, vyšetření zlomů ale bylo zřejmě v důsledku v té době probíhající léčby neúspěšné. Zvažovaný NBS nebyl prokázán, rodina dále nebyla onkologicky ani geneticky sledována. Průběh onemocnění u sestry-dvojčete ukazuje, jak je závažné nosičství dvou mutovaných alel genu BRCA2 a jak je prakticky nemožné včas nádorové onemocnění u dítěte detekovat. Dále demonstruje vysokou toxicitu onkologické léčby, byť byla modifikována s ohledem na genetický syndrom. Zajímavé výsledky přineslo u dětí cytogenetické vyšetření. U pacientů s FA se očekává nárůst chromozomálních zlomů a přestaveb v buněčných kulturách, kam byl přidán DEB. V uvedené rodině tomu bylo právě naopak. Buňky na vyšetření spontánních zlomů byly kultivovány dva dny, buňky na vyšetření indukovaných zlomů byly nejprve jeden den kultivovány, poté byl přidán DEB a pokračovala kultivace další dva dny. V buněčné kultuře s DEB byly zřejmě buňky se zlomy a přestavbami tak poškozeny, že již nedocházelo k jejich dělení a došlo k větší proliferaci buněk bez chromozomálních zlomů a přestaveb [4,5].
Preventivní opatření
V současné době nejsou stanoveny žádné specifické protokoly, jak pacienty s FANCD1 sledovat. V našem případě jsme ve spolupráci se zahraničními pracovišti stanovili plán preventivního sledování spočívající ve vyšetření CNS magnetickou rezonancí k časné detekci případného mozkového nádoru jedenkrát ročně, v tříměsíčních intervalech vyšetřování UZ břicha a krevního obrazu s mikroskopickým rozpočtem k časné detekci nádoru v dutině břišní a akutní leukemie, jedenkrát ročně vyšetření kostní dřeně.
Závěr
Cílem této práce je upozornit na možnost dědičnosti některých nádorových onemocnění v dětském věku. Péče o dítě s nádorem nezahrnuje jen údaje o současném onkologickém onemocnění dítěte, ale jedná se o komplexní pohled na zdravotní stav dítěte zahrnující zhodnocení růstových parametrů dítěte, pátrání po morfologických vrozených vadách skeletu a vnitřních orgánů, abnormálních kožních nálezech (hyperpigmentace, depigmentace, café-au-lait skvrny, které vykazují změny po ozáření nebo pobytu na slunci). Součástí vyšetření je i podrobná rodinná anamnéza se zaměřením na přítomnost onkologických onemocnění. V případě prokázání genetické etiologie nádorového onemocnění je možné v rodině provést vyšetření i dalších sourozenců či jiných členů rodiny k možnosti zahájení preventivního sledování. V případě FA skupiny D1 v důsledku bialelické mutace genu BRCA2 nám bohužel agresivita nádorů u sestry-dvojčete ukázala praktickou nefunkčnost daného sledovacího schématu. Rodina ale může v případě gravidity podstoupit prenatální vyšetření nebo preimplantační diagnostiku. V případě genu BRCA2 je vysoké riziko nádorového onemocnění i u heterozygotů, především u žen, a z tohoto důvodu je velmi důležité provést vyšetření na přítomnost mutace genu BRCA2 i u dalších příbuzných. U jedinců s heterozygotní mutací genu BRCA2 je možné nabídnout efektivní sledování včetně preventivních operací k minimalizaci rizik nádorových onemocnění spojených s nosičstvím mutace tohoto genu [12,15].
Práce byla realizována za podpory projektů NF-CZ11-PDP-3-003-2014, MZ ČR – RVO, FN v Motole 00064203 a OPPK – CZ-2.16.//3.1.00/24022.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Alena Puchmajerová
Ústav biologie a lékařské genetiky
2. LF UK a FN v Motole
V Úvalu 84
150 06 Praha 5
e-mail: alena.puchmajerova@fnmotol.cz
Obdrženo: 14. 7. 2015
Přijato: 6. 12. 2015
Sources
1. Alter BP, Kupfer G. Fanconi anemia. GeneReviews, last update: February 7, 2013.
2. Meyer S, Tischkowitz M, Chandler K et al. Fanconi anaemia, BRCA2 mutations and childhood cancer; a developmental perspective from clinical and epidemiological observations with implications for genetic counselling. J Med Genet 2014; 51(2): 71 – 75. doi: 10.1136/ jmedgenet-2013-101642.
3. Hirsch B, Shimamura A, Moreau L et al. Association of biallelic BRCA2/ FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood 2004; 103(7): 2554 – 2559.
4. Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/ BRCA pathway. Genes Dev 2012; 26(13): 1393 – 1408. doi: 10.1101/ gad.195248.112.
5. Shukla P, Solanki A, Ghosh K et al. DNA interstrand cross-link repair; understanding role of Fanconi anemie pathway and therapeutic implicatins. Eur J Haematol 2013; 91(5): 381 – 393. doi: 10.1111/ ejh.12169.
6. Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FACD1/ BRCA2. J Med Genet 2007; 44(1): 1 – 9.
7. D’Andrea AD, Grompe M. The Fanconi anaemia/ BRCA pathway. Nature Rev Cancer 2003; 3(1): 23 – 24.
8. Dewire MD, Ellison DW, Patay Z et al. Fanconi anemia and biallelic BRCA2 mutation diagnosed in a young child with an embryonal CNS tumor. Pediatr Blood Cancer 2009; 53(6): 1140 – 1142. doi: 10.1002/ pbc.22139.
9. Howlett NG, Taniguchi T, Olson S et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002; 297(5581): 606 – 609.
10. Meyer K, Davies SM, Harris RE et al. The clinical phenotype of children with Fanconi anemia caused by bialelic FANCD1/ BRCA2 mutations. Pediatr Blood Cancer 2012; 58(3); 462 – 465. doi: 10.1002/ pbc.23168.
11. Ried S, Renwick A, Seal S et al. Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumor. J Med Genet 2005; 42(2); 147 – 151.
12. Wagner JE, Tolar J, Levran O et al. Germline mutations in BRCA2; shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood 2004; 103(8): 3226 – 3229.
13. Meyer K, Fergusson WD, Oostra AB et al. A cross-linker sensitive myeloid leukemia cell line from a 2-year-old boy with severe Fanconi anemia and biallelic FANCD1/ BRCA2 mutations. Genes Chromosomes Cancer 2005; 42(4): 404 – 415.
14. Miele E, Mastronuzzi A, Po A et al. Characterization of medulloblastoma in Fanconi Anemia; a novel mutation in the BRCA2 gene and SHH molecular subgroup. Biomarker Res 2015; 3 : 13. doi: 10.1186/ s40364-015-0038-z.
15. Plevová P, Novotný J, Petráková K et al. Syndrom hereditárního karcinomu prsu a ovárií. Klin Onkol 2009; 22 (Suppl): 8 – 11.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2016 Issue Supplementum 1
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- PALB2 as Another Candidate Gene for Genetic Testing in Patients with Hereditary Breast Cancer in Czech Republic
- Hepatoblastoma, Etiology, Case Reports
- Genetics of Colorectal Tumorigenesis (Possibilities of Testing and Screening Prediction of Hereditary Form of Colorectal Cancer – Lynch Syndrome)
- Fanconi Anemia, Complementation Group D1 Caused by Biallelic Mutations of BRCA2 Gene – Case Report