
Diagnostika a možnosti léčby Niemann-Pickovy choroby typ C
Diagnosis and Treatment Options for Niemann-Pick Disease Type C
Niemann-Pick disease type C is an autosomal recessive lysosomal disorder clinically characterized by age-specific symptomatology with possible manifestation any time from neonatal age to late adulthood. Except for its neonatal form with cholestasis, respiratory failure and hepatosplenomegaly, the disease presents as a neurodegenerative disorder, frequently with splenomegaly. Pathophysiology involves dysfunction of the late endosome/lysosome membraneous system with accumulation of unesterified cholesterol and glycosphingolipids due to mutations in the NPC1 and NPC2 genes coding the corresponding lysosomal proteins. Results of clinical examination must be confirmed by specific loading tests in cultivated fibroblasts and/or molecular genetic analysis of the concerned genes. In the last years the disease is treated with a reversible glucosylceramide synthase inhibitor miglustat, potentially stabilizing its course. Recently, clinical testing of cyclodextrin has been initiated. In the Czech Republic, comprehensive diagnostics is available at The Institute of Inherited Metabolic Disorders (67 confirmed patients including 42 Czechs).
Key words:
Niemann-Pick disease type C – unesterified cholesterol – NPC1 gene – NPC2 gene – miglustat – cyclodextrin
:
H. Jahnová 1,2; L. Dvořáková 1; H. Hůlková 1; M. Hřebíček 1; P. Ješina 1
:
Ústav dědičných metabolických poruch 1. LF UK a VFN v Praze
1; Klinika dětí a dorostu 3. LF UK a FN Královské Vinohrady v Praze
2
:
Cesk Slov Neurol N 2012; 75/108(3): 303-308
:
Review Article
Niemann-Pickova choroba typ C je autozomálně recesivně dědičné lysozomální onemocnění, klinicky charakterizované věkově specifickou symptomatologií, s možností manifestace od novorozeneckého věku do pozdní dospělosti. S výjimkou neonatální formy s cholestázou, respiračním selháním a hepatosplenomegalií jde v dalších věkových kategoriích o nemoc vysloveně neurodegenerativní, často doprovázenou splenomegalií. Příčinou je funkční porucha membránového systému pozdního endozomu/lysozomu s akumulací neesterifikovaného cholesterolu a glykosfingolipidů v důsledku mutací v genech NPC1 (95 % případů) nebo NPC2 (5 % případů), kódujících lysozomální membránový protein NPC1 a solubilní protein NPC2. Základem diagnostiky je správné zhodnocení klinických symptomů s přihlédnutím k věku nemocného. Podezření potvrdí specifické testy v tkáňové kultuře fibroblastů a/nebo molekulárně genetická analýza uvedených genů. Aktuální možnost léčby spočívá v podávání reverzibilního inhibitoru glukosylceramidsyntázy (miglustat), potenciálně stabilizujícího průběh nemoci, experimentálně slibná aplikace cyklodextrinu vstupuje do fáze klinického testování. Komplexní diagnostika je v ČR dostupná prostřednictvím Ústavu dědičných metabolických poruch (diagnóza stanovena u 67 pacientů, z toho 42 českých).
Klíčová slova:
Niemann-Pickova choroba typ C – neesterifikovaný cholesterol – gen NPC1 – gen NPC2 – miglustat – cyklodextrin
Práci věnujeme prof. MUDr. Milanu Ellederovi, DrSc. (5. 12. 1938–24. 9. 2011), jednomu z předních evropských odborníků na problematiku lysozomálních chorob, dlouholetému přednostovi Ústavu dědičných metabolických poruch 1. LF UK a VFN v Praze a výjimečnému člověku. Rádi bychom panu profesorovi poděkovali za obrazovou dokumentaci pro tento článek, zejména však za inspiraci a podporu, kterou všem autorům dlouhá léta poskytoval.
Děkujeme rovněž MUDr. Věře Malinové z KDDL 1. LF UK a VFN v Praze, která zajišťuje léčebný program Zavescou® u našich pacientů s Niemann-Pickovou chorobou typ C, a prof. MUDr. Soně Nevšímalové, DrSc., za spolupráci v péči o tyto pacienty.
Úvod
Rozvoj diagnostiky a možnost potenciálně stabilizující léčby podnítily v posledních letech zájem o problematiku Niemann-Pickovy choroby typ C (NPC). Výsledkem mezinárodní odborné spolupráce je mimo jiné podrobná klinická charakterizace nemoci [1–4], i návrh „guidelines“ pro diagnostiku a eventuální léčbu [5]. Díky lepší informovanosti kliniků a dostupnosti diagnostických metod je správná diagnóza stanovena u stále většího množství pacientů, takže původně odhadovaná panetnická incidence NPC 1 : 150 000 se ukazuje jako podhodnocená. Incidence v ČR je podle počtu pacientů diagnostikovaných na našem pracovišti 1 : 109 000 [6], přesto jsme přesvědčeni, že u řady pacientů, zejména s adultní formou nemoci, ke stanovení správné diagnózy nedojde, což mimo jiné znamená, že postiženým rodinám není možné nabídnout efektivní genetické poradenství a do budoucna nelze uvažovat ani o potenciální léčbě.
Definice a patofyziologie
Definice
Původní název Niemann-Pickova choroba označoval skupinu nemocí s projevy střádání v retikuloendoteliálním systému a většinou (ne vždy) s projevy neurodegenerace. Studie na tkáňových kulturách a modelových organizmech ukázaly, že Niemann-Pickova choroba typ A/B je typické střádavé onemocnění způsobené deficitem lysozomální hydrolázy – kyselé sfingomyelinázy. Naproti tomu Niemann-Pickova choroba typ C (MIM 257220 a MIM 607625) je patofyziologicky i klinicky samostatná jednotka charakterizovaná na subcelulární úrovni poruchou transportu cholesterolu s hromaděním neesterifikovaného cholesterolu v lysozomu/pozdním endozomu [1–3,7,8]. Uvedené studie, potvrzené později i molekulárně geneticky, rovněž prokázaly, že dříve uváděná Niemann-Pickova choroba typ D (tzv. typ Nova Scotia) neexistuje. Jde o relativně izolovanou populaci pacientů s Niemann-Pickovou chorobou typ C s identickým genotypem na podkladě tzv. efektu zakladatele (mutace p.G992W v NPC1 genu).
Patofyziologie
Patofyziologický mechanizmus NPC není stále plně objasněn. Podle současných poznatků je příčinou nemoci funkční deficit membránového proteinu NPC1 (asi v 95 % případů) nebo solubilního lysozomálního proteinu NPC2 (v méně než 5 % případů, převážně u neonatálních nebo časně infantilních forem nemoci). Uvedené proteiny se ve vzájemné spolupráci podílejí v extraneurálních tkáních na intracelulárním metabolizmu převážně exogenního cholesterolu, endocytovaného prostřednictvím LDL receptoru, umožňují jeho transport z lysozomu/pozdního endozomu do endoplazmatického retikula, Golgiho aparátu a na plazmatickou membránu. Projevem deficitu NPC proteinů v tkáňové kultuře fibroblastů je akumulace neesterifikovaného cholesterolu v perinukleárních lysozomech prokazatelná fluorescentním barvením filipinem (obr. 1). V lysozomech neuronů se výrazněji než cholesterol hromadí glykosfingolipidy, zejména GM2 a GM3 gangliosid, ale také sfingosin/sfinganin a fosfolipidy. I zde je však primární poruchou defekt intracelulárního transportu neesterifikovaného cholesterolu, na rozdíl od periferie převážně endogenního původu. Výzkum zaměřený na objasnění patofyziologie NPC stále pokračuje. Jeho výsledky lze zjednodušeně interpretovat v tom smyslu, že se v patogenezi neurodegenerace u NPC uplatňuje nejen lysozomální střádání, ale zejména porucha intracelulárních regulačních mechanizmů navozená insuficiencí lysozomálního exportu cholesterolu, fungujícího v buňce jako důležitá signální molekula. Jde o procesy zajišťující normální funkci membránového systému neuronů, ale také jejich proliferaci a/nebo apoptózu. V současnosti probíhá na tkáňových a myších modelech NPC řada studií zaměřených jednak na identifikaci těchto mechanizmů, jednak na využití různých postupů, jejichž cílem je kompenzovat deficit NPC proteinů, a tak zlepšit funkci neuronu [7–15].

Histopatologie
Morfologickým korelátem poruchy jsou neuropatologické nálezy meganeuritů a extenzivního růstu ektopických dendritů s maximem v některých specifických oblastech centrálního nervového systému (CNS), zejména v Purkyněho buňkách mozečku. Ultramikroskopicky je prokazatelné v neuronech i glii určitých oblastí CNS nakupení denzních neurofibril. Tento nález není specifický pro NPC, u jiných neurodegenerativních poruch, např. Alzheimerovy demence, která ultramikroskopicky NPC připomíná, je však maximum denzních neurofibril v jiných oblastech [10].
Molekulární genetika
Transmembránový NPC1 glykoprotein je kódován rozsáhlým NPC1 genem (18q11–q12, 25 exonů), ve kterém bylo až dosud popsáno více než 300 kauzálních mutací (databáze HGMD) a 60 polymorfizmů měnících proteinový zbytek. Malý solubilní NPC2 protein je kódován NPC2 genem (14q24.3, 5exonů) s 20 popsanými sekvenačními variantami (databáze HGMD). U některých pacientů s nepochybným klinickým a biochemickým fenotypem NPC nebyly mutace na obou alelách NPC1 či NPC2 genu prokázány, což vede k úvaze o možné účasti dalších genů kódujících proteiny nezbytné pro normální subcelulární transport cholesterolu [3].
Klinický obraz
Obecná charakteristika
Heterogenita klinického obrazu společně s absencí jednoduše vyšetřitelného a spolehlivého laboratorního markeru jsou zřejmě hlavní příčinou obtížné diagnostiky. Doufáme, že znalost klinických projevů nemoci v jednotlivých věkových údobích a informace o dostupných diagnostických metodách mohou tuto situaci zlepšit.
Jde o neuroviscerální onemocnění, kde se na jednom pólu klinického spektra nachází neonatální forma s fatálním respiračně hepatálním postižením, na druhém adultní forma, řadu let probíhající jako poměrně mírná psychiatrická porucha nebo jako asymptomatická splenomegalie. Jakmile se u pacienta s NPC začne rozvíjet neurologická symptomatologie, je nemoc zatím vždy letální, přičemž čím nižší je věk rozvoje neurologických příznaků, tím je průběh nemoci rychlejší.
Didakticky lze klinické spektrum nemoci rozdělit na formu neonatální, časně infantilní, pozdně infantilní, juvenilní (klasickou), adolescentní a adultní. S výjimkou neonatální formy je pro klinické zařazení rozhodující věk rozvoje neuropsychiatrické symptomatologie, nikoliv věk detekce jinak asymptomatické splenomegalie.
Neonatální (perinatální) forma
Je většinou diagnostikována neonatology/pediatry (pokud na ni pomyslí). Může být přítomen hydrops plodu, typický je prolongovaný cholestatický ikterus, který může po druhém měsíci regredovat, ale trvá hepatosplenomegalie. Asi 10 % těchto pacientů umírá, část z nich (s mutacemi v NPC2 genu) pod obrazem těžké respirační insuficience. Histopatologicky byla u těchto pacientů prokázána akumulace lipoproteinů v plicních alveolech [3]. Neurologická symptomatologie není u těchto pacientů popisována. U 90 % přežívajících kojenců trvá po odeznění cholestázy organomegalie, která je po různě dlouhé době následována rozvojem neurodegenerativních příznaků typických pro jednotlivé věkové formy nemoci.
Časně infantilní forma
Je charakterizována rozvojem neurologické symptomatologie mezi dvěma měsíci až dvěma lety věku. Zpočátku nenápadné zpomalení psychomotorického vývoje (PMV) s centrální hypotonií je doprovázeno hepatosplenomegalií. Během druhého roku věku je již patrný regres PMV s rozvojem spasticity a cerebellární symptomatologií, někdy poruchou sluchu. Vertikální supranukleární pohledová obrna (Vertical Supranuclear Gaze Palsy, VSGP) je někdy špatně rozpoznatelná. Na rozdíl od neuronálních ceroidlipofuscinóz typu 1 a typu 2 (NCL1, NCL2) nebývají epileptické záchvaty. Většina dětí umírá do pěti let věku.
Pozdně infantilní forma
Rozvoj neurologické symptomatologie nastává mezi 2 a 6 lety, část dětí je již předtím sledována pro splenomegalii. Častý je pomalejší rozvoj řeči, neobratnost, nejistá chůze. VSGP může na počátku ujít pozornosti pediatra i neurologa, je totiž často patrná jen při rychlých fixačních pohybech očí (Saccadic Eye Movements, SEM). Někdy je prvním nápadnějším příznakem kataplexie, následovaná mentální deteriorací a rozvojem epilepsie. V odstupu 1–2 let se rozvíjí spasticita, výrazná dysfagie a dysartrie. Většinou je nutná gastrostomie. K úmrtí zpravidla dochází v důsledku sekundárních, hlavně respiračních komplikací na přelomu 1. a 2. životní dekády.
Juvenilní (klasická) forma
Neurologické symptomatologii, která se objeví mezi 6 a 15 lety (školní věk), předchází spleno - nebo hepatosplenomegalie, někdy od novorozeneckého věku. Asi 10 % pacientů však organomegalii nemá. Onemocnění začíná často velmi nenápadně jako porucha pozornosti a aktivity, specifické poruchy učení, dyspraxie, mírná porucha artikulace. U řady pacientů mohou být tyto projevy stacionární asi do 12–15 let, kdy se rychle rozvíjí závažnější neurologické příznaky. V tomto stadiu lze poměrně brzy odhalit VSGP, ale nemusí být patrná v každé denní době (častější je po ránu) nebo ve všech polohách (vsedě, vleže, ve stoji). Zpočátku mohou váznout pouze rychlé fixační pohyby očí všemi směry. Běžná je kataplexie indukovaná smíchem, někdy s narkolepsií. Následuje rychle progredující ataxie, dysfagie, dále se zhoršuje dysartrie, rozvíjí se akční dystonie, nápadná při chůzi. Mohou se objevit epileptické záchvaty, obvyklá je postupná nerovnoměrná mentální deteriorace, posléze spasticita i těžší dystonie s dyskinézami. Těžké poruchy polykání při progresi nemoci často vyžadují gastrostomii. Délka přežití se odvíjí od kvality symptomatické péče a závažnosti sekundárních komplikací, například v souvislosti s aspirací.
Adolescentní a adultní forma
Do této skupiny jsou zařazováni pacienti s rozvojem neurologické, přesněji neuropsychiatrické symptomatologie po 15. roce věku. U většiny adolescentních forem je průběh podobný jako u formy juvenilní, pouze s pozdějším nástupem neurologických příznaků mezi 15. a 18. rokem, poměrně vzácná je kataplexie. Specifickou skupinu tvoří pacienti s adultní formou. Mnoho z nich je po řadu let v péči psychiatrů pod diagnózou schizofrenní či maniodepresivní psychózy. Průběh může být progresivní, ale také velmi mírný nebo s relapsy. Často je popisována špatná tolerance neuroleptik a katatonie. Z dalších psychiatrických poruch jsou uváděny poruchy spánku, agresivita, anxiózně-depresivní syndrom, obsedantně-kompulzivní porucha nebo přechodné zrakové halucinace. Po několika, někdy i více než 10 letech, dochází k relativně rychlému rozvoji neurologické symptomatologie – cerebellární ataxie, dysartrie, dysfagie, akční dystonie. VSGP je většinou přítomna, ale nemusí být prokazatelná v každé denní době, v každé poloze, častěji je možné ji zaznamenat při rychlých fixačních pohybech očí. Někteří pacienti ani při pokročilé ataxii a dystonii VSGP nemají. Mentální deteriorace má specifický charakter s rychlým a výrazným postižením exekutivních funkcí a relativně déle uchovanou kognicí. Splenomegalie nemusí být přítomna, u některých pacientů se splenomegalie vyskytla v dětství, ale v době rozvoje neuropsychiatrické symptomatologie není prokazatelná. Byli také popsáni náhodně diagnostikovaní dospělí pacienti s NPC pouze s izolovanou splenomegalií, bez známek neuropsychické deteriorace [16]. Od rozvoje neurologických příznaků i u dospělých pacientů onemocnění poměrně rychle progreduje k obrazu známému u pozdně juvenilních a adolescentních forem nemoci.
Další klinická a paraklinická vyšetření
Elektrofyziologie
Elektrofyziologické metody většinou k diagnóze výrazněji nepřispějí. Užitečné mohou být spíše pro monitorování průběhu nemoci. V elektroencefalogramu je možné v symptomatickém období nemoci nalézt nespecifickou i epileptiformní abnormitu, evokované kmenové potenciály bývají u symptomatických pacientů patologické. Vzácně byla u dospělých pacientů popsána i elektromyograficky potvrzená periferní neuropatie.
Zobrazení CNS
MR mozku může být zcela normální nebo s nevýraznou cerebellární a kortikální atrofií. Pouze u rychle probíhající časně infantilní formy byly popsány leukodystrofické změny s výraznější atrofií.
U diagnostikovaných pacientů s NPC je možné využít některé cílené metody MR (volumometrie, DTI) k monitorování průběhu nemoci a efektu terapie [17].
Také protonová MR spektroskopie může být užitečná stanovením poměrů cholin/kreatin a N-acetylaspartát/kreatin ve specifických oblastech mozku jak v diagnostice, tak při monitorování průběhu nemoci.
Ultrasonografie
Ultrazvukové vyšetření břicha by mělo proběhnout u každého pacienta s podezřením na neurodegenerativní onemocnění. U většiny dětí s NPC prokážeme stacionární hepatosplenomegalii nebo izolovanou splenomegalii se známkami střádání. Absence splenomegalie, zejména u dospívajících a dospělých pacientů však diagnózu nevylučuje.
Oftalmologické vyšetření
Zkušený oftalmolog by měl zaznamenat poruchu (opožďování) rychlých fixačních pohybů očí (sakád) při jinak normálním oftalmoskopickém nálezu, často však ujde při standardním vyšetření pozornosti. Pokročilá vertikální pohledová obrna (většinou dříve směrem dolů) by měla být detekovatelná jak při oftalmologickém, tak neurologickém vyšetření. Retinopatie do obrazu NPC nepatří.
Psychologické vyšetření
Opakované psychologické, eventuálně neuropsychologické vyšetření odhalí zejména u juvenilních, adolescentních a adultních pacientů nerovnoměrnou deterioraci mentálních schopností v popředí s časným postižením exekutivních funkcí. Prvním příznakem u dospělých pacientů může být ztížená adaptace na nové požadavky v zaměstnání, problémy s rekvalifikací, apod. Senzitivitu psychologického vyšetření ovlivňuje vhodný výběr testovacích nástrojů a zkušenost psychologa.
Biochemické nálezy
Standardní biochemické nálezy jsou většinou nespecifické s výjimkou nálezů typických pro cholestázu u neonatální formy. U řady pacientů je snížená koncentrace HDL cholesterolu, možná je mírná elevace AST a u pacientů se splenomegalií bývá prokazatelná mírná trombocytopenie.
Nespecifickým, ale užitečným a nenáročným testem je vyšetření aktivity chitotriosidázy v plazmě nebo séru. Zvýšená hodnota svědčí pro onemocnění s aktivací retikuloendoteliálního systému, extrémně vysoké hodnoty jsou spíše typické pro Gaucherovu nemoc. Mírně až středně zvýšené hodnoty nacházíme u většiny dětských pacientů s NPC, normální hodnota v žádném případě onemocnění nevylučuje. Asi u 6 % populace nelze test pro vrozeně nízkou aktivitu chitotriosidázy využít [18].
Nátěr kostní dřeně
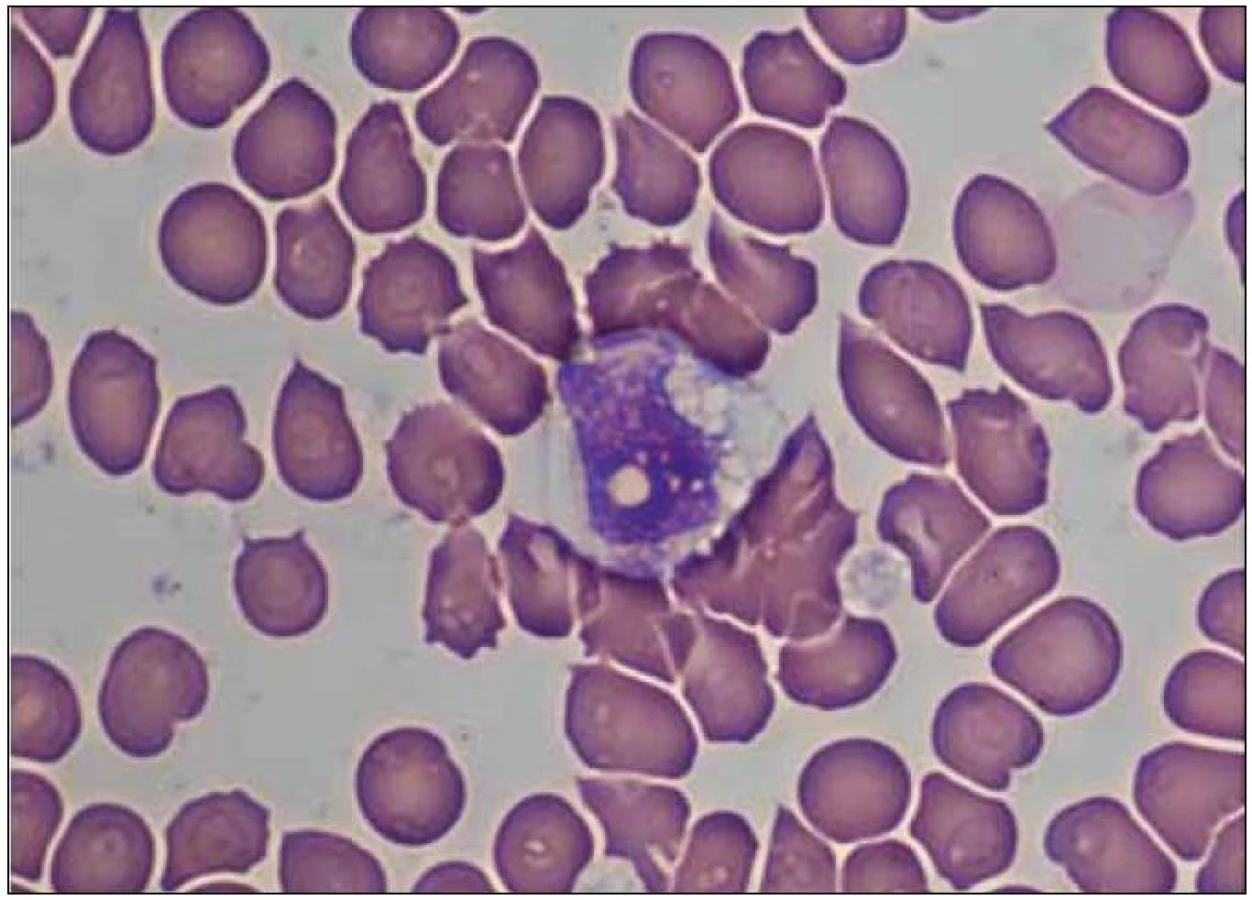
Pokud jsou v nátěru kostní dřeně (cílená indikace nebo je zákrok proveden hematology pro chronickou splenomegalii) prokázány specifické „sea-blue“ histiocyty (obr. 2), je diagnóza NPC vysoce suspektní a je také zpravidla potvrzena zátěžovými testy v kultivovaných fibroblastech a/nebo DNA diagnostikou (viz další text). Nátěr může také obsahovat pěnové buňky, ve kterých lze prokázat akumulaci neesterifikovaného cholesterolu při barvení filipinem (jde však o specifické histochemické vyšetření, nikoliv o běžně dostupnou diagnostickou metodu). Specifické histiocyty se dají prokázat i v bioptickém materiálu ze sleziny, jater, plic a lymfatických uzlin. Vzhledem k riziku nezastižení specifických buněk v konkrétním vzorku negativita nálezu zcela diagnózu NPC nevylučuje.
Elektronoptické vyšetření tkání
Pokud je u pacientů (nejčastěji u dětí s cholestatickým ikterem) provedena jaterní nebo při podezření na NCL kožní biopsie, vždy by měla být část tkáně adekvátně fixována ve 4% paraformaldehydu pro elektronoptické vyšetření, realizované zkušeným patologem, které může u NPC prokázat patognomická polymorfní cytoplazmická tělíska (PCB). Jejich nález je rovněž indikací k přímé sekvenaci NPC genů.
Stanovení diagnózy NPC
Specifické testy v tkáňové kultuře fibroblastů
Jde o filipinový test a test rychlosti esterifikace značeného LDL-cholesterolu. U filipinového testu je do vzorku tkáňové kultury fibroblastů rozpěstovaných v médiu bez lipoproteinu za standardizovaných podmínek přidán LDL cholesterol. Typická perinukleární akumulace neesterifikovaného cholesterolu je pak vizualizována antibiotikem filipinem, který se intenzivně váže na cholesterol a je dobře prokazatelný při použití fluorescenčního mikroskopu (obr. 1). Test rychlosti esterifikace značeného LDL cholesterolu detekuje množství esterifikovaného LDL cholesterolu za 4 nebo 6 hod ve fibroblastech pacienta ve srovnání s kontrolním vzorkem. Je považován za méně specifický ve srovnání s filipinovým testem.
V případě jednoznačně patologického výsledku zejména filipinového testu hovoříme o klasickém biochemickém fenotypu NPC (80–85 % pacientů). Asi 15–20 % případů se prezentuje pod obrazem variantního biochemického fenotypu NPC, kdy hodnocení nálezů není zcela jednoznačné (častěji u pozdních forem nemoci).
Molekulárně genetické vyšetření NPC1 a NPC2 genu
Mutace v NPC1 genu (18q11–q12) se nachází asi u 95 % pacientů s NPC. Analýza se většinou provádí přímým sekvenováním. V současnosti je popsáno více než 300 mutací. Jednou z častějších je v evropské populaci mutace p.I1061T, jejíž přítomnost alespoň na jedné alele většinou podmiňuje pomalejší průběh nemoci. Nejčastější mutací v populaci ČR je p.R1186H, která je spojována s klasickým biochemickým fenotypem při provedení filipinového testu [19].
Méně než 5 % případů nemoci je podmíněno mutacemi v NPC2 (HE1) genu. Jde převážně o pacienty s těžkou neonatální formou, často letální pod obrazem respiračního a/nebo jaterního selhání, nebo o pacienty s časně infantilní formou s rychle progredující neurologickou symptomatologií. Asi u poloviny pacientů byla nalezena mutace p.E20X.
Vývoj nových diagnostických metod
Absence jednoduše vyšetřitelného biochemického markeru nemoci je jednou z příčin její pozdní a nedostatečné diagnostiky. Chybí rovněž spolehlivá a snadno realizovatelná biochemická metoda použitelná pro monitorování efektu léčby. Průlom v této oblasti slibuje zavedení biochemické analýzy profilu oxysterolů (LC-MS/MS) [20,21].
Rozšíření diagnostických možností lze očekávat i od sekvenování nové generace (NGS). Tuto metodiku ve formě panelu zaměřeného na neurodegenerativní onemocnění, včetně NPC, zavádíme i na našem pracovišti.
Genetické poradenství a prenatální diagnostika
NPC je onemocnění s autozomálně recesivní dědičností. Pokud je v rodině dítě s NPC, každý z jeho biologických rodičů by měl nést patogenní variaci (mutaci) pro NPC na jedné alele. Z každého těhotenství těchto partnerů se může s 25% pravděpodobností narodit dítě s NPC. Pokud je v rodině znám patologický genotyp v některém ze dvou známých genů, je možné v každém těhotenství realizovat prenatální vyšetření v buňkách choriových klků a/nebo kultivovaných amniocytech cíleným stanovením familiárně se vyskytujících mutací. V konkrétních případech je možná i preimplantační diagnostika, předpokladem je prekoncepční vyšetření rodiny na specializovaném pracovišti, které se touto problematikou zabývá. Velkým geneticko-psychosociálním problémem u NPC je skutečnost, že se nemoc často manifestuje až v adolescenci nebo mladé dospělosti, takže v některých případech může být postiženo i více členů jedné rodiny a vzhledem k věku rodičů již další reprodukce nepřichází v úvahu.
Riziko přenašečství (heterozygocie) u sourozenců rodičů je 50 %, u zdravých sourozenců pacientů s NPC 67 %. V současnosti je možné nabídnout členům rodin s NPC vyšetření přítomnosti familiárně se vyskytujících mutací. Populační frekvence patogenních alel NPC1 genu je podobně jako u jiných lysozomálních onemocnění považována za velmi nízkou, avšak některá dosud nepublikovaná data z našeho souboru pacientů a přenašečů pro NPC vedou k podezření, že frekvence patogenních alel NPC1 genu je v populaci ČR vyšší, než se předpokládá.
Možnosti terapie a management
Specifická léčba
Pokusy o ovlivnění průběhu nemoci dietou a léky snižujícími koncentraci cholesterolu nebyly úspěšné stejně jako pokusy s transplantací kostní dřeně. V současnosti je již několik set pacientů ve světě léčeno perorálně podávaným miglustatem, obsahujícím látku deoxynojirinomycin. Jde o malou molekulu reverzibilně blokující syntézu glukosylceramidu, a tím sfingolipidů. Menší nabídka substrátu (sfingolipidů) v lysozomech vede k menší míře střádání, což by mělo zpomalit nebo stabilizovat průběh nemoci (tzv. substrát redukující terapie). Efekt uvedeného terapeutického postupu je zatím obtížné hodnotit (relativně krátká doba podávání u pacientů v různém klinickém stadiu nemoci), nicméně jsou již popsáni pacienti, u kterých se zdá terapie miglustatem úspěšná [22–25]. Podle očekávání jde o pacienty s pozdějším nástupem neurologické symptomatologie a pomalejším průběhem nemoci. Od srpna 2011 bylo zahájeno podávání miglustatu (Zavesca®) u skupiny 14 českých pacientů, léčebný program je řízen MUDr. Věrou Malinovou (KDDD 1. LF UK a VFN v Praze).
Novým nadějným léčebným postupem je subkutánní aplikace 2-hydroxypropyl-beta-cyklodextrinu a jeho homologů. Léčebné možnosti aplikace cyklodextrinu byly náhodně zjištěny při testování léčebného efektu allopregnanolonu na myším modelu, kdy byl použit jako vehikulum. Předpokládá se, že cyklodextrin může pomoci obejít blok v transportu neesterifikovaného cholesterolu z lysozomu způsobený poruchou funkce NPC1 proteinu, a tím umožnit mimo jiné jeho vstup do endoplazmatického retikula (ER), kde hraje významnou roli jako signální molekula pro tzv. SREBPs (Sterol Regulatory Element Binding Proteins). Přes problémy s aplikací (krátký biologický poločas) již v zahraničí probíhá experimentální léčba u několika pacientů se slibným efektem [26–28].
Ve fázi výzkumu jsou další léčebné možnosti, například testování efektu curcuminu (ovlivnění intracelulární homeostázy kalcia) a využití inhibitorů specifických izoforem histonových deacetyláz [14,15].
Symptomatická léčba
Většina symptomatických pacientů vyžaduje péči neurologa (podle klinického stavu jsou podávána antiepileptika, léky k ovlivnění kataplexie, někdy anticholinergika, lokálně botuloxin apod.) a psychiatra (podávání neuroleptik, antidepresiv). Progrese nemoci vede k závažným poruchám polykání s rizikem aspirace a k malnutrici, u řady pacientů v pokročilém stadiu nemoci je proto nutná gastrostomie. Důležitá je včasná a přiměřeně intenzivní terapie respiračních infekcí (antibiotika, bronchodilatancia). Nezbytná je adekvátní udržovací fyzioterapie, masáže hrudníku, využití drenážních poloh, důležité je vybavení pacientů adekvátními kompenzačními pomůckami. Velmi podstatnou součástí komplexní péče je přiměřená psychosociální podpora rodiny – jak vlastní fyzická péče, tak její psychická komponenta představují enormní a dlouhodobou zátěž pro členy rodiny, kteří o nemocné pečují (většinou rodiče).
Monitorování průběhu nemoci
Pro sledování průběhu nemoci a hodnocení efektu terapie byly vytvořeny dva skórovací systémy [29,30], jejichž použitelnost testujeme také na našem souboru pacientů. Obě škály pracují převážně s klinickými ukazateli, které se zatím jeví i v našem souboru pro hodnocení progrese nemoci jako nejpoužitelnější.
Závěr
NPC je závažné neuroviscerální onemocnění, podle našich zkušeností pravděpodobně v populaci ČR poddiagnostikované, v současnosti částečně léčebně ovlivnitelné, nicméně s perspektivou účinnější léčby v blízké budoucnosti. V ÚDMP 1. LF UK a VFN v Praze jsme schopni nabídnout komplexní diagnostiku nemoci – klinické zhodnocení stavu, vyšetření aktivity chitotriosidázy, zhodnocení nátěru kostní dřeně zkušeným patologem, DNA i RNA analýzu NPC1 a NPC2 genů. Zátěžové testy v kultivovaných fibroblastech sami neprovádíme, pokud se nám však nepodaří stanovit diagnózu molekulárně genetickými metodami, zajišťujeme jejich provedení na spolupracujících zahraničních pracovištích. V současnosti zavádíme i diagnostiku prostřednictvím NGS. Ve spolupráci s KDDL 1. LF UK a VFN v Praze je možné pacientům poskytnout jak dostupnou léčbu (pokud bude zajištěno její financování), tak všestranné sledování a genetické poradenství.
V posledních 35 letech bylo na našem pracovišti na různých úrovních diagnostikováno celkem 67 pacientů z několika zemí (ČR, SR, Polsko, Německo, Brazílie). Ze 42 dosud diagnostikovaných českých pacientů byla diagnóza na molekulárně genetické úrovni stanovena u 33. Komplexně zpracovaná data o našem souboru pacientů s NPC bychom rádi publikovali v nedaleké budoucnosti.
Práce vznikla z podpory grantu Interní grantové agentury Ministerstva zdravotnictví ČR IGA NT/12239-5 a grantového projektu RVO-VFN64165/2012.
MUDr. Helena Jahnová
Ústav dědičných metabolických poruch
1. LF UK a VFN v Praze
Ke Karlovu 2
128 08 Praha 2
e-mail: helena.jahnova@vfn.cz
Přijato k recenzi: 4. 4. 2012
Přijato do tisku: 17. 5. 2012
Sources
1. Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea A, Neufeld EB et al. Niemann-Pick disease type C: a lipid trafficking disorder. In: Scriver CR, Beaudet AL et al (eds). The Metabolic and Molecular Bases of Inherited Diseases. 8th ed. New York: McGraw Hill 2001 : 3611–3634.
2. Wraith JE, Guffon N, Rohrbach M, Hwu WL, Korenke GC, Bembi B et al. Natural history of Niemann-Pick disease type C in a multicentre observational retrospective cohort study. Mol Genet Metab 2009; 98(3): 250–254.
3. Vanier MT. Niemann-Pick disease type C. Orphanet Journal of Rare Diseases [online]. 3 June 2010. Available from: http://www.ojrd.com/content/5/1/16.
4. Sévin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT et al. The adult form of Niemann-Pick disease type C. Brain 2007; 130(Pt 1): 120–133.
5. Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, Mengel E et al. NP-C Guidelines Working Group. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab 2009; 98(1–2): 152–165.
6. Poupetová H, Ledvinová J, Berná L, Dvoráková L, Kozich V, Elleder M. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 2010; 33(4): 387–396.
7. Pentchev PG, Comly ME, Kruth HS, Vanier MT, Wenger DA, Patel S et al. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc Natl Acad Sci USA 1985; 82(23): 8247–8251.
8. Sturley LS, Patterson MC, Balch W, Liscum L. The pathophysiology and mechanism of NP-C disease. Biochim Biophys Acta 2004; 1685(1–3): 83–87.
9. Patterson MC. A riddle wrapped in a mystery: understanding Niemann-Pick disease, type C. Neurologist 2003; 9(6): 301–310.
10. Paul CA, Boegle AK, Maue RA. Before the loss: neuronal dysfunction in Niemann-Pick type C disease. Biochim Biophys Acta 2004; 1685(1–3): 63–76.
11. Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 2008; 9(2): 125–138.
12. Bartz F, Kern L, Erz D, Zhu M, Gilbert D, Meinhof T et al. Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Met 2009; 10(1): 63–75.
13. Ximing D, Hongyuan Y. Sterol-binding proteins and endosomal cholesterol transport. Front Biol 2011; 6(3): 190–196.
14. Helquist P, Wiest O. Current status of drug therapy development for Niemann-Pick type C disease. Drugs Fut 2009; 34 : 315–331.
15. Pipalia NH, Cosner CC, Huang A, Chatterjee A, Bourbon P, Farley N et al. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc Natl Acad Sci USA 2011; 108(14): 5620–5625.
16. Dvořáková L, Sikora J, Hřebíček M, Hůlková H, Boučková M, Stolnaja L et al. Subclinical course of adult visceral Niemann-Pick type C1 disease. A rare or underdiagnosed disorder? J Inherit Met Dis 2006; 29(4): 591.
17. Walterfang M, Fahey MC, Desmond P, Wood A, Seal ML, Steward C et al. White and gray matter alterations in adults with Niemann-Pick disease type C: a cross-sectional study. Neurology 2010; 75(1): 49–56.
18. Wajner A, Michelin K, Burin MG, Pires RF, Pereira ML, Giugliani R et al. Biochemical characterization of chitotriosidase enzyme: comparison between normal individual and patients with Gaucher and with Niemann-Pick diseases. Clin Biochem 2004; 37(10): 893–897.
19. Park WD, O’Brien JF, Lundquist PA, Kraft DL, Vockley CW, Karnes PS et al. Identification of 58 novel mutations in Niemann-Pick disease type C: correlation with biochemical phenotype and importance of PTC1-like domains in NPC1. Hum Mutat 2003; 22(4): 313–325.
20. Ory D, Porter F, Scherrer D, Lanier M, Langmade S, Mologu V et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Mol Genet Metab 2010; 99: S28.
21. Jiang X, Sidhu R, Porter FD, Yanjanin NM, Speak AO, te Vruchte DT et al. A sensitive and specific LC-MS//MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J Lipid Res 2011; 52(7): 1435–1445.
22. Patterson MC, Platt F. Therapy of Niemann-Pick disease, type C. Biochim Biophys Acta 2004; 1685(1–3): 77–82.
23. Lachmann RH, te Vruchte DT, Lloyd-Evans E, Reinkensmeier G, Sillence DJ, Fernandez-Guillen L et al. Treatment with miglustat reverses the lipid-trafficking defect in Niemann-Pick disease type C. Neurobiol Dis 2004; 16(3): 654–658.
24. Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol 2007; 6(9): 765–772.
25. Pineda M, Wraith JE, Mengel E, Sedel F, Hwu WL, Rohrbach M et al. Miglustat in patients with Niemann-Pick disease type C (NP-C): a multicenter observational retrospective cohort study. Mol Genet Metab 2009; 98(3): 243–249.
28. Rosenbaum AI, Zhang G, Warren JD, Maxfield FR. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc Natl Acad Sci USA 2010; 107(12): 5477–5482.
29. Iturriaga A, Pineda M, Fernández-Valero EM, Vanier MT, Coll MJ. Niemann-Pick C disease in Spain: clinical spectrum and development of a disability scale. J Neurol Sci 2006; 249(1): 1–6.
30. Yanjanin NM, Vélez JI, Gropman A, King K, Bianconi SE, Conley SK et al. Linear clinical progression, independent of age of onset, in Niemann-Pick disease, type C. Am J Med Genet B Neuropsychiatr Genet 2009; 153B(1): 132–140.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2012 Issue 3
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Neurosyphilis
- Surgical Treatment of a Tarsal Tunnel Syndrome
- Bilateral Phrenic Nerve Lesion Manifesting as an Orthopnea – Three Case Reports
- Diagnosis and Treatment Options for Niemann-Pick Disease Type C