
Pozdní manifestace Wilsonovy choroby: kazuistika
Late Manifestation of Wilson’s Disease: A Case Report
A case of a woman examined repeatedly at different medical workplaces because of a progressive tremor of the head is presented. This symptom started at the patient’s age of 55 years. The investigations have not revealed etiology of the clinical state so far. Previous therapy was also ineffective. The patient was sent for an examination to our department seven years after the symptoms had appeared. The liver biopsy showed an increased copper amount in the liver, and that led to the diagnosis of Wilson’s disease. The symptoms of the disease were atypical and occurred late. This case-report has confirmed possibility of a rare occurrence of this disease in elderly patients with dyskinesia without clear etiology.
Key words:
Wilson´s disease – tremor – liver biopsy – ceruloplasmin – magnetic resonance
Authors:
M. Vališ 1; R. Taláb 1; J. Bártová 2; P. Hůlek 2; J. Žižka 3; J. Masopust 4
Authors‘ workplace:
Neurologická klinika LF UK a FN
Hradec Králové
1; I. interní klinika LF UK a FN Hradec
Králové
2; Radiologická klinika LF UK a FN
Hradec Králové
3; Psychiatrická klinika LF UK a FN
Hradec Králové
4
Published in:
Cesk Slov Neurol N 2007; 70/103(3): 328-331
Category:
Case Report
Overview
Popisujeme případ ženy, která byla opakovaně vyšetřována na různých pracovištích pro progredující tremor hlavy. Obtíže začaly v 55 letech věku. Provedená vyšetření nevedla k etiologickému objasnění stavu. Terapie byla bez efektu. Po 7 letech od nástupu obtíží byla nemocná vyšetřena na našem pracovišti. Stanovení zvýšeného obsahu mědi v játrech na základě bioptického vyšetření vedlo k definitivnímu určení diagnózy Wilsonovy choroby. Výskyt prvních příznaků choroby byl v uvedeném případě pozdní a také klinický obraz byl atypický. Kazuistika potvrzuje možnost raritního výskytu tohoto onemocnění i u starších pacientů s etiologicky nejasným dyskinetickým syndromem.
Klíčová slova:
Wilsonova nemoc – tremor – jaterní biopsie – ceruloplazmin – magnetická rezonance
Úvod
Wilsonova choroba (hepatolentikulární degenerace) je geneticky podmíněná porucha metabolizmu mědi s autozomálně recesivním typem dědičnosti. Označení choroby je odvozeno od jména amerického neurologa Samuela Alexandra Kinniera Wilsona, který popsal v roce 1912 jako první familiární onemocnění postihující játra a nervový systém [1]. Již v roce 1902 Kayser a Fleischer popsali typický prstenec na rohovce, který vzniká ukládáním mědi do rohovky. Pro poměrně nízký výskyt onemocnění s prevalencí 1 případ na 25 000-40 000 narozených ji řadíme mezi vzácnější choroby běžné neurologické praxe [2].
Jedná se o lyzozomální onemocnění způsobená deficitem ATPázy transportující měď (ATP 7B) vedoucí ke kumulaci mědi v orgánech, a to především v játrech a mozku. Deficit ATPázy vede k poruše exkrece mědi do žluče a inkorporaci do apoceruloplazminu. Následkem je hromadění mědi v játrech a dále v mozku, v endoteliálním povrchu Descemetovy membrány rohovky i dalších tkáních [3]. U postižených jedinců nacházíme v játrech 20-50krát větší depozitum mědi než u zdravé populace. Později dochází k predilekčnímu ukládání mědi v mozku (ncl. caudatus, putamen, ncl. subthalamicus Luysi, thalamus, ncl. ruber), v rohovce (Kayserův-Fleischerův prstenec), v cévních stěnách a v myelinizovaných nervových vláknech. Typická je manifestace mezi 5. až 40. rokem života, v polovině případů před 15. rokem [4]. Kumulace mědi v orgánech vede k nadbytku volných radikálů a k poklesu redukovaného glutathionu. Následkem toho dochází ke komplexu poškození DNA, peroxidace membránových lipidů a také poškození proteinů bohatých na SH skupiny. Klinický obraz Wilsonovy choroby je velmi různorodý a je to onemocnění především mladšího věku. Maximum výskytu spadá do 2. až 3. desetiletí života. Vzácný je výskyt onemocnění před 5. rokem anebo po 50. roce života.
Klinický obraz je dán postižením různých orgánů, odlišným průběhem a různou formou první manifestace nemoci [5]. Dle dominující symptomatiky rozeznáváme jaterní, neurologicko - psychiatrickou, fulminantní a asymptomatickou formu (tab. 1). Jaterní forma je typická pro dětský věk a adolescentní období. Dominující formou manifestace zůstává forma neurologicko-psychiatrická představující více než 50 % všech případů. Forma jaterní tvoří asi 30 % všech případů. Asi v 15 % je onemocnění zachyceno jako forma asymptomatická. Přibližně 30 % nemocných je léčeno primárně psychiatrem pro poruchy chování, depresi nebo emoční labilitu. Dále se mohou vyskytnout další příznaky, jako jsou úzkost, desinhibice chování, zvýšená dráždivost nebo agresivita, mánie, změny osobnosti, progredující kognitivní postižení a psychotické příznaky [2]. Později někdy dochází také k alteraci endokrinního systému. U žen se objevuje amenorea a opakované aborty. Můžeme nacházet poruchy srdečního rytmu, artralgie a artritidy, renální poškození a hemolytickou anémii.

Diagnostika se opírá zejména o nutnost vyslovení podezření na Wilsonovu chorobu dle klinického obrazu. Definitivní diagnóza se opírá o histologické, genetické, biochemické a oftalmologické vyšetření. Izotopová vyšetření radioaktivní mědí nejsou u nás rutinně prováděna Diagnostika je v mnoha případech obtížná a pozdní ve fázi ireverzibilních změn. Nejproblematičtější je v asymptomatické fázi, v níž se častěji se setkáváme s normálními hodnotami ceruloplazminu a mědi v séru i s normální exkrecí mědi močí, rovněž Kayserův - Fleischerův prstenec často chybí. Výrazné zvýšení obsahu mědi v játrech je však přítomno u všech asymptomaticky nemocných. I v dnešní době část pacientů není diagnostikována a zůstává skryta pod obrazem fulminantní hepatitidy či juvenilní cirhózy nebo jako nejasné extrapyramidové syndromy a progredující demence.
Za základní screening jsou považována biochemická vyšetření, a to zejména stanovení hladiny ceruloplazminu v séru. Snížení je přítomno u více než 80 % všech nemocných. U většiny nemocných je snížení velmi výrazné pod 0,1 g/l. U další části pacientů nalézáme hodnoty, které jsou jen lehce pod normálním rozmezím. Snížení sérové mědi je rovněž konstantním nálezem, ale pokles je méně průkazný než pokles hladiny ceruloplazminu. Exkrece mědi do moči vyšší než 1,5 mmol/24 hod je konstantním nálezem u symptomatické formy onemocnění. Vyšetření je možno zpřesnit penicilaminovým testem, který zahrnuje vyšetření exkrece mědi močí po podání penicilaminu – 3 dny podáváme 1 200 mg penicilaminu a sledujeme odpady moči. Za signifikantní je považován minimálně 5násobný vzestup exkrece (5-7 mmol/24 hod). Výsledek je často falešně pozitivní.
Konfirmačním testem je stanovení mědi v játrech. Normální hodnoty jsou nízké 25-50 mg Cu/g suché jaterní tkáně. Hodnoty u nemocných s Wilsonovou chorobou se nejčastěji pohybují mezi 450-1 200 mg/g.
Oftalmologické vyšetření je zahrnuto do vyšetřovacího algoritmu pro typický nález hnědozeleného Kayserova-Fleischerova prstence na rohovce. Je viditelný štěrbinovou lampou a jeho průkaz vyžaduje určité praktické zkušenosti vyšetřujícího. Prstenec je tvořen komplexy mědi na povrchu Descemetovy membrány. Velmi často se vyskytuje u neuropsychiatrické manifestace choroby, ale ani zde to neplatí absolutně [6].
Molekulární genová analýza je další dostupnou vyšetřovací metodou ve specializovaných laboratořích v Praze a v Brně. Hlavní indikací je vyšetření na průkaz mutace v rodinách s prokázaným výskytem Wilsonovy choroby. Vyšetření je limitované možností výskytu jiné mutace, což v praxi znamená, že negativní výsledek genetického vyšetření nevylučuje přítomnost onemocnění.
Vyšetření mozku zobrazením magnetickou rezonancí (MRI) v T2 váženém obrazu
ukazuje symetrické ložiskové změny bazálních ganglií, asymetrická ložiska bílé hmoty frontálních, temporálních a okcipitálních laloků, mezencefala, pontu a hlubokých jader mozečku [7]. Bývá popisován typický obraz příznaku „hlavy medvídka Pandy“, který je způsoben na jedné straně zvýšením intenzity signálu v bílé hmotě mezencefala na T2 vážených obrazech, na druhé straně pak akcentací hypointenzit v jádrech šedé hmoty. Tento obraz bývá dominující u pacientů s převládající extrapyramidovou symptomatikou. Variabilita signálu závisí na charakteru ložisek, edému, demyelinizaci, glióze a paramagnetickém efektu depozit mědi.
Vyšetření výpočetní tomografií (CT) má nižší senzitivitu. Nejčastěji zachytí atrofii mozku a ložiskové změny v oblasti talamu a bazálních ganglií [8]. Neurofyziologická vyšetření mají nespecifický nález. Nejcennější je vyšetření kognitivních evokovaných potenciálů (EP) - prodloužení latence vlny P300.
Kauzální terapie Wilsonovy choroby s reparací genetického defektu zatím není možná. Pomocným opatřením zejména v prvním roce léčby je dieta s omezením mědi. Mezi „rizikové“ potraviny řadíme ořechy, čokoládu, játra, kuřecí maso, kávu a luštěniny.
Základní léčbou první linie je stále penicilamin (beta-beta dimethylcystein), který vytváří s mědí chelát jenž je následně vylučován močí. Navíc má schopnost reaktivovat enzymy obsahující SH skupiny. Léčba penicilaminem je celoživotní a, pokud možno, kontinuální. Při chronické léčbě je nejčastěji podáváno 900-1 200 mg/den. Základní zásadou je začínat malou dávkou 150-300 mg/den. Titrace léčebné dávky je dosahováno po několika měsících léčby. Nástup účinku terapie je pomalý. Největším problémem této léčby je poměrně vysoký výskyt nežádoucích účinků, jakou jsou poruchy krvetvorby, hypersenzitivní kožní reakce a v pozdějších fázích renální komplikace. Častým jevem je přechodné zhoršení neurologické symptomatiky po zahájení terapie penicilaminem.Nesmíme opomenout podávat malou dávku pyridoxinu vzhledem k jeho zvýšené spotřebě.
Při nesnášenlivosti penicilaminu podáváme zinek, který snižuje resorpci mědi ze střeva a zvyšuje intestinální exkreci mědi.Všeobecně je spíše považován za léčbu druhé linie. Běžná léčebná dávka je 150 mg zinku denně. Nástup účinku je také pomalý a efekt je srovnatelný s penicilaminem. Všeobecně má tato terapie nižší výskyt nežádoucích reakcí než léčba penicilaminem.
Mezi nové preparáty patří tetrathiomolybdenát, který se jeví jako účinnější než trietylentetramin u pacientů s neurologickou symptomatikou [9]. Tyto preparáty dosud
nejsou dosud běžně dostupné v klinické praxi. Při fulminantním jaterním selhání je poslední možností jaterní transplantace s velmi dobrými klinickými výsledky.
Po objevu účinné terapie došlo k zásadní změně prognózy tohoto dříve infaustního onemocnění. Při zahájení léčby v asymptomatické fázi je možno zcela zabránit manifestaci onemocnění. Léčbou se také daří výrazně zlepšit i velmi pokročilé případy. Obtížně reagují pozdě diagnostikované formy onemocnění a případy s masivně rozvinutou neurologicko-psychiatrickou symptomatikou.
Kazuistika
62letá pacientka, která je léčená pro hypotyreózu a monoklonální gamapatii. Její bratr zemřel ve 27 letech na Wilsonovu chorobu. Od roku 1999 byla (ve věku 55 let) postupně vyšetřena na několika neurologických pracovištích pro pozvolna progredující izolovaný tremor hlavy. Po 3 letech se přidružil mírný akční tremor horních končetin. Z psychických obtíží byla přítomna emoční labilita, pokleslá nálada a úzkost.
Screeningová biochemická vyšetření ceruloplazminu a mědi byla opakovaně negativní včetně jaterních testů. První MRI vyšetření mozku, provedené v lednu 2004 na jiném pracovišti, zobrazilo signálové T2 hyperintenzní změny v oblasti bazálních ganglií a mezencefalicky. V diferenciálně diagnostické rozvaze se objevila progresivní supranukleární paralýza, nebo Hallervordenova-Spatzova nemoc. Následné vyšetření hustoty a distribuce dopaminových transportérů jednofotonou emisní tomografií (123J-FP-CIT SPECT) v roce 2005 prokázalo symetrické vychytávání radiofarmaka v bazálních gangliích. Provedené elektromyografické vyšetření vyznělo pro„suspektní dystonický tremor“. Diferenciálně diagnosticky byl také zvažován esenciální tremor staršího věku. Veškerá farmakoterapie (klonazepam, L dopa, primidon, anticholinergika) byla bez efektu.
Opakované vyšetření MRI v prosinci 2005 prokázalo symetrické neexpandující zóny T2 hyperintenzity v centrálních partiích obou talamů a v dorzálních partiích mozkového kmene (obr. 1). Přítomna byla difuzní mozková atrofie. Nález na MRI byl uzavřen jako značně suspektní onemocnění metabolické etiologie, v diferenciálně diagnostické rozvaze figurovala Wilsonova choroba a Wernickeova encefalopatie, případně multisystémová atrofie. Terapie klonazepamem, primidonem a L-dopa byla nadále bez efektu. Jen nepatrné zlepšení přinesla léčba botulotoxinem.Vyšetření štěrbinovou lampou neprokázalo patologický nález. Byla znovu provedena screeningová biochemická vyšetření. Hladina ceruloplazminu v séru byla 0,11 g/l a mědi 11,8 µmol/l. Hodnoty se nacházely pod dolní hranicí referenčních norem.Vyšetření exkrece mědi močí nebylo u pacientky provedeno. Při sonografickém vyšetření jater byl shledán obraz chronické hepatopatie a vícečetné hemangiomy v obou jaterních lalocích.V únoru 2006 byla provedena necílená jaterní biopsie pod USG kontrolou jako definitivní vyšetření. Histologický nález svědčil pro chronickou aktivní hepatitidu. Obsah mědi v jaterní sušině byl zvýšený: 633,57 µg Cu/g (norma do 50 ug/g). Pro histologicky potvrzenou diagnózu nebyla provedena molekulární genová analýza. Pacientka byla vyšetřena také psychiatrem. Z psychopatologie byla přítomná emoční labilita a pokleslá nálada. Neuropsychologické vyšetření neprokázalo postižení kognitivních funkcí. Patrná byla pouze snížená pozornost, pomalejší psychomotorické tempo a tendence k perseveracím v odpovědích.
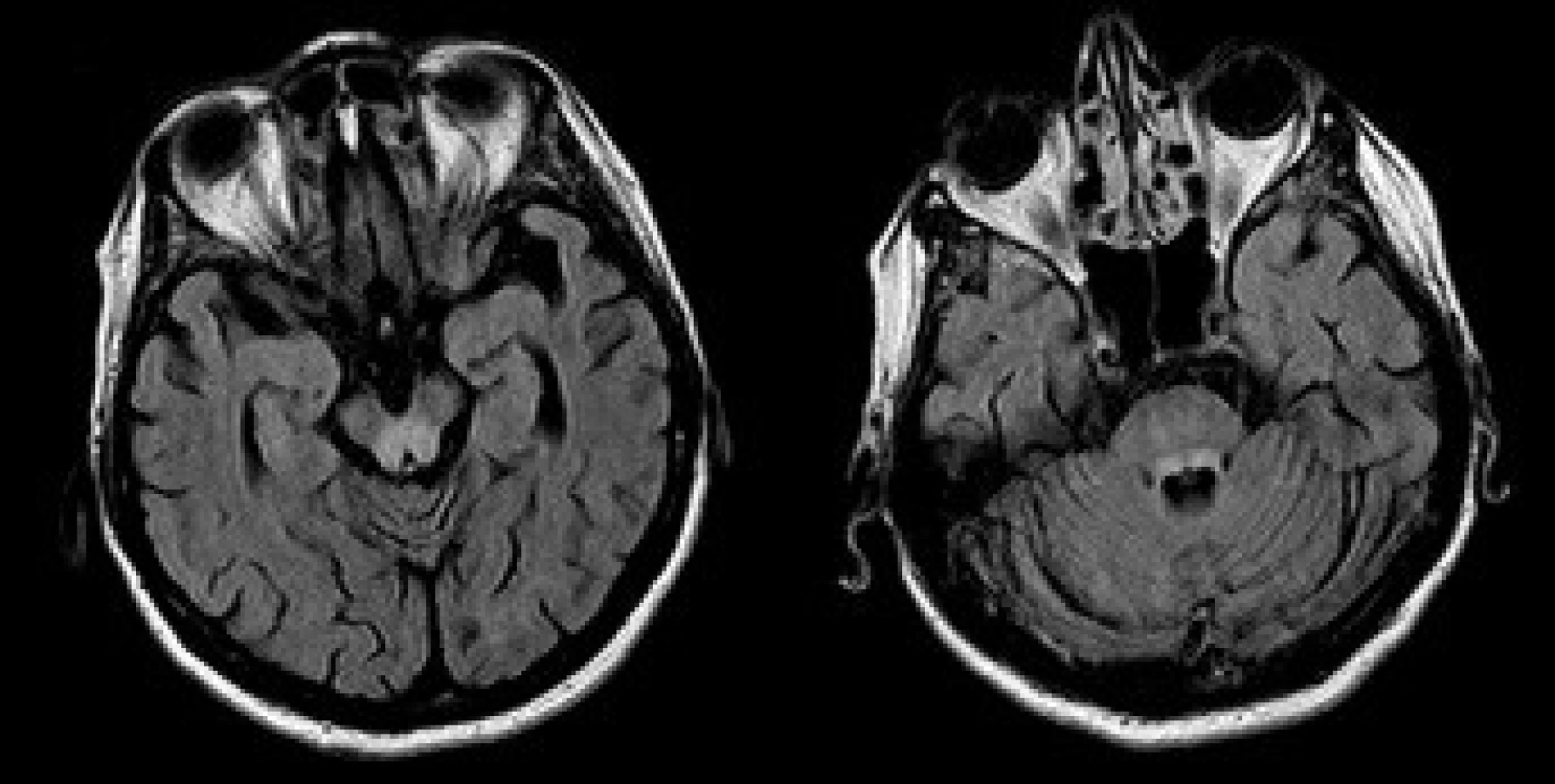
Na základě klinického obrazu a uvedených vyšetření byla stanovena diagnóza Wilsonovy nemoci s pozdním nástupem. Byla zahájena terapie penicilaminem s pozvolnou titrací na dávku 900 mg pro die a stanoven příslušný dietní režim. Po 1 měsíci terapie došlo k předvídatelnému přechodnému zhoršení neurologické symptomatiky. Nejvíce se zvýraznil třes hlavy. V květnu 2006 byl klinický obraz již mírně zlepšen: došlo ke zmírnění tremoru hlavy, který byl od počátku dominujícím neurologickým projevem onemocnění. Nemocná nadále užívá penicilamin ve stejné dávce.
Diskuse
Výskyt prvních projevů Wilsonovy nemoci před 3. nebo po 50. roce věku je raritní [10]. V popsaném případě obtíže nemocné začaly v 55 letech. Správně diagnostikováno a léčeno bylo onemocnění až o 7 let později.
Diagnostické rozpaky podpořil opakovaně negativní laboratorní screening odběru ceruloplazminu a mědi. Ani vyšetření štěrbinovou lampou neprokázalo typický Kayserův-Fleischerův prstenec. Zpočátku byla zvažována možnost esenciálního tremoru staršího věku, a to jeho pomalá varianta. Projevuje se zejména třesem hlavy o frekvenci 4-8 Hz o hrubší amplitudě a špatnou odezvou na léčbu. Proti této diagnóze svědčil negativní alkoholový test. Elektromyografický (EMG) nález poukazoval spíše na dystonický tremor, i když nebyla splněna diagnostická kritéria podle Jankovice. Vyloučena byla Parkinsonova nemoc. L-dopa test byl negativní a vyšetření hustoty a distribuce dopaminových transportérů neukázalo patologii.
Vyšetření MRI mozku ukazovalo na možnost metabolického, případně neurodegenerativního onemocnění. Přes jednoznačně patologický morfologický nález na MRI a dominující neurologickou symptomatiku nebyl přítomen Kayserův-Fleischerův prstenec. Snížené sérové hladiny ceruloplazminu a mědi spolu s USG a MRI nálezem nás znovu přivedly k úvaze o Wilsonově nemoci. Na tuto chorobu navíc zemřel bratr nemocné. Definitivně se podezření potvrdilo po vyšetření vzorku z jaterní biopsie s následným histologickým vyšetřením a stanovením zvýšeného obsahu mědi v jaterní sušině.
Kazuistika upozorňuje na možnost výskytu Wilsonovy nemoci i u starších pacientů s etiologicky nejasným dyskinetickým syndromem.Ukazuje také na konfirmační význam stanovení obsahu mědi v játrech na základě punkční jaterní biopsie pro diagnostiku této choroby. Wilsonova choroba přesahuje rámec neurologie. Na stanovení diagnózy a další péči o nemocnou se podíleli vedle neurologa také gastroenterolog, radiolog, psychiatr a psycholog.
Přijetí k recenzi: 18. 10. 2006
Přijetí do tisku: 4. 1. 2007
Adresa pro korespondenci:
MUDr. Martin Vališ
Neurologická klinika FN a LF UK
500 05 Hradec Králové
e-mail: valismar@mediclub.cz
Sources
1. Wilson SAK. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain 1912; 34 : 295–507.
2. Mitchell AJ. Neuropsychiatry and behavioural neurology explained. London: Saunders 2004.
3. Sternlieb I, Van den Hamer CJ, Morell AG, Alpert S, Gregoriadis G, Scheinberg IH. Lysosomal defect of hepatic copper excretion Wilson’s disease (hepatolenticular degeneration). Gastroenterology 1973; 64 : 99–105.
4. Gow PJ, Smallwood RA, Angus PW, Smith AL, Wall AJ, Sewell RB. Diagnosis of Wilson’s disease: an experience over three decades. Gut 2000; 46 : 415–419.
5. Pulkrábek J. Wilsonova nemoc. In: Rektor I, Rektorová I et al. Centrální poruchy hybnosti v praxi. Praha: Triton 2003 : 141–152.
6. Ross ME, Jacobsen IM, Dienstag JL, Martin JB. Late-onset Wilson’s disease with neurological involvement in the absence of Kayser - Fleischer rings. Ann Neurol 1985; 17 : 411–413.
7. Saatci I, Topcu M, Baltaoglu FF, Kose G, Yalaz K, Renda Y et al. Cranial MR findings in Wilson’s disease. Acta Radiol 1997; 38 : 250–258.
8. Vymazal J, Kvíčala V, Nevšímalová S, Mareček Z. Význam CT diagnostiky u Wilsonovy choroby. Česk Slov Neurol N 1982; 45 : 148–153.
9. Brewer GJ, Askari F, Lorincz MT, Carlson M, Schilsky M, Kluin KJ et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol 2006; 63 : 521–527.
10. Campos Franco J, Dominguez Santalla MJ, Tome Martinez de Rituerto S, Otero Anton E, Gonzalez Quintela A. Late-onset Wilson’s disease. An Med Interna 2003; 20 : 416–418.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2007 Issue 3
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Chiariho malformace – vlastní zkušenosti
- Osmotický demyelinizační syndrom – diagnostika magnetickou rezonancí: kazuistika
- MRI zobrazení mozku u pacientů s myotonickou dystrofií DM 1
- Osteoplastická dekompresivní kraniotomie