
Hepatoblastom, etiologie, kazuistiky
Hepatoblastoma, Etiology, Case Reports
Hepatoblastoma is an uncommon malignant neoplasm in general, yet, it is the most common liver malignancy in children with the incidence about one per milion children. This type of liver tumor usually occurs before the age of three years. The etiology of hepatoblastoma remains unknown. However, there are some genetic conditions known to be associated with an increased risk of developing hepatoblastoma such as Beckwith-Wiedemann syndrome, hemihypertrophy, APC-associated polyposis, α-1-antitrypsin defficiency and some metabolic disorders including tyrosinemia, galactosemia and glycogen storage disease type 1. There is a higher risk of hepatoblastoma in children with very low birthweight, children who acquire hepatitis B at an early age and children with congenital biliary atresia.
Key words:
hepatoblastoma – α-fetoprotein – Beckwith-Wiedemann syndrome – APC-associated polyposis
This work was supported by grant from Norway NF-CZ11-PDP-3-003-2014, MH ČR –RVO, UH Motol 00064203 and OPPK – CZ-2.16.//3.1.00/24022.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
6. 8. 2015
Accepted:
9. 9. 2015
:
A. Puchmajerová 1; A. Křepelová 1; J. Indráková 2; R. Sítková 2; I. Balaščak 3; J. Kruseová 4; K. Švojgr 4; R. Kodet 5; M. Kynčl 6; A. Vícha 4; M. Macek Jr. 1
:
Ústav biologie a lékařské genetiky, 2. LF UK a FN v Motole, Praha
1; Úsek molekulární biologie, Laboratoř lékařské genetiky, Laboratoře AGEL a. s., Nový Jičín
2; Novorozenecké oddělení, Gynekologicko-porodnická klinika 2. LF UK a FN v Motole, Praha
3; Klinika dětské hematologie a onkologie 2. LF UK a FN v Motole, Praha
4; Ústav patologie a molekulární medicíny, 2. LF UK a FN v Motole, Praha
5; Klinika zobrazovacích metod 2. LF UK a FN v Motole, Praha
6
:
Klin Onkol 2016; 29(Supplementum 1): 78-82
:
Review
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amko2016S78
Hepatoblastom je nejčastější maligní nádor jater u dětí, jehož incidence se uvádí okolo jednoho na milion dětí. Tento typ nádoru jater je obvykle diagnostikován u dětí do tří let věku. Etiologie hepatoblastomu není zcela jasná, avšak některé genetické jednotky jsou asociovány se zvýšeným rizikem vývoje hepatoblastomu, nejčastěji Beckwith-Wiedemannův syndrom včetně izolované hemihypertrofie, APC asociovaná polypóza, deficit α-1-antitrypsinu a některá metabolická onemocnění, jako jsou tyrozinemie, galaktosemie a glykogenóza 1. typu. Vyšší riziko hepatoblastomu je také u dětí s velmi nízkou porodní hmotností, u těch, které prodělaly v raném věku hepatitidu B a dětí s vrozenou atrézií žlučových cest.
Klíčová slova:
hepatoblastom – α-fetoprotein – Beckwith-Wiedemannův syndrom – APC asociovaná polypóza
Úvod
Hepatoblastom je vzácný maligní tumorjater dětského věku, jeho incidence seuvádí okolo 1 : 1 000 000 dětí. Jednáse o nejčastější typ nádoru jater u dětí.Obvykle je diagnostikován u dětí do tří let věku. Pro tento typ nádoru je typický vzestup sérové hladiny α-fetoproteinu (AFP). Pokud je tato hladina normální, znamená to špatnou prognózu. Etiologie hepatoblastomu nebyla dosud zcela objasněna. Rizikovými faktory rozvoje hepatoblastomu jsou velmi nízká porodní hmotnost, hepatitida B, kterou děti prodělají v raném věku, vrozená atrézie žlučových cest, deficit α-1-antitrypsinu, některá metabolická onemocnění, jako jsou tyrozinemie, galaktosemie a glykogenóza 1. typu. U některých genetických syndromů je též nutné počítat se zvýšeným rizikem hepatoblastomu. Jedná se o Beckwith-Wiedemannův syndrom (BWS) a APC asociovanou polypózu [1,2]. Předmětem sdělení jsou dvě kazuistiky dětí s hepatoblastomem, u kterých byl prokázán některý z výše uvedených genetických syndromů.
Beckwith-Wiedemannův syndrom
Etiologie a typ dědičnosti, klinický obraz
Tento syndrom vzniká v důsledku epigenetických a genomických alterací chromozomu 11p15 zahrnujících maternální delece, uniparentální paternální disomie, mutace v imprintingovém centru či maternální alely genu CDKN1C [3–6].Nejčastěji se jedná o sporadický výskyt v důsledku epigenetických změn, vzácně je dědičnost autozomálně dominantní v důsledku mutace či delece maternální alely [7]. Alelickým onemocněním k BWS je Silver-Russellův syndrom.
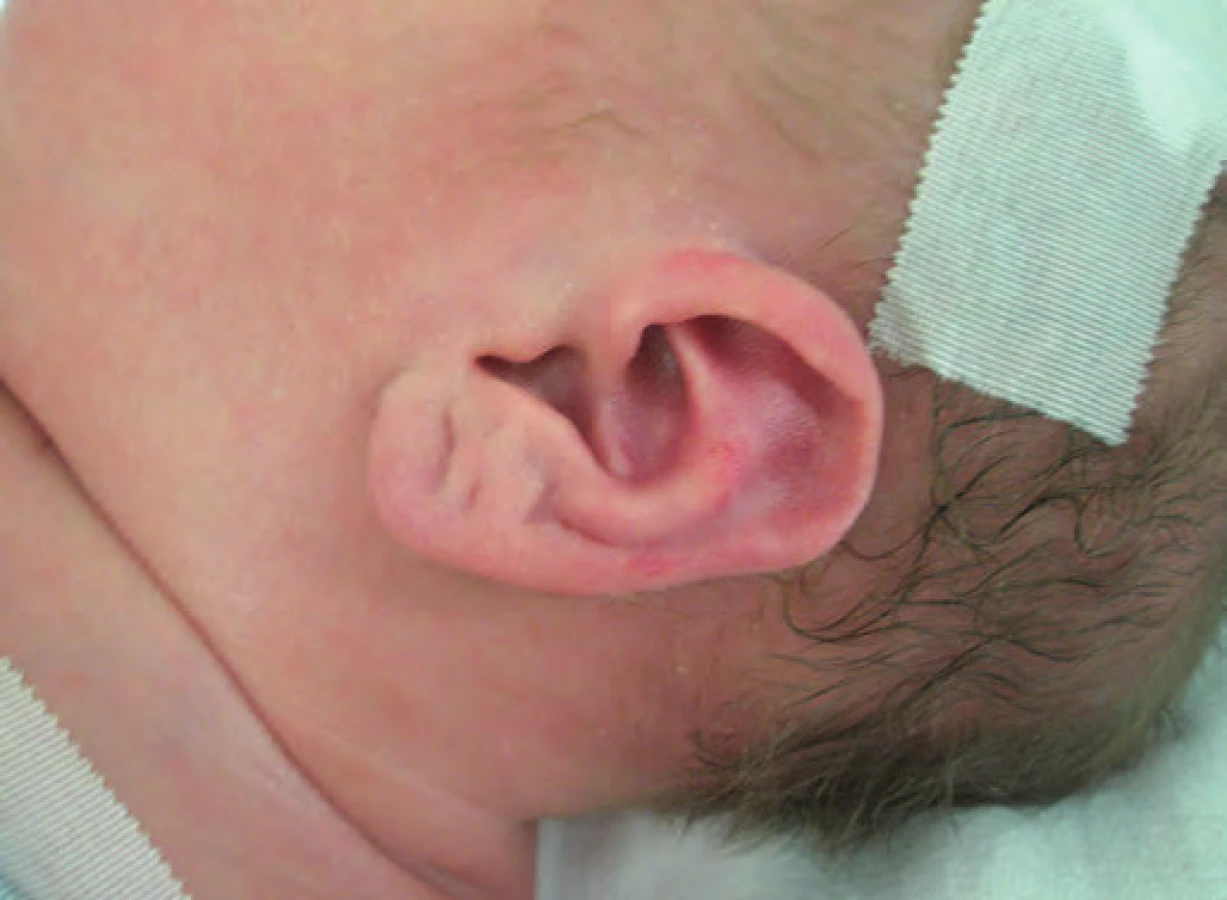
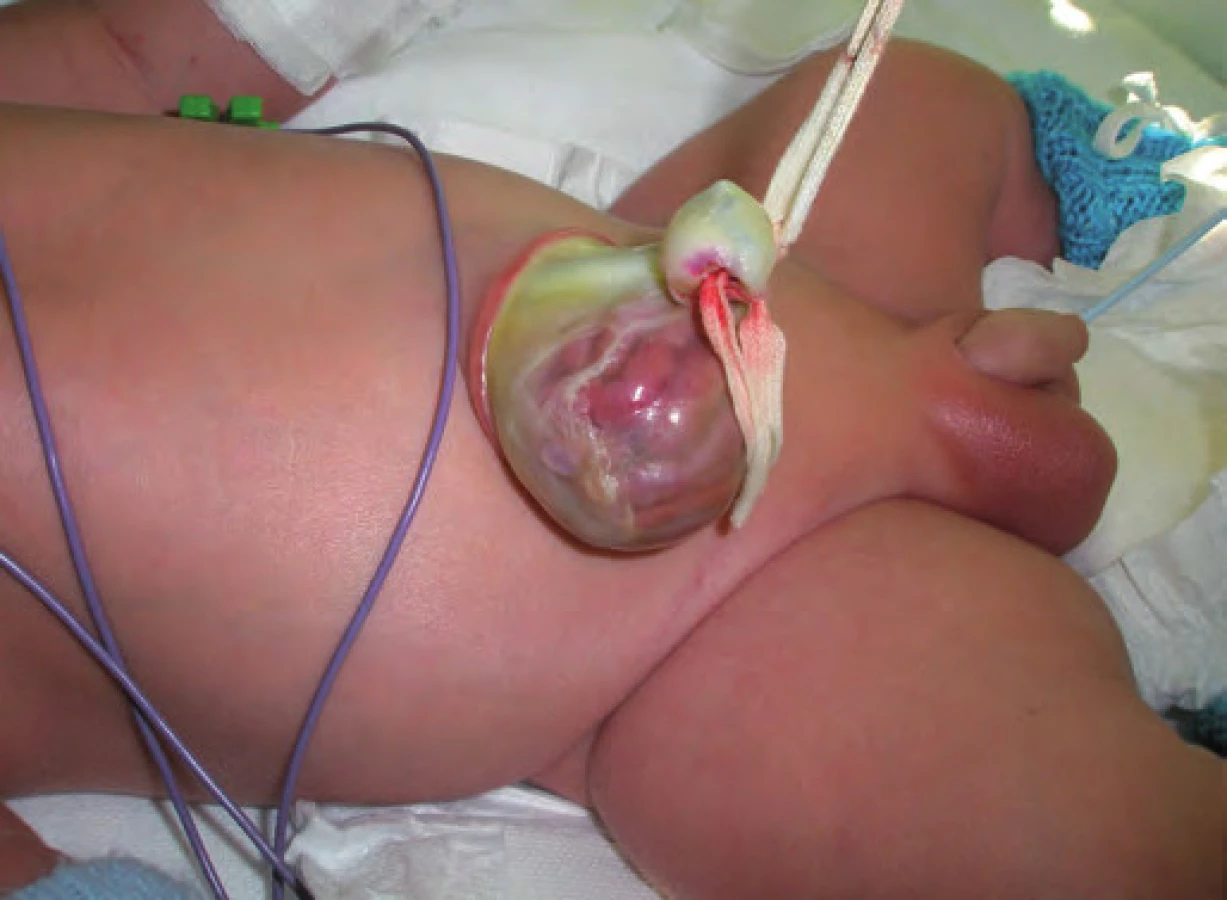
Typickými projevy u pacientů s BWS jsou makrosomie, makroglosie, hemihypertrofie, visceromegalie (játra, slezina, ledviny, nadledviny, pankreas), omfalokéla, atypické rýhy a dolíčky na ušních lalůčcích, neonatální hypoglykemie, renální anomálie zahrnující strukturální anomálie, nefromegalii, nefrokalcinózu, později rozvoj spongiózní ledviny, kardiomegalie (vzácně kardiomyopatie), faciální névus flammeus a jiné vaskulární malformace (obr. 1, 2) [7]. U pacientů s BWS je zvýšené riziko tumorů v dětském věku [8], jedná se o embryonální tumory (nefroblastom, hepatoblastom, neuroblastom, rhabdomyosarkom). Toto riziko je zvýšené především u dětí s BWS v důsledku uniparentální paternální isodisomie [7].
Kazuistika 1
Ke genetickému vyšetření byl doporučen třídenní hypertrofický novorozenec pro široký nezasychající pupek. Chlapec byl narozen ve 38. t. t. s porodní hmotností 3 930 g (nad 97. percentilem), poporodní adaptace byla protrahovaná, několik dní po porodu bylo nutné korigovat hypoglykemii. Klinicko-genetickým vyšetřením byla dále zjištěna asymetrie obličeje a makroglosie. Molekulárně-genetickým vyšetřením byla prokázána paternální uniparentální disomie 11p15.5 (20 % případů BWS). Čtvrtý den po porodu bylo provedeno UZ vyšetření bříška, kde byl zjištěn suspektní útvar v pravém jaterním laloku velikosti 12 × 14 mm, podle MRI vyšetření (10. den) se jednalo u solitární ložisko v játrech o průměru 18 mm. Vzhledem k charakteru tohoto ložiska bylo vysloveno podezření na hepatoblastom. Toto podpořila i vysoká hladina AFP v krvi (126 000 µg/ l, norma pro novorozence do 18 700 µg/ l). Chlapec byl předán k dovyšetření a terapii na Kliniku dětské hematologie a onkologie FN v Motole, kde byla prokázána rychlá progrese nádoru (22. den bylo UZ vyšetřením jater zjištěno již ložisko velikosti cca 30 mm v průměru). Byla proto provedena segmentektomie pravého jaterního laloku. Histopatologické vyšetření prokázalo hepatoblastom se strukturami fetálního a embryonálního typu.
Doporučení pro děti s BWS
Po stanovení diagnózy jsou ke zjištění rozsahu choroby a prevenci komplikací doporučena v případě makroglosie opatření k zajištění dostatečné průchodnosti dýchacích cest a zajištění dostatečného příjmu potravy, sledování a korekce novorozenecké hypoglykemie, event. v případě přetrvávání hypoglykemie vyšetření endokrinologem, UZ vyšetření břicha k detekci organomegalie, strukturálních anomálií a detekci nádorů, event. MRI nebo CT, kardiologické vyšetření zahrnující EKG, echokardiografické vyšetření, vyšetření α-fetoproteinu k vyloučení hepatoblastomu.
Doporučené sledování k časné detekci embryonálních tumorů zahrnuje UZ břicha každé tři měsíce do osmi let věku, stanovení hladiny sérového AFP každé 2–3 měsíce prvních pět let života [9]. V případě zvýšené hladiny AFP bez podezřelé léze a přetrvávání zvýšené hladiny AFP se doporučuje pátrat po skrytém tumoru. Od osmi let do rané dospělosti je doporučováno jedenkrát ročně vyšetření ledvin a vyšetření poměru kalcium/ kreatinin jedenkrát za 1–2 roky. Nebyl popsán vyšší výskyt sekundárních malignit, ale je doporučeno sledování v ambulanci dětského onkologa podle schématu poléčebného vyšetřování dle příslušného terapeutického protokolu.
APC asociovaná polypóza
Etiologie, typ dědičnosti, klinický obraz
APC asociovaná polypóza v sobě zahrnuje několik genetických jednotek: familiární adenomatózní polypózu (FAP), atenuovanou FAP, Gardnerův syndrom a Turcotův syndrom [10,11]. Tyto jednotky se navzájem prolínají, co se týče klinického obrazu a etiologie. Jedná se o autozomálně dominantně dědičné onemocnění v důsledku mutace genu APC (chromozom 5q21-q22). U cca 20 % případů se jedná o mutace de novo nebo mozaicizmus. Pro klasickou FAP jsou typické především adenomatózní polypy tlustého střeva, které se začínají tvořit okolo 15. roku života, v některých případech i dříve. Postupně se vytvoří stovky až tisíce takových polypů, z nichž každý může malignizovat. Polypy se mohou tvořit též v žaludku, duodenu a v dalším průběhu tenkého střeva, u atenuované formy FAP se tvoří méně adenomatózních polypů než v případě klasické FAP (v průměru okolo 30), riziko kolorektálního karcinomu (colorectal cancer – CRC) je ale stejně jako u klasické FAP významné. Onemocnění může být spojeno i s extrakolickými projevy zahrnujícími vrozenou hypertrofii pigmentového listu sítnice (congenital hypertrophy of the retinal pigment epithelium – CHRPE), osteomy, nadpočetné zuby, odontomy, desmoidy, epidermoidní cysty. Extrakolické projevy: osteomy a tumory měkkých tkání jsou typické především pro Gardnerův syndrom [12]. Kromě CRC se u pacientů mohou objevit i jiné malignity, jako jsou meduloblastom, papilární karcinom štítné žlázy, hepatoblastom do pěti let věku, karcinom pankreatu a žaludku [10]. Tumory CNS jsou asociovány hlavně s Turcotovým syndromem. Byla vypozorována fenotyp-genotypová korelace [13]. Nejčastěji je patogenní varianta lokalizována v kodonu 1309, v tomto případě dochází k mnohočetné polypóze již v mladém věku. K nejtěžším formám polypózy (cca 5 000 polypů) dochází v případě mutace v kodonech 1250–1464. V případě mutace na 5’ nebo 3’ konci genu APC (kodony 1–163 a 1860–1987) nebo v exonu 9 dochází k tzv. atenuované familiární adenomatózní polypóze, kdy se tvoří méně než 1 000 adenomatózních polypů. U nosičů patogenní varianty v kodonech 1395–1493 je významně zvýšené riziko desmoidních tumorů, osteomů a epidermoidních cyst, jedinci s patogenní variantou v kodonech 457–1309 mají zvýšené riziko hepatoblastomu [14,15] a mozkových tumorů. CHRPE bývá asociována s patogenní variantou mezi kodony 148 a 2043 a u delecí celého APC genu [10].
Kazuistika 2
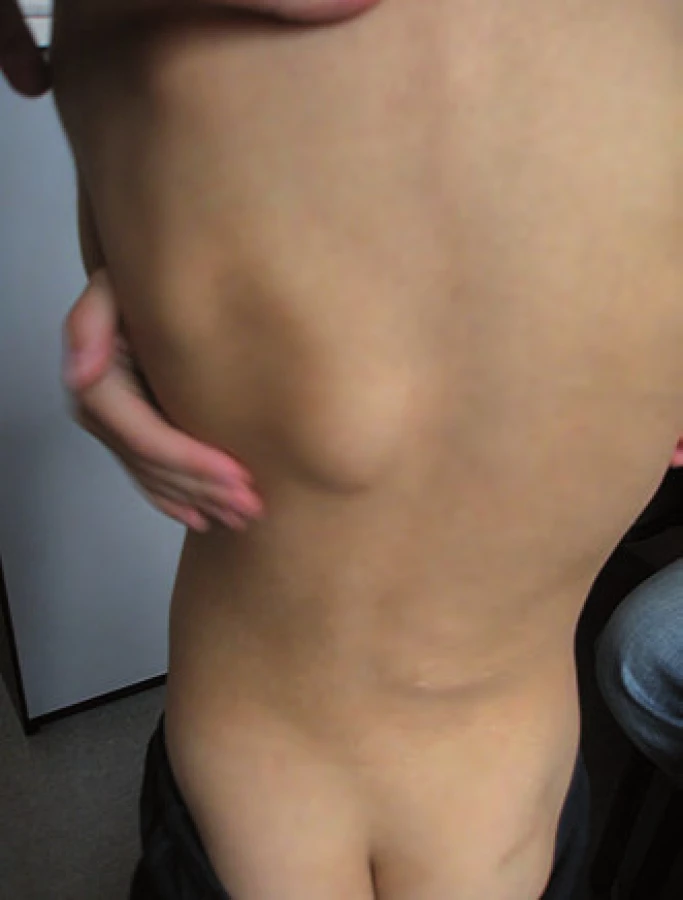
Ke genetickému vyšetření byl doporučen šestiletý chlapec pro dvě rezistence na zádech a hepatoblastom v anamnéze (obr. 3). Perinatologická data v normě, chlapec rostl a prospíval dobře, psychomotorický vývoj odpovídal věku. Ve věku šest měsíců se objevilo první ložisko na zádech v Th oblasti, další ložisko se objevilo v LS oblasti v jednom roce věku. Zvažovány lipomy, v rámci vyšetření rozsahu ložisek bylo provedeno vyšetření MRI zad a jako náhodný nález zjištěna expanze v oblasti pravého laloku jater. Biopsií byl prokázán fetální hepatoblastom a ve věku 1,5 roku byla provedena lobektomie pravého laloku jater. Následně byla provedena biopsie ložisek na zádech, histopatologicky byla prokázána kosterní svalovina s jizevnatou fibrózní tkání, diferenciálně diagnosticky zvažován desmoplastický fibrózní tumor. Při genetickém vyšetření ve věku šesti let bylo u chlapce kromě dvou kulovitých ložisek na zádech o velikosti cca 5 cm zjištěno i difuzně několik rezistencí na lebečních kostech, v.s. osteomy, histologicky nebylo verifikováno. V rodinné anamnéze se ze strany matky a otce podobné onemocnění neobjevilo, onkologicky rodina negativní. Diferenciálně diagnosticky zvažována APC asociována polypóza, i když se toto onemocnění v rodině nevyskytuje [16]. Vzhledem k věku dítěte kolonoskopie neprovedena. Molekulárně-genetickým vyšetřením byla u chlapce prokázána de novo mutace c.4438C>T (p.Gln1480*) APC genu. Tím byla u chlapce prokázána APC asociovaná polypóza, nejspíše Gardnerův syndrom. U rodičů dítěte tato mutace nebyla prokázána.
Doporučení pro pacienty s APC asociovanou polypózou
Polypy tlustého střeva
U pacientů s klasickou formou FAP je indikována kolektomie. Její načasování je stanoveno individuálně podle velikosti a počtu adenomatózních polypů. Většinou je doporučovaná, když se vyvinou mnohočetné adenomy nebo více než 20–30 adenomů s pokročilým histologickým nálezem ve smyslu dysplazie. U jedinců s atenuovanou FAP může být nezbytná, ale u přibližně jedné třetiny pacientů stačí pravidelné kolonoskopické polypektomie [11,17]. Tam, kde zatím není indikována kolektomie, je doporučena pravidelná sigmoideoskopie nebo kolonoskopie každé 1–2 roky se začátkem sledování ve věku 12 let, v případě nálezu polypů při sigmoideoskopii je dále indikována kompletní kolonoskopie a dále každoroční kolonoskopie. Po totální kolektomii by měl být ileální pouch vyšetřen každé dva roky, v případě subtotální kolektomie by mělo být vyšetřeno každých 6–12 měsíců rektum, podle počtu vytvořených polypů. U pacientů s atenuovanou FAP je kolonoskopie doporučována každé 2–3 roky, počínaje věkem 18–20 let.
Polypy tenkého střeva a žaludku
U klasické i atenuované FAP je od 25 let doporučována ezofagogastroduodenoskopie každé 1–3 roky, frekvence je stanovena podle velikosti a stupně dysplazie, součástí vyšetření by mělo být i vyšetření duodenální papily a v některých případech endoskopická retrográdní cholangiopankreatografie k detekci adenomů žlučovodu. V případě vilózních změn adenomů, jejich těžké dysplazie nebo velikosti nad 1 cm či v případě klinických projevů je indikováno endoskopické odstranění adenomů, v případě těžkých duodenálních adenomů je indikována i pankreatoduodenektomie. V případě nálezu duodenálních adenomů je indikováno každé 1–3 roky vyšetření tenkého střeva.
Extrakolické projevy
Osteomy se odstraňují spíše z kosmetického důvodu. Dostupná léčba desmoidních tumorů zahrnuje chirurgické excize s vysokým rizikem rekurence. Pro zvýšené riziko karcinomu štítné žlázy je doporučováno každoroční palpační vyšetření štítné žlázy spolu s UZ vyšetřením. Pokud jsou přítomny uzly ve štítné žláze, je doporučována aspirační biopsie [18].
Hepatoblastom
Screening hepatoblastomu u pacientů s mutací APC genu zahrnuje každé tři měsíce UZ vyšetření břicha a každé 2–3 měsíce vyšetření sérového AFP od narození nebo stanovení diagnózy do věku pěti let [9].
Indikace k vyšetření genu APC
Molekulárně-genetické vyšetření je doporučeno u všech forem difuzní střevní adenomatózní polypózy a Gardnerova syndromu. V případě průkazu mutace genu APC by mělo být nabídnuto genetické poradenství rodině pacienta. Prediktivní testování je vhodné provést u všech členů rodiny včetně malých dětí, jakmile je v rodině mutace genu APC prokázána. Je možná prenatální diagnostika, rodina se může rozhodnout graviditu ukončit v případě nálezu mutace genu APC u plodu, i když se u něho nepředpokládá těžké postižení vrozenými vadami či mentálním deficitem. Vhodnější je preimplantační genetická diagnostika, která je prováděna v úzce specializovaných centrech asistované reprodukce. Vzhledem k nízké incidenci hepatoblastomu v populaci a možné asociaci hepatoblastomu s mutací APC genu je vhodné zvážit provedení molekulárně-genetického vyšetření tohoto genu u dítěte s hepatoblastomem i přes negativní rodinnou anamnézu familiární adenomatózní polypózy z důvodu možné dosud latentní FAP u některého z rodičů nebo de novo vzniklé mutace u dítěte.
Jako preventivní opatření tvorby desmoidů a polypů je uváděno podávání nesteroidních antiflogistik (celecoxib, Sulindac). Randomizované studie potvrdily sníženou tvorbu polypů a prodloužení intervalu ke vzniku high-grade dysplastických změn polypů, a tím k oddálení nutné kolektomie [19–21]. Nevýhodou jejich dlouhodobého podávání (nejčastěji uváděno 3–6 měsíců) je však zvýšené riziko kardiovaskulárních komplikací, především u pacientů, kteří se pro kardiovaskulární onemocnění léčili ještě před zahájením podávání colecoxibu [19–23]. Terapeutické ovlivnění počtu a velikosti polypů bylo prokázáno při použití léčby anti-estrogeny (Tamoxifen), cytotoxickou chemoterapií a radioterapií [20,24].
Diskuze
Na dvou pacientech s hepatoblastomem ukazujeme různou možnou etiologii rozvoje hepatoblastomu. Jedná se o dvě zcela odlišné klinické jednotky. BWS lze rozpoznat na základě určitých fenotypových projevů již po narození dítěte, a hepatoblastom byl zachycen až na základě indikovaných preventivních vyšetření po stanovení diagnózy BWS. Diagnóza APC asociované polypózy, v našem případě nejspíše Gardnerova syndromu, může být dlouho skryta, pokud v rodině není nikdo sledován pro polypózu, protože typické projevy této choroby –polypy GIT – se objevují až v pozdějším věku. V našem případě byl chlapec doporučen ke genetickému vyšetření na základě diagnózy hepatoblastomu a diagnóza byla stanovena indikovaným molekulárně-genetickým vyšetřením v rámci diferenciální diagnózy hepatoblastomu. Molekulárně-genetickým vyšetřením byla u chlapce prokázána jednoznačně kauzální de novo mutace c.4438C>T (p.Gln1480*) APC genu. Ve shodě s dostupnými publikacemi, které se zabývají korelací genotyp-fenotyp, tato mutace může být asociována se zvýšeným rizikem desmoidních tumorů. Zvýšené riziko hepatoblastomu je ale uváděno v souvislosti s mutací v kodonech 457–1309, a toto se s nalezenou mutací neshoduje. Spektrum mutací APC genu, které zvyšují riziko hepatoblastomu, je zřejmě širší, než je dosud uváděno.
Závěr
Cílem práce je poukázat na možnou dědičnou etiologii hepatoblastomu. Hepatoblastom je vzácný maligní nádor raného dětského věku, jehož etiologie není zcela jasná, ale vždy by mělo být pomýšleno i na možnou genetickou etiologii, především tam, kde jsou přítomny i další abnormální nálezy. Současně je cílem poukázat na důležitost včasného syndromologického zařazení a následného molekulárně-genetického vyšetření při podezření na BWS a význam preventivního sledování těchto dětí již od narození. V případě hepatoblastomu jako jednoho z prvních klinických projevů APC asociované polypózy je možné díky včasné diagnostice zahájit u dítěte gastroenterologické sledování k včasné detekci závažných prekancerózních lézí v trávicím traktu ještě před jejich klinickými projevy. Následně je možná extrakce těchto lézí nebo preventivní kolektomie jako preventivní opatření významného snížení rizika CRC. Jsou zvažovány i možnosti farmakologické léčby u pacientů s APC asociovanou polypózou, které dle některých dostupných studií snižují počet polypů a růst desmoidů. Tato léčba se ale vzhledem ke kardiovaskulárním komplikacím dlouhodobě nedoporučuje.
Práce byla podpořena projekty NF-CZ11--PDP-3-003-2014, MZ ČR – RVO, FN v Motole 00064203 a OPPK – CZ-2.16./3.1.00/24022.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Alena Puchmajerová
Ústav biologie a lékařské genetiky
2. LF UK a FN v Motole
V Úvalu 84
150 06 Praha 5
e-mail: alena.puchmajerova@fnmotol.cz
Obdrženo: 6. 8. 2015
Přijato: 9. 9. 2015
Sources
1. Spector LG, Birch J. The epidemiology of hepatoblastoma. Pediatr Blood Cancer 2012; 59(5): 776–779. doi: 10.1002/ pbc.24215.
2. Venkatramani R, Spector LG, Georgieff M et al. Congenital abnormalities and hepatoblastoma: a report from the Children‘s Oncology Group (COG) and the Utah Population Database (UPDB). Am J Med Genet A 2014; 164A(9): 2250–2255. doi: 10.1002/ ajmg.a.36638.
3. Cohen MM Jr. Beckwith-Wiedemann syndrome: historical, clinicopathological, and etiopathogenetic perspectives. Pediatr Dev Pathol 2005; 8(3): 287–304.
4. Cooper WN, Luharia A, Evans GA et al. Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur J Hum Genet 2005; 13(9): 1025–1032.
5. Enklaar T, Zabel BU, Prawitt D. Beckwith-Wiedemann syndrome: multiple molecular mechanisms. Expert Rev Mol Med 2006; 8(17): 1–19.
6. Ilencikova D, Cizmarova M, Krajciova A et al. Clinical dysmorphic syndromes with tumorigenesis. Klin Onkol 2012; 25 (Suppl): S39–S48. doi: 10.14735/amko2012S39.
7. Shuman CB, Smith AC, Weksberg R. Beckwith-Wiedemann Syndrome. Initial posting: March 3, 2000; Last Update: December 14, 2010. Available from: http:/ / www.ncbi.nlm.nih.gov/ books/ NBK1394/ .
8. Rump P, Zeegers MP, van Essen AJ. Tumor risk in Beckwith-Wiedemann syndrome: a review and meta-analysis. Am J Med Genet A 2005; 136(1): 95–104.
9. Clericuzio CL, Chen E, McNeil DE et al. Serum alpha-fetoprotein screening for hepatoblastoma in children with Beckwith-Wiedemann syndrome or isolated hemihyperplasia. J Pediatr 2003; 143(2): 270–272.
10. Jasperson K, Burt RW. APC-associated polyposis conditions. Initial posting: December 18, 1998; Last Update: March 27, 2014. Available from: http:/ / www.ncbi.nlm.nih.gov/ books/ NBK1345/ .
11. Plevova P, Stekrova J, Kohoutova M et al. Familial adenomatous polyposis. Klin Onkol 2009; 22 (Suppl): S16–S19.
12. Lanckohr C, Debiec-Rychter M, Muller O et al. Gardner fibroma: case report and discussion of a new soft tissue tumor entity. Pathologe 2010; 31(2): 97–105. doi: 10.1007/ s00292-009-1260-y.
13. Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol 2007; 61(2): 153–161.
14. Giardiello FM, Petersen GM, Brensinger JD et al. Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 1996; 39(6): 867–869.
15. Hirschman BA, Pollock BH, Tomlinson GE. The spectrum of APC mutations in children with hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr 2005; 147(2): 263–266.
16. Aretz S, Uhlhaas S, Goergens H et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006; 119(4): 807–814.
17. Guillem JG, Wood WC, Moley J et al. ASCO/ SsO review of current role of risk-reducing surgery in common hereditary cancer syndromes. J Clin Oncol 2006; 24(28): 4642–4660.
18. Herraiz M, Barbesino G, Faquin W et al. Prevalence of thyroid cancer in familial adenomatous polyposis syndrome and the role of screening ultrasound examinations. Clin Gastroenterol Hepatol 2007; 5(3): 367–373.
19. Bertagnolli MM, Eagle CJ, Zauber AG et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med 2006; 355(9): 873–884.
20. Cooper K, Squires H, Carroll C et al. Chemoprevention of colorectal cancer: systematic review and economic evaluation. Health Technol Assess 2010; 14(32): 1–206.
21. Solomon SD, Wittes J, Finn PV et al. Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial safety analysis. Circulation 2008; 117(16): 2104–2113. doi: 10.1161/ CIRCULATIONAHA.108.764530.
22. Giardiello FM, Yang VW, Hylind LM et al. Primary chemoprevention of familial adenomatous polyposis with sulindac. N Engl J Med 2002; 346(14): 1054–1059.
23. Lynch PM, Ayers GD, Hawk E et al. The safety and efficacy of celecoxib in children with familial adenomatous polyposis. Am J Gastroenterol 2010; 105(6): 1437–1443.
24. Gega M, Yanagi H, Yoshikawa R et al. Successful chemotherapeutic modality of doxorubicin plus dacarbazine for the treatment of desmoid tumors in association with familial adenomatous polyposis. J Clin Oncol 2006; 24(1): 102–105.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2016 Issue Supplementum 1
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- PALB2 as Another Candidate Gene for Genetic Testing in Patients with Hereditary Breast Cancer in Czech Republic
- Hepatoblastoma, Etiology, Case Reports
- Genetics of Colorectal Tumorigenesis (Possibilities of Testing and Screening Prediction of Hereditary Form of Colorectal Cancer – Lynch Syndrome)
- Fanconi Anemia, Complementation Group D1 Caused by Biallelic Mutations of BRCA2 Gene – Case Report