
Tuberózní skleróza: optimalizace postupu její DNA di agnostiky
Tuberous Sclerosis: Optimisati on of its DNA Di agnosing Procedure
The go al of the study is to inform abo ut clinical and genetic aspects of tuberuos sclerosis, to po int o ut the benefit of genetic testing as well as its limits, and to inform abo ut the optimised genetic testing procedure. Tubero us sclerosis is an a utosomally dominant hereditary ne urocutaneo us dise ase with extremely vari able clinical manifestati ons. The dise ase results from mutati on in one of the two tumo ur - suppressor genes, i.e. TSC1 or TSC2. A wide range of ca usal mutati ons with random distributi on in the responsible genes has been described. The proposed DNA examinati on procedure was designed on the basis of knowledge obtained from the analysis of 65 samples from Czech pati ents and by tests performed on 53 positive control samples from partner centres abro ad. The basic criteri a for the selecti on of the best procedure for DNA di agnosis in TSC pati ents was economy, effici ency and laboratory capacity.
Key words:
tubero us sclerosis – tumo ur - suppressor gene – mutati on screening – sequence analysis – epilepsy – psychomotor retardati on
Authors:
R. Vrtěl 1; G. Vo utsinas 2; R. Vodička 1; H. Filipová 1; P. Kotlárová 1; V. Smutná 1; D. Šimková 1; D. Konvalinka 1; A. Šantavá 1; J. Šantavý 1
Authors‘ workplace:
Ústav lékařské genetiky a fetální medicíny, LF UP a FN Olomo uc
1; Laboratory of Environmental Mutagenesis and Carcinogenesis, Institute of Bi ology, NCSR “Demokritos”, Aghi a Paraskevi Attikis, Greece
2
Published in:
Cesk Slov Neurol N 2008; 71/104(4): 478-482
Category:
Short Communication
Zdroj podpory a poděkování: Práce byla řešena jako so učást projektu MŠMT česko- řecké vědecko- technické spolupráce kontaktu č. 7- 2006- 20, ME 923. TSC1 and TSC2 genetic alterati ons in tubero us sclerosis (Genetické změny TSC1 a TSC2 u tuberózní sklerózy, 2006– 2007). Za radu a spolupráci děkujeme dr. D. Halley a dr. A. van Ouweland z pracoviště Klinické genetiky Erasmovy Univerzity Rotterdam, Holandsko.
Overview
Cílem práce je uvést klinickogenetické aspekty u tuberózní sklerózy, po ukázat na přínos genetického testování i jeho limity a seznámit s optimalizovaným postupem při genetickém vyšetřování. Tuberózní skleróza je a utozomálně dominantně dědičné ne urokutánní onemocnění s extrémní vari abilito u klinických projevů. Choroba je podmíněna mutací v jednom ze dvo u tumor supresorových genů, TSC1 nebo TSC2. Bylo popsáno široké spektrum ka uzálních mutací s náhodno u distribucí v zodpovědných genech. Navrhovaný postup vyšetření DNA byl vytvořen na základě zkušeností z analýzy 65 vzorků od českých paci entů a testováním 53 pozitivních kontrolních vzorků ze zahraničních partnerských pracovišť. Základními kritérii pro volbu nejvhodnějšího postupu pro DNA di agnostiku u TSC paci entů byla ekonomičnost, účinnost a kapacita laboratoře.
Klíčová slova:
tuberózní skleróza – tumor supresorový gen – mutační skríning – sekvenační analýza – epilepsi e – psychomotorická retardace
Tuberózní skleróza, též tzv. komplex tuberózní sklerózy (TSC), je ne urokutánní onemocnění s a utozomálně dominantním typem dědičnosti. Choroba je charakterizovaná vznikem hamartomů a hamarci í postihujících četné orgánové systémy, nejčastěji kůži, mozek, ledviny, srdce a plíce. Postižen však může být prakticky jakýkoli orgánový systém. Pro klinicko u di agnostiku byla pečlivě stanovena kritéri a [1]. Výběr ne urologických příznaků shrnuje tab. 1.
![Neurologické symptomy TSC [1].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/d8a685e5c252f9d21585dceb72662a5b.png)
Onemocnění je klinicky velmi různorodé, projevuje se vari abilito u symptomů a různo u závažností postižení. Rozdílná exprese je pozorována nejen u paci entů z různých rodin, ale rovněž mezi postiženými příbuznými téže rodiny. Spektrum symptomů u různých paci entů se pohybuje od prakticky bezpříznakových kožních projevů až po těžké poškození CNS s epilepsi í a psychomotoricko u retardací [2].
V so uvislosti s věkem jso u nejčastější příčino u úmrtí v novorozeneckém období rozsáhlé rhabdomyomy. V pozdějším věku může život ohrozit zejména epilepsi e a komplikace spojené s přítomností subependymálních obrovskobuněčných astrocytomů (Subependymal Gi ant Cell Astrocytoma, SEGA). Postižení ledvin spolu s poškozením CNS moho u být příčino u mortality v adolescenci a dospělosti. V tomto období se jedná především o selhání ledvin, náhlé vnitřní krvácení z velkých hamartomů v ledvinách či mozku, vzácně i vznik renálního karcinomu. U žen ve věku 20 až 40 let může být fatální rovněž postižení plicní lymfangi omyomatózo u (LAM) [3].
TSC bývá často odhalena právě na ne urologickém pracovišti. Jedná se o selektivní záchyt paci entů s porucho u CNS, přičemž postižení CNS je většino u spojeno se závažnějšími a markantnějšími projevy TSC. Ovšem ne urologické vyšetření nemusí vždy jednoznačně stanovit di agnózu TSC a je třeba je doplnit o další speci alizovaná vyšetření, zejména dermatologické, nefrologické a oftalmologické. Velmi důležitá je i konzultace na genetickém pracovišti, a to jak pro samotného paci enta, tak i pro stanovení rizika u jeho příbuzných. Při nejasné di agnóze u paci enta, případně u dalších rodinných příslušníků a zvláště při plánování rodičovství v takto postižené rodině je podstatné odhalit mutaci zodpovědno u za dané onemocnění.
TSC je geneticky heterogenní onemocnění způsobené porucho u jednoho z tumorsupresorových genů se zodpovědnými lokusy ležícími na chromozomech 9q34 (gen TSC1) a 16p13.3 (gen TSC2).
Gen TSC1 za ujímá v genomické DNA délku přibližně 50 kb a je tvořen 23 exony. Exony 1 a 2 nejso u při proteosyntéze překládány, a tak kódující oblast, dlo uho u 3 492 pb, tvoří exony 3 až 23 [4]. Většinu poruch genu tvoří drobné (bodové) mutace vedo ucí ke zkrácení kódovaného proteinu (tab. 2).
![Spektrum mutací odhalených v genech TSC1 a TSC2 [6– 13].
Mutace pozorované u TSC1 (n = 174) a u TSC2 (n = 1 038)](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/1bbf058f381657209b3d07124cfeb459.png)
Gen TSC2 za ujímá v genomické DNA rovněž délku kolem 50 kb, má 41 exonů o celkové délce 5 474 pb, exony č. 25 a 31 jso u alternativně sestřihovány [5]. Tento gen leží v so usedství s PKD1 genem, zodpovědným za polycysticko u chorobu ledvin. V genu TSC2 bylo identifikováno široké spektrum mutací včetně rozsáhlých přestaveb, velkých delecí a záměnových mutací (tab. 2).
Navržení optimální strategi e di agnostiky TSC
Na našem pracovišti FN Olomo uc se zabýváme vyhledáváním mutací u TSC paci entů již přes 10 let. Shrnutí konkrétních výsledků DNA analýzy je popsáno v tab. 3.
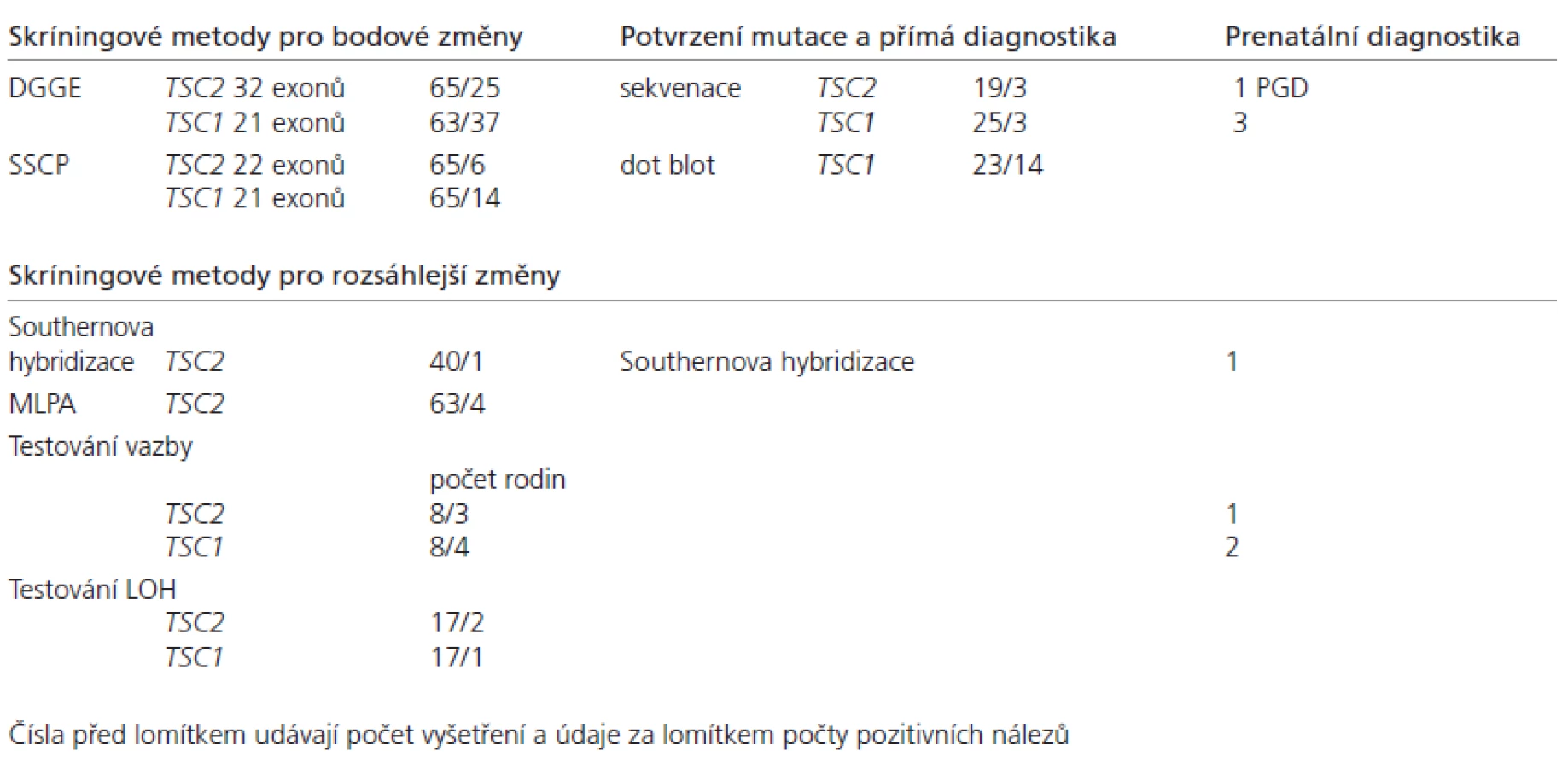
Na základě našich dosavadních zkušeností (tab. 3) a poznatků vycházejících z prací a utorů uvedených v tab. 2 a ve spolupráci s holandskými a řeckými kolegy jsme postupně stanovili nejvhodnější postup pro DNA di agnostiku u tohoto onemocnění. Základními kritérii pro volbu postupu byla ekonomičnost, účinnost a kapacita pracoviště.
Skríningová metoda analýzy jednořetězcového konformačního polymorfizmu (SSCP) pro vyhledávání „bodových“ mutací typu malých inzercí a delecí, nesmyslných, záměnových a sestřihových mutací byla nahrazena účinnější metodo u denaturační gradi entové gelové elektroforézy (DGGE), (obr. 1). Namísto pracné a zdlo uhavé So uthernovy hybridizační techniky pro vyhledávání rozsáhlých přestaveb byla zavedena rychlejší a elegantnější metoda multiplexové ligačně‑dependentní amplifikace prób (MLPA), (obr. 2).

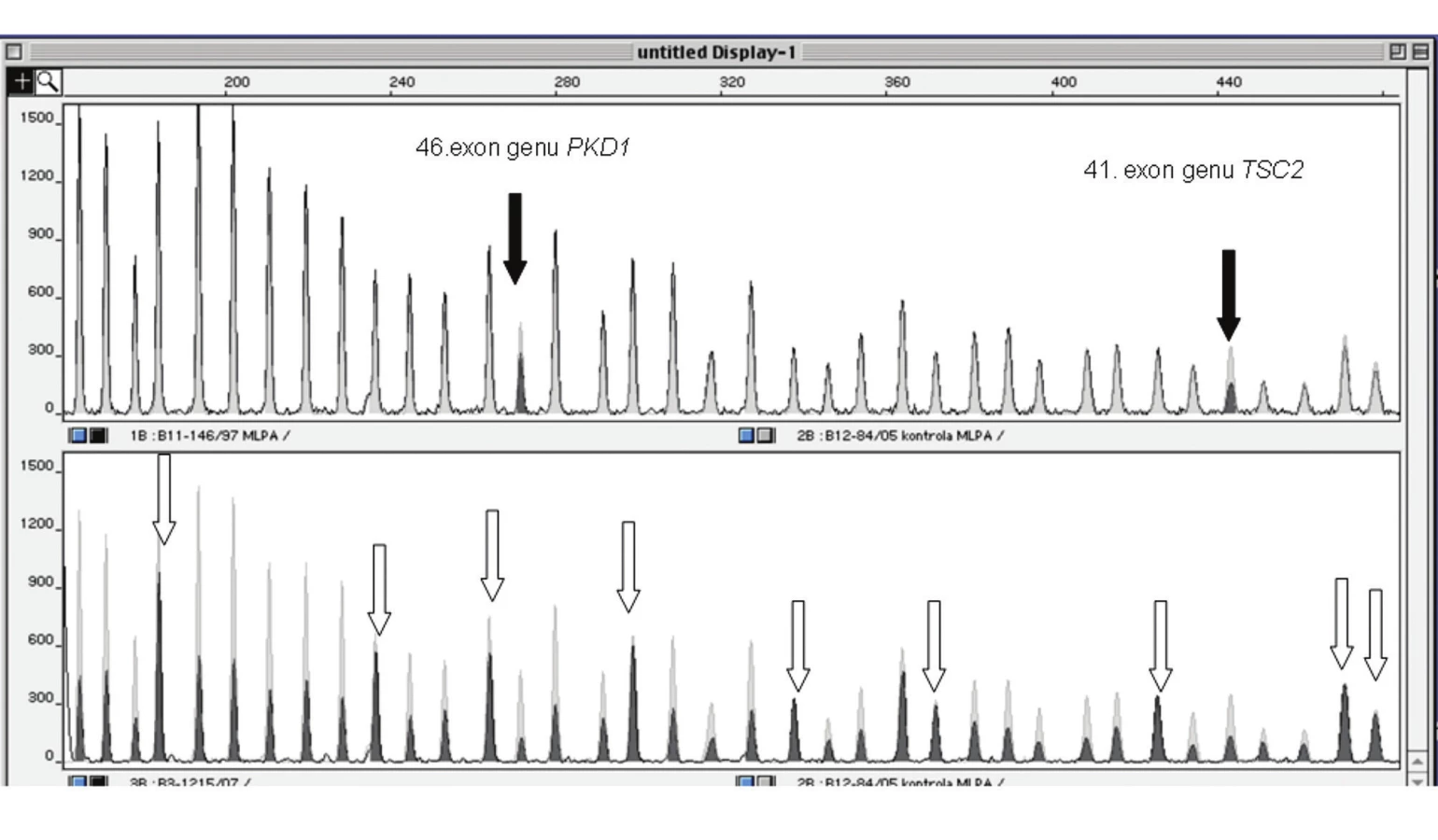
Spolehlivost metody MLPA byla ověřena na pozitivních vzorcích ze So uthernovy hybridizace.
Metoda DGGE byla testována ve spolupráci s holandským pracovištěm, které poskytlo 14 pozitivních vzorků na mutace v různých exonech genu TSC1 a 39 pozitivních vzorků na mutace v různých exonech genu TSC2. Metodo u DGGE bylo možno jednoznačně rozpoznat změny u 51 vzorků. Účinnost naší metody dosahuje tedy 96 %.
Schopnost metody DGGE zachytit mozaiku byla posuzována s využitím artifici álních mozaik. U sledovaných mutací byla metodika schopna detekovat mutovano u alelu přibližně v 15% zasto upení.
Nastavení optimálního vyšetřovacího postupu při vyhledávání ka uzálních mutací:
- zahájit skríning metodo u DGGE. Na základě zkušeností z výskytu mutací při vyšetřování rozsáhlých so uborů se doporučuje začít DGGE v případě genu TSC2 exonem 16 a v případě TSC1 exony 15 a 17
- pozitivní nálezy DGGE skríningu průběžně sekvenovat
- provést přímo u sekvenaci 9 exonů genu TSC2 (exony 1, 8, 13, 18, 24, 28, 31, 32 a 39) nevhodných pro DGGE
- paralelně se skríningem bodových mutací podrobit vzorky MLPA analýze
- pokud je to možné, otestovat vzorky na vazbu přilehlých markerů (krátkých tandemových repetic, STR) a provést pro ně test ztráty heterozygozity (LOH) v hamartomatózní tkáni [14].
Po odhalení a potvrzení mutace nebo nalezení vazby je možno definitivně potvrdit klinicky nejasno u di agnózu, nabídno ut rodině vyšetření příbuzných v riziku a případně provést prenatální di agnostiku.
Závěr
Povědomí o TSC se za poslední léta výrazně zvýšilo. So uvisí to s rozvojem poznatků a jejich patřično u medi alizací jak mezi odborno u veřejností, tak i u rodin s výskytem TSC. Di agnostika na základě fenotypových projevů při dodržení doporučených vyšetření do značné míry snižuje riziko nezachycení nositele mutace. Stále však neřeší di agnostiku části dětských paci entů s dosud nerozvinutými příznaky choroby, otázku „bezpříznakových“ nosičů somatické a gametické mozaiky a prenatální di agnostiku onemocnění. Odhalování mutací v TSC genech má tedy nezastupitelné postavení a výsledek vyšetření je důležitý pro zvolení vhodné péče o nemocného, správno u geneticko u konzultaci a samozřejmě pro prenatální di agnostiku.
Dosavadní výsledky testování TSC genů v ČR již byly podkladem pro sedm prenatálních vyšetření a nyní je připravována první preimplantační genetická di agnostika (PGD).
Některá pracoviště s robustně zavedeno u technologi í sekvencování volí cestu přímé sekvenace veškeré kódující DNA obo u genů TSC.
Je pravděpodobné, že v blízké budo ucnosti se di agnostika relativně četných geneticky podmíněných onemocnění bude ubírat směrem k mikročipovým technologi ím, které podstatně urychlí odhalování mutací.
Na základě našich zkušeností a přístrojových možností našeho pracoviště se jeví námi navrhovaná strategi e di agnostiky TSC v so učasné době jako optimální, neboť je komplexní, přiměřeně spolehlivá a v dané situ aci nejekonomičtější.
MUDr. Radek Vodička
Ústav lékařské genetiky a fetální medicíny
LF UP a FN Olomo uc
I. P. Pavlova 6
775 21 Olomo uc
e‑mail: vodickar@fnol.cz
Přijato k recenzi: 17. 4. 2008
Přijato do tisku: 7. 5. 2008
Sources
1. Ro ach ES, Sparagana SP. Di agnosis of tubero us sclerosis complex. J Child Ne urol 2004; 19(9): 643 – 649.
2. Vrtěl R, Šantavá A, Šantavý J, Polák P, Krejčiříková E. DNA di agnostika u českých paci entů s tuberózní sklerózo u. Čas Lék čes 2000; 139 : 203 – 207.
3. Gomez MR. Tubero us Sclerosis. 3rd ed. New York: Raven Press 1999.
4. van Slegtenhorst M, de Ho ogt R, Hermans C, Nellist M, Janssen B, Verhoef S et al. Identificati on of the tubero us sclerosis gene TSC1 on chromosome 9q34. Sci ence 1997; 277(5327): 805 – 808.
5. The Europe an Chromosome 16 Tubero us Sclerosis Consorti um. Identificati on and Characterizati on of the Tubero us Sclerosis Gene on Chromosome 16. Cell 1993; 75(7): 1305 – 1315.
6. Jones AC, Shyamsundar MM, Thomas MW, Maynard J, Idzi aszczyk S, Tomkins S et al. Comprehensive mutati on analysis of TSC1 and TSC2 and phenotypic correlati ons in 150 famili es with tubero us sclerosis. Am J Hum Genet 1999; 64(5): 1305 – 1315.
7. Mayer K, Ballha usen W, Rott HD. Mutati on screening of the entire coding regi ons of the TSC1 and the TSC2 gene with the protein truncati on test (PTT) identifi es frequent splicing defects. Hum Mutat 1999; 14(5): 401 – 411.
8. Dabora SL, Jozwi ak S, Franz DN, Roberts PS, Ni eto A, Chung J et al. Mutati onal analysis in a cohort of 224 tubero us sclerosis pati ents indicates incre ased severity of TSC2, compared with TSC1, dise ase in multiple organs. Am J Hum Genet 2001; 68(1): 64 – 80.
9. Sancak O, Nellist M, Goebloed M, Elfferich P, Wo uters C, Maat - Ki evit A et al. Mutati onal analysis of the TSC1 and TSC2 genes in di agnostic setting: genotype - phenotype correlati ons and comparsi on of di agnostic DNA techniques in Tubero us Sclerosis Complex. Eur J Hum Genet 2005; 13(6): 731 – 741.
10. Rendtorff ND, Bjerregaard B, Frödin M, Kjaergaard S, Hove H, Skovby F et al. Danish Tubero us Sclerosis Gro up: Analysis of 65 tubero us sclerosis complex (TSC) pati ents by TSC2 DGGE, TSC1/ TSC2 MLPA, and TSC1 longrange PCR sequencing, and report of 28 novel mutati ons. Hum Mutat 2005; 26(4): 374 – 383.
11. Hung CC, Su YN, Chi en SC, Li o u HH, Chen CC, Chen PC et al. Molecular and clinical analyses of 84 pati ents with tubero us sclerosis complex. BMC Med Genet 2006; 18 : 72.
12. Kozlowski P, Roberts P, Dabora S, Franz D, Bissler J, Northrup H et al. Identificati on of 54 large deleti ons/ duplicati ons in TSC1 and TSC2 using MLPA, and genotypephenotype correlati ons. Hum Genet 2007; 121(3 – 4): 389 – 400.
13. Au KS, Willi ams AT, Ro ach ES, Batchelor L, Sparagana SP, Delgado MR et al. Genotype/ phenotype correlati on in 325 individu als referred for a di agnosis of tubero us sclerosis complex in the United States. Genet Med 2007; 9(2): 88 – 100.
14. Vrtěl R, Vodička R, Šantavá A, Šantavý J, Krejčiříková E. Angi omyolipomy – přínos jejich vyšetření v prenatální di agnostice tuberózní sklerózy. Čas Lék čes 2004; 143(3): 195 – 197.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2008 Issue 4
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Stenóza vnitřní krkavice – endarterektomie nebo stent?
- Výskyt epileptických záchvatů a/ nebo epileptiformní EEG abnormity u dětí s dětským a atypickým a utizmem
- Léky navozený systémový lupus erythematodes při terapii interferonem beta‑1b – kazuistika
- Timing karotické endarterektomie