
Sporadický „guamský parkinsonský komplex“ nebo koincidence více neurodegenerativních onmocnění?
Sporadic Guam Parkinsonian Complex or the Co-incidence of Several Neurodegenerative Conditions?
Background:
The parkinsonian complex of Guam is an endemic neurodegenerative condition, which has been described only in the islands of Guam archipelago and at the Kii peninsula of Japan. Up to now, only one „sporadic“ case has been described (including the autopsy) in Japan. Study objective: To describe the clinical, laboratory and neurophysiological characteristics of the neurodegenerative disorder presenting in 4 patients with the complex syndrome of parkinsonism, amyotrophic lateral sclerosis, and dementia.
Patients and Methods:
4 consecutive patients of caucasian and Czech origin, presenting with the complex syndrome of slowly progressive parkinsonism, amyotrophic lateral sclerosis and dementia were completely examined clinically, including neuropsychological examination, and they were assessed using MRI, EMG and EP. The blood and CSF samples were also examined, and the levels of inflammatory and neurodegenerative markers (beta-amyloid, cystatin C and tau-proteins) were established.
Results:
The clinical phenotype in all four patiens was corresponding to that one described in the parkinsonian complex of Guam, including the presence of cognitive deficit at the level of mild to severe dementia. The findings of EMG examination in all cases were those typically seen in ALS, and they met the El Escorial criteria. CSF levels of neurodegenerative markers (tau-protein) were elevated in all four patiens. CSF levels of inflammatory markers were normal.
Conclusion:
The unique appearance of the syndrome typical for the endemic Guam complex in patients of caucasian origin in Europe raises a question of endemicity and heredity of the Guam complex and deserves further rersearch.
Key words:
parkinsonism – dementia – motor neuron disease
Authors:
K. Farníková; P. Kaňovský; I. Nestrašil; P. Otruba
Authors‘ workplace:
Neurologická klinika LF UP a FN Olomouc
Published in:
Cesk Slov Neurol N 2008; 71/104(3): 342-351
Category:
Case Report
Overview
Úvod:
Guamský parkinsonský komplex, nazývaný také „lytico-bodig“, je neurodegenerativní onemocnění, považované dosud za endemické pro ostrov Guam v Pacifiku. Manifestuje se obvykle v mladším středním věku postupnou progresí příznaků typických pro parkinsonizmus, amyotrofickou laterální sklerózu a demenci.
Pacienti a metodika:
U 4 pacientů z České republiky, léčených pro Parkinsonovu nemoc po dobu více než 5 let, byla zjištěna porucha kognitivních funkcí a současně (nebo s mírným časovým odstupem) se u nich objevily příznaky typické pro amyotrofickou laterální sklerózu.
Výsledky:
U všech pacientů byl zjištěn hyperintenzní pás v putamen oboustranně při MRI vyšetření mozku. U všech pacientů byl pozitivní L-DOPA nebo apomorfinový test. Výsledky běžného likvorologického vyšetření byly u všech pacientů normální. Při podrobném likvorologickém vyšetření bylo zjištěno, že hladiny neurodegenerativních markerů byly u všech pacientů zřetelně vyšší, hladiny markerů zánětu byly v normě. Zároveň byl u všech pacientů diagnostikován kognitivní deficit (různého stupně). Při EMG vyšetření byl u všech přítomen typický nález pro diagnózu amyotrofické laterální sklerózy splňující kritéria El Escorial.
Závěr:
Koincidence neurodegenerativních onemocnění není sice častá, ale je ve světovém písemnictví dostatečně dokumentována. Nicméně, výše popsaná opakovaná koincidence neurodegenerativních onemocnění, která fenotypicky odpovídá guamskému komplexu, dosud referována nebyla. Vzhledem k dosud nejasnému původu buď endemického nebo familiárního onemocnění se nabízí otázka, zda toto specifické postižení nervové soustavy nemůže existovat i ve sporadické formě.
Klíčová slova:
parkinsonizmus – demence – onemocnění motoneuronu
Úvod
Guamský parkinsonský komplex (GPK), nazývaný také „lytico-bodig“, tj. v chamorštině „blbý a líný“, je neurodegenerativní onemocnění považované dosud za endemické pro ostrov Guam a přilehlé atoly v Marianách v Pacifiku. Manifestuje se obvykle v mladším středním věku postupnou progresí příznaků typických pro parkinsonizmus (PD), amyotrofickou laterální sklerózu (ALS) a demenci (PDC). Podle starých španělských záznamů se soudí, že se GPK na ostrově vyskytuje již asi 150 let. Prevalence nemoci je asi 420 nemocných na 100 tis. obyvatel.
Existuje několik hypotéz týkajících se etiologie GPK. Vzhledem k měnícím se trendům v prevalenci tohoto onemocnění, od jejího maxima po druhé světové válce s postupným snižováním během dalších let, byla v etiologii zvažována především spoluúčast enviromentálních faktorů. Z těchto byl hlavní důraz kladen na hypotetickou chronickou intoxikaci selektivním neurotoxinem obsaženém v plodech místního cykasu, které domorodci s oblibou konzumují. Předpokládalo se, že neurotoxin navozuje změny DNA, vyúsťující v abnormální genový produkt či abnormální genovou regulaci [2]. Další z hypotéz tvrdila, že liposolubilní toxin fytosterol, taktéž obsažený v cykasech, při perzistenci v mozkové tkáni navozuje řetězec událostí, které vedou k expresi hyperfosforylovaného tau proteinu v neuronech, jehož výsledkem je široký výskyt neurofibrilárních klubek [2]. Některými autory byl rovněž zvažován možný vliv patologie genu TRPM7 související s deficitem magnézia a kalcia, jenž byl zaznamenán u velké části chamorské populace [2]. Vzhledem k familárnímu výskytu onemocnění řada jiných pramenů naopak uváděla, že onemocnění má genetický původ. Byly proto zahájeny četné studie k nalezení lokusu, z nichž mnohé ještě probíhají, lokus však dosud odhalen nebyl [5].
Nemoc postihuje zhruba stejně často obě pohlaví. Průměrný věk vzniku onemocnění je 50 let u žen a 60 let u mužů. V dosud popsaných případech se v klinickém obraze pacientů vyskytovala zejména hypomimie, zpomalení psychomotorického tempa, hypokineza, bradykineza, někdy až akineza. Později docházelo k rozvoji dysartrie a axiální rigidity. Tremor se většinou nevyskytoval. Pokud byl přítomen, tak nebyl dominantním znakem, byl střední intenzity s frekvencí 4–7 Hz, většinou klidový, někdy i posturální nebo akční. Dále byla přítomna hyperreflexie, obraz charakteristický pro postižení charakteru motor neuron disease (MND) s pozitivním Babinského příznakem a různý stupeň demence. Byla přítomna dezorientace, poruchy recentní i dlouhodobé paměti, neschopnost jednoduchého počítání. Běžně byla přítomna deprese. Autoři popisů uváděli, že mentální obraz byl nejspíše psychoorganický, ve smyslu organického psychosyndromu, nikoliv funkčního, emočního či psychotického typu. V pozdějších stadiích se pacienti stávali apatickými, abulickými až mutistickými, byli imobilní, nakonec docházelo k vyústění v progresivní vegetativní stav a terminální, život ukončující infekci [1]. Průběh nemoci byl (a je) popisován jako velmi variabilní, její rozvoj až do terminálního stadia trvá 5–7 let. V současné době není známa žádná efektivní léčba. Podávání levodopy a dopaminergních agonistů mívá jen nevýrazný a krátkodobý efekt.
Z neuropatologického hlediska je GPK tauopatií s neurofibrilárními klubky, vyskytujícími se bez neuritických plak, jinak typických pro Alzheimerovu demenci [2]. Dále bývají přítomna tzv. Hiranova a Lewyho tělíska. U 215 pacientů byla v minulosti provedena autopsie. U všech byly nalezeny znaky jasně vyjádřené neurofibrilární degenerace [1]. V různém stupni šlo především o postižení globus pallidus, substantia nigra, locus coeruleus, periakveduktální šedé hmoty a různých oblastí retikulární formace. Dále byla zjištěna přítomnost alfa synukleinopatie v amygdale a mozečku pacientů [7,8].
Neuroradiologická vyšetření pacientů trpících GPK ukazovala většinou různý stupeň atrofie frontálních a temporálních laloků na CT a MRI mozku. Při vyšetření SPECT bylo u všech případů detekováno snížení perfuze v uvedených lokalitách. Progresivní atrofie frontálních a temporálních laloků byla přítomna u všech pacientů s GPK, společně se snížením perfuze v těchto lokalitách na SPECT. U pacientů s dominantním syndromem ALS byla na CT a MRI mozku atrofie méně vyjádřena, někdy dokonce chyběla, ale SPECT vždy ukazoval významné snížení perfuze ve frontálních i temporálních lalocích. Toto snížení perfuze bylo přítomno od časných stadií u obou, PDC i ALS, dokonce i když pacienti ještě nejevili klinické známky demence nebo zatím neměli přítomnou atrofii na zobrazení CT či MRI mozku [4]. Bylo taktéž provedeno několik dalších korelačních neuroradiologicko-neuropatologických studií [9]. Při MRI spektroskopii s volumetrií hipokampu byl zjištěn jeho významně redukovaný objem, který koreloval s tíží paměťového deficitu a neuropatologicky s výskytem velkého množství depozit neurofibrilárních klubek. Při vyšetření PET (pozitronová emisní tomografie) bylo přítomno signifikantně nižší vychytávání 18F-6-fluorodopy ve striatu, a to jak u pacientů s PDC, tak u pacientů s ALS. U pacientů s ALS byla intenzita vychytávání o něco menší, ale pohybovala se přibližně ve středu mezi skupinou pacientů s PDC a skupinou kontrolní [4].
Vlastní soubor
U 4 pacientů z České republiky, u kterých byla diagnostikována buď Parkinsonova nemoc nebo progredující kognitivní deficit, byla v průběhu nemoci a její léčby nově zjištěna buď progredující porucha kognitivních funkcí nebo postupně se rozvíjející parkinsonský syndrom. U všech se dále v určitém stadiu onemocnění objevily taktéž postupně progredující příznaky typické pro ALS. Jednalo se o smíšenou parézu na končetinách a bulbární symptomatologii doprovázenou přítomností svalových fascikulací, s pozitivním EMG nálezem svědčícím pro přítomnost syndromu onemocnění motorického neuronu. U všech bylo provedeno podrobné klinické, likvorologické, neuroradiologické, neurofyziologické a psychologické vyšetření a byla taktéž provedena foto - a videodokumentace jednotlivých případů.
Pacientka č. 1
V prvním případě šlo o ženu ve věku 52 let. V osobní anamnéze měla vaginální hysterektomii s adnexotomií bilaterálně pro karcinom a resekci žaludku pro vředovou chorobu. Rodinná anamnéza byla bez pozoruhodností.
Od roku 2001, tj. od věku 46 let, pozorovala progredující třes levostranných končetin, zvláště levé horní končetiny (LHK). Jednalo se o klidový třes, horšící se při psychickém rozrušení. Udávala potíže při psaní, byla přítomna porucha jemné motoriky. Při chůzi zakopávala o levou dolní končetinu (LDK). Současně se rozvinula porucha řeči, řeč byla pomalá, tichá a monotonní. Pro tyto potíže byla poprvé hospitalizována v roce 2005. V objektivním neurologickém nálezu byla přítomna rigidita a bradykineza převažující na levostranných končetinách, jemný klidový a statický tremor levostranných končetin a vyšší elementární reflexy posturální (ERP) vlevo. Chůze byla se semiflexí trupu, drobnými krůčky, chyběla synkineza LHK. Na MRI mozku byla přítomna lehká atrofie frontálně a temporálně oboustranně a stejně tak oboustranně hyperintenzní proužek v putamen (obr. 1). Likvorologické vyšetření nemohlo být provedeno, protože pacientka s ním opakovaně nesouhlasila. Psychologické vyšetření detekovalo izolovaný kognitivní deficit s počínající poruchou krátkodobé paměti, hodnota Mini Mental State Examination (MMSE) byla 25 bodů. Byl pozitivní L-DOPA test.
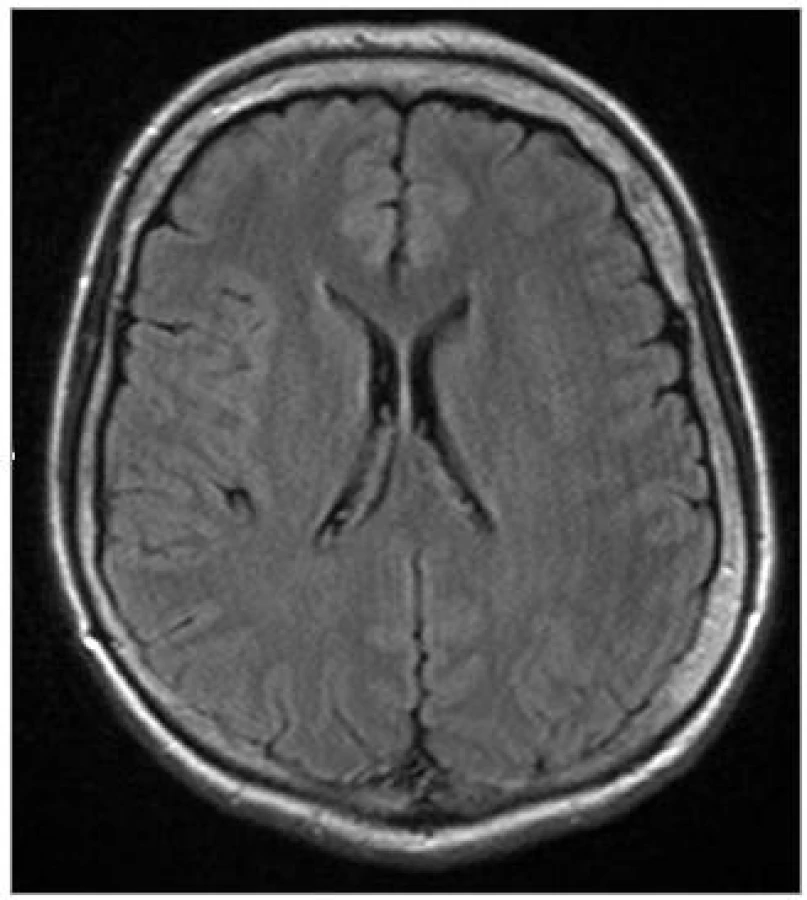
Od května 2006 začala pozorovat progresivní hubnutí s váhovým úbytkem 6 kg za 3 měsíce. Zhoršila se řeč, došlo k rozvoji těžké dysartrie. Porucha polykání nebyla. Zhoršila se chůze.
V srpnu 2006 byla znovu hospitalizována. V objektivním neurologickém nálezu byla výrazná axiální rigidita, řeč byla těžce dysartrická, byl zvýšený maseterový reflex, obleněná elevace patra při fonaci, snížený patrový reflex, na jazyku byly pozorovány ojedinělé fascikulace. Na horních končetinách (HKK) byl přítomen statický a klidový třes bilaterálně, byly zachyceny fascikulace v pletencovém svalstvu oboustranně, pozitivní abnormní kožní reflexy oboustranně s kvadruhyperreflexií. Současně byla přítomna bradykineza s oboustranným zvýšením ERP. Na dolních končetinách (DKK) byla hyperreflexie s pozitivitou abnormních kožních reflexů a zvýšením ERP oboustranně. Chůze byla drobnými kroky, s anteflexí trupu, chyběly synkinezy horních končetin oboustranně.
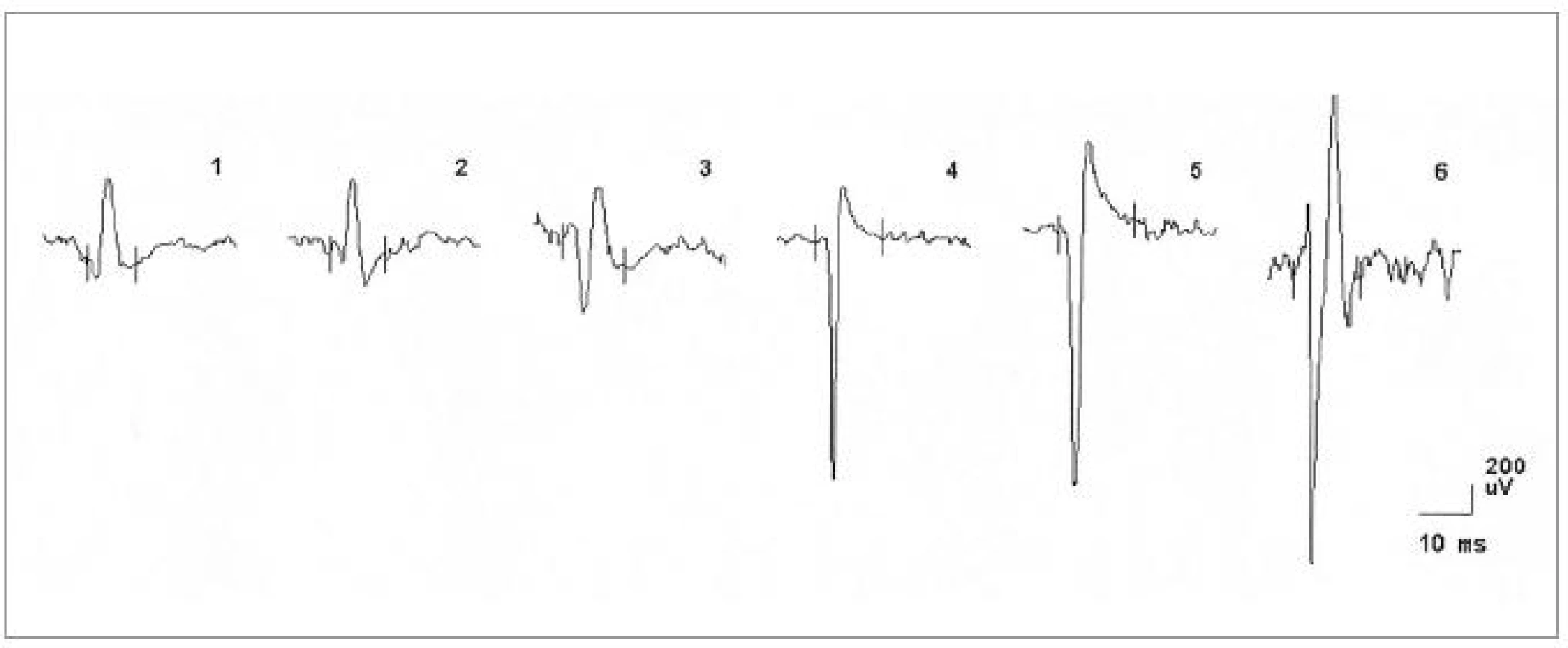
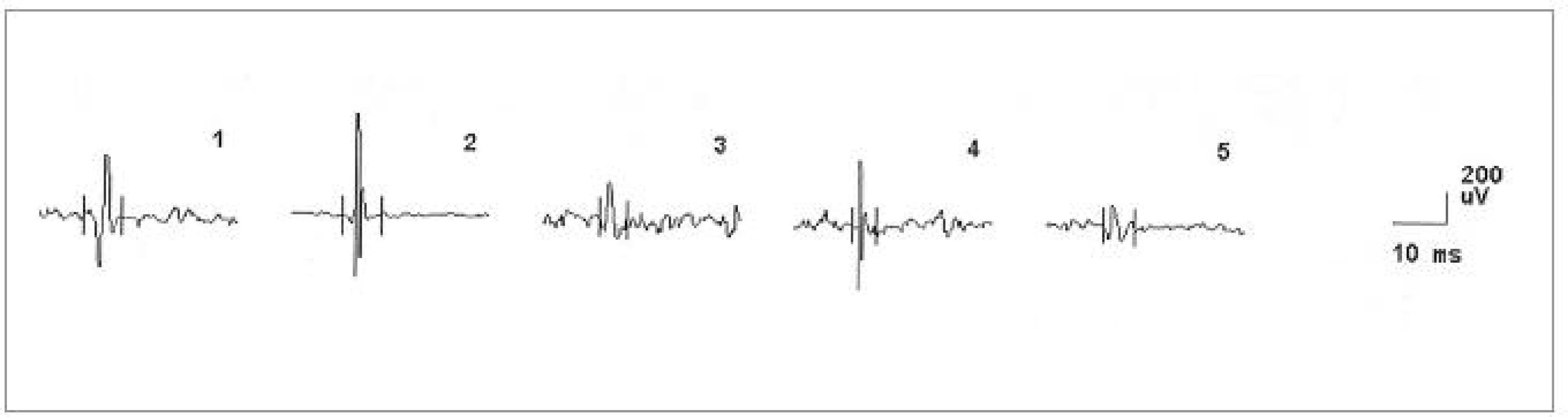
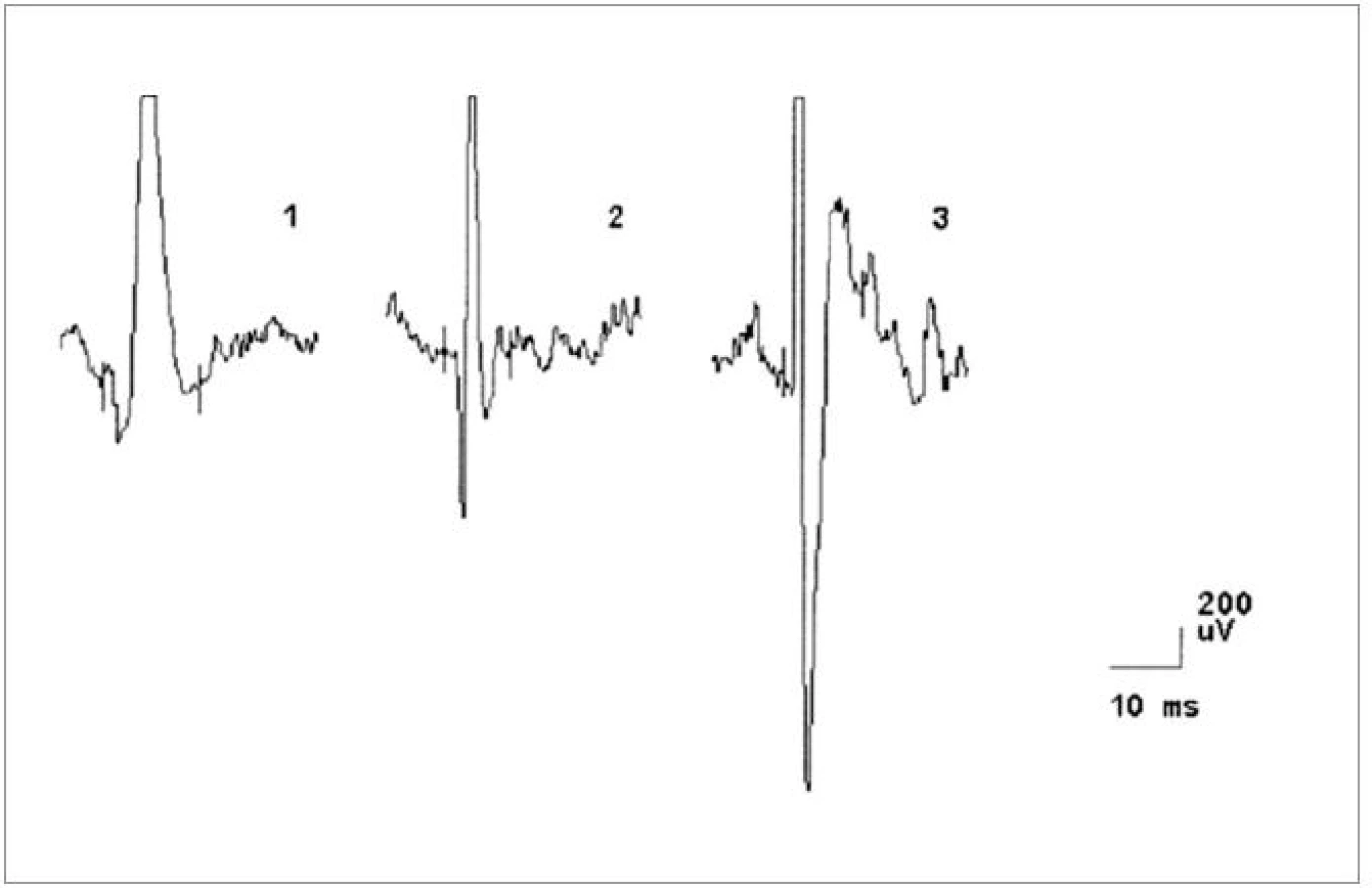
Za hospitalizace byla provedena následující vyšetření. MRI krční (C) páteře s nálezem degenerativních změn skeletu s protruzí plotének C4-7. Nález při vyšetření motorických evokovaných potenciálů (MEP) svědčil pro možné difuzní postižení pyramidové dráhy. Nálezy somatosenzorických (SEP), zrakových (VEP), a sluchových (BAEP) evokovaných potenciálů a při vyšetření vlny P300 byly v normě. EEG bylo s normálním nálezem. Při neurosonologickém vyšetření byla popsána hyperechogenní substantia nigra a normální nález na intrakraniálních i extrakraniálních tepnách. Při psychologickém vyšetření byl detekován progredující kognitivní deficit charakteru demence středního stupně, spíše kortikálního typu. Paměť byla v pásmu středního podprůměru, intelekt v pásmu podprůměru s převahou názorové složky. Nález při EMG vyšetření svědčil pro onemocnění z okruhu motor neuron disease (tab. 2, obr. 8). Diagnóza idiopatické Parkinsonovy nemoci byla přehodnocena a v léčbě byl nově podán riluzol.
Pacient č. 2
V druhém případě se jednalo o 62letého muže. Jeho rodinná i osobní anamnéza byly bez pozoruhodností.
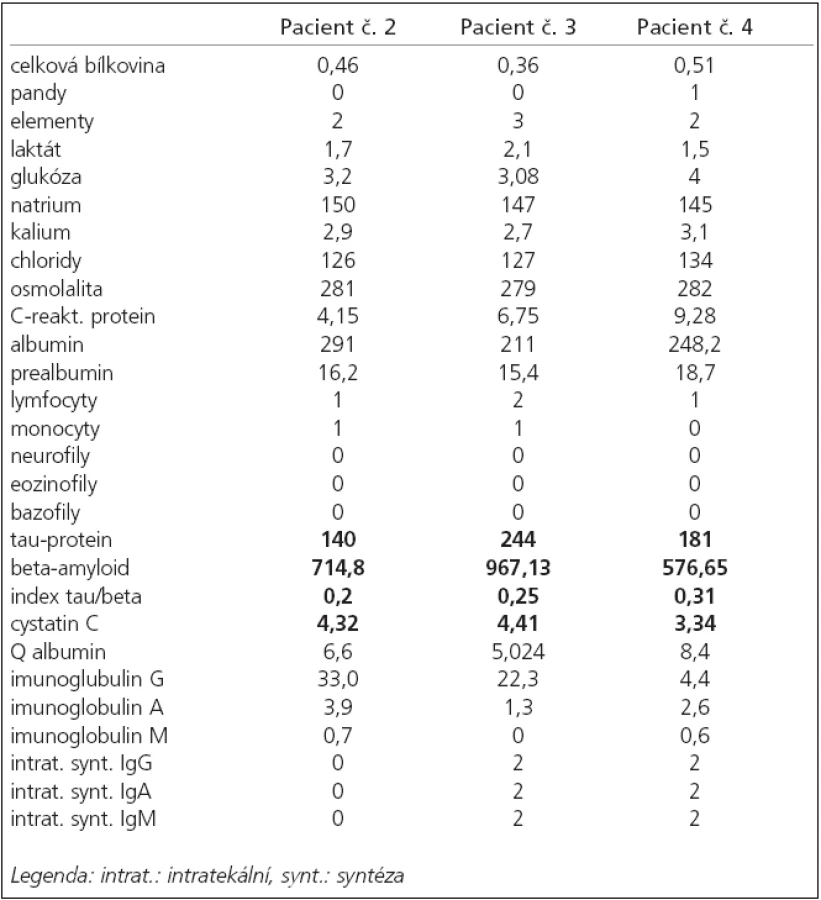
Od června 2004, tj. od věku 59 let, se u něj začala rozvíjet porucha řeči charakteru dysartrie, kterou logoped označil za hypokinetickou dysartrii. V lednu 2005 byl poprvé hospitalizován. V objektivním neurologickém nálezu byly přítomny pozitivní iritační pyramidové jevy na pravé dolní končetině (PDK) a již přítomné omezení synkinezy LHK při chůzi. Bylo provedeno MRI vyšetření mozku s nálezem difuzní atrofie mozku a mozečku oboustranně, dále byly popsány drobné hyperintenzity periventrikulárně oboustranně (obr. 2, 3). Elektrofyziologická vyšetření, tj. EMG a vyšetření evokovaných potenciálů, byla s normálními nálezy. Byla vyloučena Wilsonova choroba (penicilaminový test a biopsie jater). Při psychologickém vyšetření byl detekován lehký kognitivní deficit, MMSE 24 bodů. Kompletní likvorologický nález byl v normě, byla provedena vyšetření cytologická, biochemická, vyšetření zánětlivých markerů a markerů neurodegenerace (tab. 1).

Od prosince 2005 se začala rozvíjet porucha polykání, progredovala porucha řeči ve smyslu zhoršení dysartrie a nově se objevila dysfonie. Přidala se také celková bradykineza a progredující váhový úbytek. V květnu 2006 byl pacient znovu hospitalizován. Objektivně byl při přijetí zjištěn váhový úbytek 14 kg, byla přítomna dysfagie. Pacient byl již prakticky neschopen řeči, byla přítomna pouze dysfonická vokalizace, avšak dávivý reflex zůstával výbavný. Dále byla přítomna hypomimie a apraxie jazyka. Na horních a dolních končetinách byly přítomny difuzní hypotrofie svalstva s akrálním maximem, svalová síla byla snížena na 4. stupeň oboustranně. Byly detekovány ojedinělé fascikulace v pletencových svalech a kvadruhyperreflexie s pozitivitou abnormních kožních reflexů na dolních končetinách oboustranně. Stoj a chůze byly v normě. EMG nález svědčil pro onemocnění z okruhu motor neuron disease (tab. 2). Nález při vyšetření MEP taktéž odpovídal kombinovanému postižení I. a II. motoneuronu. Vyšetření SSEP bylo s normálním nálezem. Kondukční studie byly v normě. Při logopedickém vyšetření byla shledána labioglosolaryngeální insuficience. Neuropsychologické vyšetření bylo proveditelné jen omezeně pro prakticky anartrii, z provedených testů byl zřejmý významný kognitivní deficit, charakter demence bylo tedy obtížné stanovit.




Pacientka č. 3
Třetím případem byla žena ve věku 74 let. V osobní anamnéze měla arteriální hypertenzi, Sjögrenův syndrom a polytopní vertebrogenní algický syndrom. Rodinná anamnéza byla bez pozoruhodností. K rozvoji potíží u ní docházelo od roku 2001, tj. od věku 68 let. V tomto roce začala být pacientka depresivní. V roce 2003 se objevila zhoršená výslovnost s pocitem neobratnosti jazyka. Řeč byla setřelá, nesrozumitelná. Porucha řeči se stupňovala při psychické zátěži. Od roku 2004 se objevila porucha polykání, zejména pro tekutiny, současně udávala nestabilitu při chůzi s tahem do stran a opakovanými pády.
Pro tyto potíže byla v roce 2005 hospitalizována. V objektivním neurologickém nálezu byla přítomna hypomimie, dysartrie, řeč byla tichá a monotonní. Při vyšetření mozkových nervů byl zvýšený patrový reflex, jinak byl nález v normě. Na horních a dolních končetinách byly zvýšeny šlachookosticové reflexy, dále byly přítomny počínající atrofie pletencového svalstva na horních končetinách a pozitivní iritační pyramidové jevy na dolních končetinách. Chůze byla s lehce rozšířenou bazí a omezením synkinez horních končetin oboustranně. Byla provedena MRI mozku s nálezem drobných hypersignálních změn v oblasti sulcus centralis oboustranně, drobných hyperintenzivních změn v bílé hmotě frontoparietálně oboustranně, hyperintenzních proužků v putamen oboustranně, atrofie šedé hmoty frontálních a temporálních laloků a mozečku (obr. 4, 5). EMG nález svědčil pro onemocnění ze skupiny motor neuron diseases. Kondukční studie byly v normě. Likvorologický nález byl normální kromě hladiny tau proteinu, která byla zřetelně zvýšená (tab. 1). Při neurosonologickém vyšetření byla popsána zvýšená echogenita substantia nigra oboustranně, jinak byl nález v normě. Psychologickým vyšetřením byly detekovány izolované kognitivní deficity, MMSE 25b. Genetickým vyšetřením byla vyloučena spinocerebellární ataxie (SCA) typu 1–3, 6, 7.

V dalším průběhu docházelo k postupnému zhoršování stavu, progredovala svalová slabost, horšila se řeč a nestabilita při chůzi. Z toho důvodu byla pacientka v roce 2006 opět hospitalizována. V objektivním neurologickém nálezu bylo již přítomno povšechné zvýšení ERP, lehký klidový třes pravé horní končetiny (PHK) a hypokineza s hypomimií. Na kontrolní MRI mozku byl stacionární nález. V roce 2007 došlo k dalšímu zhoršení slabosti končetin a šíjového svalstva, zhoršila se bulbární symptomatologie a objevily se fascikulace v pletencovém svalstvu horních i dolních končetin a na jazyku. V objektivním neurologickém nálezu přetrvával parkinsonský syndrom a bulbární syndrom s fascikulacemi na jazyku, maseterový reflex byl přiměřený. Na horních končetinách byly zvýšeny šlachookosticové reflexy s pozitivitou iritačních pyramidových jevů, hypotrofie interoseálních svalů, oslabení svalové síly akrálně a vyšší ERP bilaterálně, lehký klidový třes PHK. Na dolních končetinách byly taktéž vysoké šlachookosticové reflexy s pozitivitou pyramidových iritačních jevů, oslabení svalové síly na 3/5 ve všech segmentech a rigidita oboustranně. EMG nález svědčil pro motor neuron disease (tab. 2).
Pacient č. 4
Ve čtvrtém případě šlo o muže ve věku 42 let, jehož osobní i rodinná anamnéza byly bez pozoruhodností. Od 40 let věku se u něj začala rozvíjet porucha paměti. Rok poté se objevila porucha řeči, která byla drmolivá a nesrozumitelná. Špatně se mu polykalo. Zároveň začal pozorovat slabost aker horních končetin. Také se začal pomočovat. V roce 2007 byl poprvé hospitalizován. V objektivním neurologickém nálezu bylo přítomno výrazně zpomalené psychomotorické tempo, chudé nepřiléhavé odpovědi, již z orientačního klinického vyšetření byla patrná těžká porucha kognitivních funkcí včetně dezorientace v místě a času. Řeč byla dysfonická se zřetelnou nazolalií. Maseterový reflex byl přiměřený. Byla přítomna dysfagie s nevýbavným patrovým reflexem. Na jazyku byly zachyceny jemné fascikulace. Horní a dolní končetiny byly bez paréz, oboustranně byly zvýšeny ERP, byla přítomna rigidita končetinového svalstva, byla přítomna hypokineza při pohybech končetin a dále byla přítomna kvadruhyperreflexie s pozitivitou iritačních pyramidových jevů bilaterálně. Pacient se pohyboval pomalou, šouravou chůzí s krátkými krůčky, s omezenými synkinezami horních končetin, otáčení bylo obtížné a byly přítomny festinace. Pull test byl pozitivní. Byly přítomny fascikulace v pletencovém svalstvu horních končetin a atrofie pletencového svalstva, interoseálních svalů a obou tenarů. Psychologickým vyšetřením byla detekována středně těžká až těžká demence smíšeného typu s významným narušením exekutivních funkcí, oslabením mnestických funkcí, osobnostními a behaviorálními změnami a narušením sociálních vztahů. Při vyšetření MMSE bylo skóre 17 bodů. Na MRI mozku byla popsána drobná, vícečetná hyperintenzivní ložiska frontálně bilaterálně, pokročilá atrofie frontálně a temporálně oboustranně a hyperintenzní proužky v putamen oboustranně (obr. 6, 7). Likvorologické vyšetření bylo s normálními nálezy včetně markerů neurodegenerace (tab. 1). Výsledek EMG vyšetření odpovídal diagnóze motor neuron disease (tab. 2, obr. 9, 10).

Diskuse
Neurolog, který se věnuje pacientům postiženým neurodegenerativními onemocněními, si je nepochybně vědom sice vzácných, ale pravidelně se objevujících průniků a více či méně plynulých přechodů z jednoho fenotypu do druhého. Tento fenomén je vídán u progresivní supranukleární paralýzy (PSP), která se i několik let může jevit jako dopa-responzivní parkinsonizmus typu Parkinsonovy nemoci, a symptomy typické pro PSP se objevují postupně [10]. Stejně tak se může fenotyp typický pro některou z forem SCA „překlopit“ do typického fenotypu multisystémové atrofie (MSA). Vzácně lze pozorovat toto „překlopení“ u fenotypu parkinsonského, kdy výsledným klinickým obrazem je MSA-P [11]. Ostatně, původní Quinnova idea o existenci MSA se zrodila na základě pochopení vzájemného průniku fenotypů neurodegenerativních onemocnění [12,13]. Z tohoto hlediska tedy postupný přechod parkinsonského fenotypu k fenotypu demence s Lewyho tělísky (DLBD) a dále k fenotypu kombinace parkinsonské s DLBD a ALS není nepochopitelný. Co je zarážející, je podobnost tohoto „kombinovaného“ fenotypu s fenotypem onemocnění považovaným dosud za endemické s výskytem omezeným na pacifickou oblast. Ani tato situace však není žádné novum, jako příklad lze uvést jen sporadický výskyt von Economovy encefalitidy, která byla po dlouhá léta považována pouze za záležitost evropské epidemie na začátku 20. století [14]. Lze tedy konstatovat, že i tato skutečnost existuje a je dostatečně dokumentována v medicinské literatuře.
Stačí však opravdu výše uvedená evidence k úvaze o možné existenci sporadické formy GPK? Jak již uvádíme v úvodu, John Steele charakterizuje GPK jako neurodegenerativní onemocnění manifestující se obvykle ve středním věku postupnou progresí příznaků typických pro parkinsonizmus, amyotrofickou laterální sklerózu a demenci. Dosud bylo a nadále je považováno za endemické pro ostrov Guam, přilehlé atoly v Pacifiku a japonský poloostrov Kii. Největší prevalence nemoci byla popisována po II. světové válce, ale to může být nepochybně artefakt způsobený zvýšeným zájmem o strategické souostroví (z Guamu mj. startovaly bombardéry B-29, které svrhly atomové pumy na Hirošimu a Nagasaki). V průběhu dalších 50 let došlo podle literárních údajů k podstatnému snížení prevalence nemoci. Nicméně, nemoc je popisována již ve španělských záznamech starých skoro 200 let, které byly pořizovány laiky. Muselo jít tedy o jev nápadný. Jinými slovy, je možné, že před 200 lety byla prevalence ještě vyšší než po II. světové válce. Její pokles mohl být potom důsledkem jak eliminace genu z populace (snížení partnerské atraktivity jedinců z postižených rodin), tak i změnou stravovacích návyků, tj. snížením množství (již zmiňovaných) cykasových plodů v potravě.
Od konce 60. let 20. století, kdy začal být GPK blíže zkoumán (NINCDS dokonce v průběhu 60. let zde založila polní výzkumnou stanici, která je dnes plnohodnotným výzkumným centrem, které vede John Steele) bylo jasné, že vzhledem k jeho klinickému a neuropatologickému obrazu jde o různé klinické manifestace jednoho a téhož onemocnění. Proběhlo několik studií zaměřených na průkaz jeho etiologie [2,3,6,8]. Familiární výskyt onemocnění nabízel otázku přítomnosti genetických faktorů, jež by mohly hrát roli v etiologii tohoto onemocnění. V dosud popsaných studiích zaměřených na detekci genového lokusu pro GPK nebyl žádný jednoznačně identifikován [1,3]. Největší pozornost přitom byla věnována genu pro abnormální hyperfosforylovaný tau protein, který byl označen jako zodpovědný za neurofibrilární degeneraci. Jeho mutace lokalizované na chromozomu 17 (FTDP-17) byly identifikovány v případech dědičných autozomálně dominantních tauopatií s frontotemporální demencí a parkinsonizmem, dále je tau polymorfizmus asociován s progresivní supranukleární paralýzou. Byl vyšetřen rovněž jako kandidátní gen pro GPK, ale nebyla nalezena žádná abnormalita [1]. Dále byl zvažován vliv enviromentálních faktorů, z nichž byl hlavní důraz kladen na stravovací návyky Chamořanů. Velmi intenzivně byla zkoumána role již zmiňovaných cykasových plodů obsahujících selektivní neurotoxin, ale ani jejich vliv na vznik choroby nebyl dosud jednoznačně prokázán a je řadou autorů zpochybňován [2,6]. Hypotézy týkající se prionové infekce, vlivu socioekonomických faktorů či deficitu kalcia a magnézia byly vyvráceny. V současné době tedy nezbývá než se domnívat, že se jedná o familiární onemocnění, jehož podstatou může být dědičná zvýšená susceptibilita k působení selektivního neurotoxinu obsaženého v potravě, možná právě v cykasových plodech (ale stejně možná také ne). Nicméně, stejný může být patofyziologický podklad dalších neurodegenerativních onemocnění, u kterých se dosud nepodařilo identifikovat lokus působící právě specifický fenotyp neurodegenerace. Potom v případě GPK může jít o tento typ neurodegenerativního onemocnění, který se díky vzájemným příbuzenským spojením (inbreedingu, jež je v uzavřených komunitách pacifických společenství běžný, to připouštějí všichni autoři) rozšířilo až k prevalenci popisované ve španělských a amerických pramenech. Zrušení dogmatu endemického výskytu v důsledku působení místně specifického neurotoxinu k takovéto úvaze nutně musí vést.
Poslední otázkou je fenotyp onemocnění. Zatím si každý badatel, zabývající se GPK, kladl otázku, zda se jedná opravdu o unikátní, specifický typ neurodegenerace, nebo o koincidenci 2–3 různých (a dosud různě popisovaných a nozologicky řazených) fenotypů neurodegenerace. Koincidence neurodegenerativních onemocnění není sice častá, ale je ve světovém písemnictví dostatečně dokumentována. Již před více než 10 lety byly publikovány úvahy o jednotném patologickém a patofyziologickém podkladu (do té doby) zcela odlišně klasifikovaných nemocí [15]. Nicméně, u našich pacientů výše popisovaná, opakovaná koincidence neurodegenerativních onemocnění, která fenotypicky odpovídá guamskému komplexu, dosud kromě GPK samotného zatím popisována nebyla. Vzhledem k dosud nejasnému endemickému nebo familiárnímu původu GPK je proto zcela namístě uvažovat o tom, že pokud se GPK může vyskytovat endemicky a možná bez vazby na specifický neurotoxin, může existovat i jeho forma sporadická. Klinická manifestace této sporadické formy by potom vypadala stejně jako onemocnění u našich pacientů. Další úvahy jsou však čirou hypotézou, nejméně do doby, kdy bude možno srovnat výsledky ultrastrukturálního zkoumání mozkové tkáně. Jediné, co lze zatím morfologicky srovnat, je hladina tau-proteinu, typického markeru tauopatické neurodegenerace, v likvoru. Ta byla u 2 našich pacientů v mezích běžné normy a u pacientky č. 2 byla zvýšená. Údaje o tomto vyšetření u pacientů trpících GPK chybí a je velmi pravděpodobné, že nebyly prováděny [16].
Domníváme se, že sporadický výskyt neurodegenerativních onemocnění, považovaných dosud za endemická, je možný [17], a považujeme proto za smysluplné po existenci „koincidence“ různých fenotypů neurodegenerace u našich pacientů cíleně pátrat.
prof. MUDr. Petr Kaňovský, CSc.
Neurologická klinika LF UP a FN
I. P. Pavlova 6
775 20 Olomouc
e-mail: petr.kanovsky@fnol.cz
Přijato k recenzi: 11. 1. 2008
Přijato do tisku: 28. 2. 2008
Sources
1. Steele JC, Caparros-Lefebvre D, Lees AJ, Sacks OW. Progressive supranuclear palsy and its relation to pacific foci of the parkinsonism–dementia complex and Gaudeloupean parkinsonism. Parkinsonism Relat Disord 2002; 9(1): 39–54.
2. Friedlant RP, Armon C. Tales of Pacific tangles. Neurology 2007; 68(21): 1759–1761.
3. Morris HR, Steele JC, Crook R, Wavrant-De Vrieze F, Wood NV, Onstead-Cardinale L et al. Genome-Wide. Analysis of the Parkinsonism-Dementia Complex of Guam. Arch Neurol 2004; 61(12): 1889–1897.
4. Kokubo Y, Kuzuhara S. Neuroradiological Study of Patients with Amyotrophic Lateral Sclerosis and Parkinsonism – Dementia Complex on the Kii Peninsula of Japan. Arch Neurol 2003; 60(9): 1257–1261.
5. Plato CC, Garruto RM, Galasko D, Craig U, Plato M, Gamst A et al. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of Guam: changing incidence rates during the past 60 years. Am J Epidemiol 2003; 157(2): 149–157.
6. Borenstein AR, Mortimer JA, Schofield E, Wu Y, Salmon DP, Gamst A et al. Cycad exposure and risk of dementia, MCI and PDC in the Chamorro population of Guam. Neurology 2007; 68(21): 1764–1771.
7. Forman MS, Schmidt ML, Kasturi S, Perl DP, Lee VM, Trojanowski JQ. Tau and alpha-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol 2002; 160(5): 1725–1731.
8. Ince PG, Codd GA. Return of the cycad hypotesis – does the amyotrophic lateral sclerosis/parkinsonism dementia complex (ALS/PDC) of Guam have new implications for global health? Neuropathol Appl Neurobiol 2005; 31(4): 345–353.
9. Cox PA, Banack S, Murch S, Sacks O. Commentary on: Return of the cycad hypotesis – does the amyotrophic lateral sclerosis/Parkinsonism dementia complex (ALS/PDC) of Guam have nex implications for global health? Neuropathol Appl Neurobiol 2006; 32(6): 679–682.
10. Kaňovský P. Parkinsonské syndromy. In: Růžička E, Roth J, Kaňovský P (Eds). Extrapyramidová onemocnění I. Parkinsonova nemoc a parkinsonské syndromy. Praha: Galén 2001.
11. Hughes AJ, Daniel SE, Lees AJ. Idiopathic Parkinson´s disease combined with multiple system atrophy. A clinicopathological report. Mov Disord 1991; 6(4): 342–346.
12. Quinn N. Multiple system atrophy – the nature of the beast. J Neurol Neurosurg Psychiatry 1989; Suppl: 788–789.
13. Kaňovský P, Streitová H, Bareš M, Kuba R, Pospíšilová D. Multisystémová atrofie: nový nosologický koncept. Cesk Slov Neurol N 1996; 59/92(1): 3–9.
14. Dale RC, Churchyard AJ, Surtees RA, Lees AJ, Adcock JE, Gardiny B et al. Encephalitis lethargica syndrome: 20 new cases and evidence of basal ganglia imunity. Brain 2004; 127(Pt 1): 21–33.
15. Uitti R, Calne DB. Pathogenesis of idiopathic parkinsonism. Eur Neurol 1993; 33 Suppl 1 : 6–23.
16. Shintaku M, Oyanagi K, Kaneda D. Amyotrophic lateral sclerosis with dementia showing clinical parkinsonism and severe degeneration of the substantia nigra: report of an autopsy case. Neuropathology 2007; 27(3): 295–299.
17. Lees AJ. osobní sdělení 2007.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2008 Issue 3
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Depersonalizace a derealizace – současné nálezy
- Degenerace krční meziobratlové ploténky – indikace a možnosti chirurgické léčby
- Migréna v těhotenství
- Pohybové aktivity pacientů trpících dědičnou polyneuropatií