
Sclerosis multiplex – úloha regulačných T‑lymfocytov v patogenéze a biologickej liečbe choroby
Multiple Sclerosis – a Role of Regulatory T Cells in the Pathogenesis and Biological Treatment of the Disease
Every biological system has its executive as well as control mechanisms that, by the means of feed back, maintain homeostasis within the organism. Regulatory B cells (Breg) and especially regulatory T cells (Treg) are of paramount importance in preventing auto ‑ aggressive (allergic and autoimmune) processes. Multiple sclerosis (MS) is an autoimmune disease driven by proinflammatory activities of various cell types led by TH1 and TH17 cells. Recently, it has been established that the over‑activity of the involved cells is enabled by insufficient activity of regulatory T cells. It is, therefore, natural that, in MS, therapy aims to re‑establish their physiological function. IFN‑β and glatiramer acetate, first line biological agents, are able to do so. The second line agents, natalizumab and FTY720, influence the activity of Treg cells in various ways – natalizumab does not affect T cells in any way, while FTY720 supports both, their proliferation and activity. It seems that it is an insufficient immunosuppressive activity after suspension of the 720 (FTY720) treatment that results in a development of the IRIS. With respect to the role of regulatory T cells in the development and therapy of MS it is worth mentioning that they can be induced and expanded in vitro and subsequently re‑introduced to the patient. Recently, monoclonal antibodies rituximab, alemtuzumab and daclizumab have entered clinical tests as the treatments of MS. Their mechanisms of action are different. Rituximab down ‑ regulates antigen ‑ presentation function of B cells, alemtuzumab profoundly depletes T cells, including auto ‑ reactive lines, and daclizumab induces NK cells that enter the itrathecal compartment and kill autoreactive T cells. The paper also discusses the support that laboratory immunology has to offer to physicians in terms of the diagnosis and biological treatment decision making.
Key words:
glatiramer acetate – interferon b –monoclonal antibodies – regulatory T cells – multiple sclerosis – TH1 cells – TH17 cells
Authors:
M. Buc
Authors‘ workplace:
Imunologický ústav LF UK, Bratislava
Published in:
Cesk Slov Neurol N 2013; 76/109(3): 293-299
Category:
Review Article
Overview
Každý biologický systém zahàňa v sebe nielen efektorové, ale aj kontrolné mechanizmy, ktoré spôsobom spätnej väzby udržujú homeostázu organizmu. Najdôležitejšími imunitnými mechanizmami, ktoré bránia rozvoju autoagresívnych procesov (autoimunitných a alergických), sú tie, ktoré sprostredkúvajú regulačné B‑lymfocyty (Breg), a predovšetkým regulačné T‑lymfocyty (Treg). Sclerosis multiplex je autoimunitná choroba, ktorú podmieňuje prozápalová aktivita početných buniek imunitného systému pod „vedením“ TH1 - a najmä TH17 - subpopulácie lymfocytov. V ostatnej dobe sa zistilo, že nadmernú prozápalovú aktivitu uvedených skupín lymfocytov umožňuje aj nedostatočná imunosupresívna aktivita Treg‑lymfocytov. Je prirodzené, že v liečbe SM sa snažíme ich funkciu obnoviť. Dokážu to už súčasné biologiká prvej línie, t.j. IFN‑β a glatiramér acetát. Biologiká druhej línie aktivitu Treg‑lymfocytov ovplyvňujú rôzne – FTY720 podporuje aj ich proliferáciu, aj imunosupresívnu aktivitu. Nasvedčujú tomu tiež klinické príznaky, kedy pri prerušení liečby týmito preparátmi môže dôjsť k indukcii IRIS ‑ syndrómu. V súvislosti s úlohou Treg‑lymfocytov v rozvoji a terapii SM sa poukazuje aj na možnosť indukcie a expanzie vlastných Treg‑lymfocytov pacienta in vitro a následné ich spätné podanie pacientovi. Do štádií klinických skúšok sa dostavajú aj monoklonové protilátky rituximab, alemtuzumab a daclizumab, ktorých mechanizmus na zníženie imunopatologických procesov je rozdielny. Rituximab znižuje antigénovo ‑ prezentačnú funkciu B‑lymfocytov, alemtuzumub pôsobí prostredníctvom hlbokej deplécie T‑lymfocytov, vrátane autoreaktívnych, a daclizumab indukuje vznik NK ‑ buniek, ktoré ničia autoreaktívne T‑lymfocyty. Napokon sa v článku uvádza, čo z hľadiska diagnostiky choroby a rozhodovania lekára, aký typ liečby má použiť, súčasná laboratórna imunológia môže poskytnúť.
Kľúčové slová:
glatiramér-acetát – interferón b – monoklonové protilátky – regulačné T-lymfocyty – sclerosis multiplex – TH1 a TH17-lymfocyty
Zoznam skratiek
| BLIMP ‑ 1 | B ‑ Lymphocyte ‑ Induced Maturation Protein (transkripčný represor) |
| CMV | cytomegalový vírus |
| CTLA | Cytotoxic T Lymphocyte Antigen |
| DC | dendritové bunky |
| EBV | vírus Epsteina a Barrovej |
| FOXO | Forkhead box O (transkripčný faktor) |
| FOXP3 | Forkhead box P (transkripčný faktor) |
| GITR | Glucocorticoid ‑ Induced Tumor necrosis factor Receptor family related gene |
| HBV | vírus hepatitídy B |
| HLA | hlavný histokompatibilný komplex človeka |
| HSV | vírus herpes simplex |
| IFN‑β/ IFN‑γ | interferón beta/ gama |
| IRF4 | Interferon Regulatory Factor 4 |
| MBP | myelínový bázický proteín |
| MOG | myelínový oligodendritový antigén |
| NK | NK ‑ bunky (Natural Killer cells) |
| PD | Programmed Death |
| STAT5 | Signal Transducer and Activator of Transcription 5 |
| TIGIT | T cell Ig and ITIM domain |
| TIM | T cell Immunoglobulin Mucin protein |
Principiálnou úlohou imunitného systému je likvidácia vonkajších i vnútorných narušiteľov integrity jedinca. Vonkajších „nepriateľov“ predstavujú predovšetkým mikroorganizmy, kým k vnútorným patria najmä potenciálne rakovinotvorné bunky, ktoré vznikajú v našom organizme ako výsledok poruchy v ich replikácii. Omnoho menej sa zdôrazňuje druhá, nemenej významná biologická funkcia imunitného systému, a to zábrana aktivácie autorektívnych T ‑ a B‑lymfocytov, ktoré môžu predstavovať hrozbu indukcie autoimunitných procesov. Dosahuje sa mechanizmami centrálnej (recesívnej) a periférnej (dominantnej) tolerancie. Centrálna tolerancia sa zabezpečuje deléciou autoreaktívnych T ‑ a B‑lymfocytov v týmuse, resp. v kostnej dreni počas ich dozrievania v týchto primárnych lymfoidných orgánoch [1,2]. Tak ako v každom biologickom systéme, ani mechanizmy centrálnej tolerancia nie sú 100% a časť autoreaktívnych lymfocytov tomuto procesu unikne a dostáva sa na perifériu, do sekundárnych lymfoidných orgánov, kde po stretnutí sa s autoantigénmi, krížovo ‑ reagujúcimi antigénmi, či pri dysregulácii imunitného systému sa môžu aktivovať a spustiť autoimunitné procesy, ktoré niekedy vedú aj k plne rozvinutým autoimunitným chorobám. Aby k tomu nedochádzalo, príroda vytvorila mechanizmy periférnej tolerancie, ktoré zabezpečujú predovšetkým regulačné lymfocyty. Tieto priamym kontaktom s autoreaktívnymi lymfocytmi alebo nepriamo, najmä syntézou imunosupresívnych cytokínov, zabránia ich aktivácii alebo utlmia ich efektorovú aktivitu [1,2]. Najdôležitejšie regulačné lymfocyty patria T‑lymfocytom, kým regulačné B‑lymfocyty zohrávajú síce tiež svoju významnú úlohu, ale skôr doplnkovú.
Regulačné T‑lymfocyty (Treg) zaraďujeme do dvoch skupín – prirodzené a indukované. Prirodzené regulačné T‑lymfocyty (nTreg) predstavujú samostatnú skupinu buniek, tak ako sú B‑lymfocyty, NK ‑ bunky atď. Naproti tomu indukované regulačné T‑lymfocyty (iTreg) nepredstavujú samostatnú skupinu buniek, ale ide o subpopuláciu pomocných CD4+ T‑lymfocytov, ktorá vzniká iba počas imunitnej odpovede [3 – 5].
nTreg‑lymfocyty potláčajú aktivitu T ‑ (aj αβ aj γδ), B‑lymfocytov, NK ‑ , NKT ‑ a aj dendritových buniek (DC). Vyvíjajú sa dreni týmusu ako nezávislá, samostatná populácia buniek. Na ich diferenciáciu treba, aby rozpoznávali peptidy pochádzajúce z autoantigénov, ktoré im prezentujú HLA‑molekuly dendritových buniek drene týmusu, kostimulačné interakcie medzi molekulami CD28 (nTreg) a CD80, resp. CD86 (DC) a napokon cytokíny IL‑2 alebo IL‑15. Rozpoznávanie prezentovaných peptidov je vysokoafinitné [3,6].
Diferenciácia nTreg‑lymfocytov si vyžaduje okrem iného aj aktiváciu špecifického génu – FOXP3 (Forkhead box P3; Xp11.23 – q13.3.), ktorý kóduje rovnomenný transkripčný faktor. O jeho veľkom biologickom význame svedčí skutočnosť, že ak dôjde k jeho mutácii, vznikne choroba, ktorá je nezlučiteľná so životom – IPEX (Immune dysregulation, Polyendocrinopathy, Enteropathy). Okrem FOXP3 sa na diferenciácii nTreg zúčastňujú aj iné transkripčné faktory, najmä BLIMP ‑ 1, IRF4, transkripčné faktory FOXO ‑ rodiny a STAT5, ktorý vzniká aktiváciou dráh, ktoré indukuje IL‑2 [7,8]. nTreg predstavujú 5 – 10 % CD4+CD8– tymocytov v periférii je ich približne 10 % z celej populácie CD4+‑lymfocytov. Sú to dlhodobo žijúce bunky, ktoré charakterizujú viaceré membránové antigény. Problémom však je, že nie sú špecifické iba pre nTreg; typicky majú CD4 a CD25. CD25 (α ‑ reťazec vysokoafinitného receptora pre IL‑2) však vlastnia aj aktivované T‑lymfocyty. Neskôr sa ukázalo, že nTreg majú zníženú expresiu CD127 (α ‑ reťazec receptora pre IL‑7). Z iných znakov sa treba zmieniť o CD5, CD49d, CD69, CD103, CD152 (CTLA ‑ 4), AITR (CD357; skôr je známy jeho ekvivalent pri myšiach – GITR) a neuropilín [3,9].
nTreg sú prirodzene anergické, t.j. pri in vitro stimulácii monoklonovými anti‑CD3protilátkami (CD3 je súčasťou antigénového receptora T‑lymfocytov), fytohemaglutinínom alebo alogénnymi bunkami neproliferujú a neprodukujú ani IL‑2. Výrazne však tlmia indukciu imunitnej odpovede. Mechanizmy, akým to robia, sú viaceré; v podstate ich možno rozdeliť do 4 skupín: 1. prostredníctvom inhibičných cytokínov; 2. indukciou apoptózy; 3. prostredníctvom indukcie zmien v metabolizme; 4. moduláciou dozrievania alebo funkcie DC [10 – 12].
Indukované regulačné T‑lymfocyty sa diferencujú z naivných CD4+ T‑lymfocytov počas imunitnej odpovede. Vznikajú najmä v slizničných priestoroch; najlepšie tieto udalosti poznáme v čreve. Ak na naivné CD4+ T‑lymfocyty pôsobí TGF‑β, IL‑2 a kyselina retinová, ktorú spracovaním vitamínu A produkujú dendritové bunky, diferencujú sa na iTreg [2,4,13]. Na ich difereciáciu tiež treba FOXP3 - gén, ale rozdielne transkripčné gény a odlišnú promótorovú oblasť (CNS1). Pri ich úplnom chýbaní (v experimente) nevzniká nijaká systémová choroba, ktorá by sa podobala IPEX ‑ syndrómu, ale zápalové procesy slizníc (kolitída, asthma bronchiale) [14,15]. Bol problém, ako ich odlíšiť od nTreg. Vyriešilo sa to nedávno, keď sa zistilo, že na rozdiel od nTreg majú v svojich membránach iba málo neuropilínu [16]. iTreg svoju imunosupresívnu aktivitu presadzujú podobne ako nTreg, kontaktným alebo bezkontaktným (syntézou najmä TGF‑β a IL‑10) spôsobom [4,13].
Aký je zásadný rozdiel medzi nTreg ‑ a iTreg‑lymfocytmi? Oboje potláčajú aktivitu buniek imunitného systému, avšak principiálne nTreg‑lymfocyty tlmia aktivitu autoreaktívnych T‑lymfocytov, t.j. tých, ktoré unikli svojej delécii v týmuse, napr. lymfocytov, ktoré rozpoznávajú MBP, kým iTreg‑lymfocyty obmedzujú aktivitu tých efektorových T‑lymfocytov, ktoré vznikajú počas imunitnej odpovede na antigén, napr. na komenzálne baktérie, ktoré máme v čreve. nTreg ‑ a iTreg‑lymfocyty sa nezastupujú, ale dopĺňajú svoju aktivitu [14].
Funkciu imunosupresívnych buniek môžu zastávať aj B‑lymfocyty. Svoju imunosupresívnu funkciu presadzujú či už priamym kontaktom, alebo prostredníctvom TGF‑β a najmä IL‑10, príp. indukujú apoptózu aktivovaných T‑lymfocytov. Dokážu indukovať a potenciovať prirodzené regulačné T‑lymfocyty. Charakteristiku ich membránových znakov pri myšiach už poznáme, u človeka zatiaľ nie. Označujú sa skratkou Breg ‑ alebo B10‑lymfocyty [17 – 19].
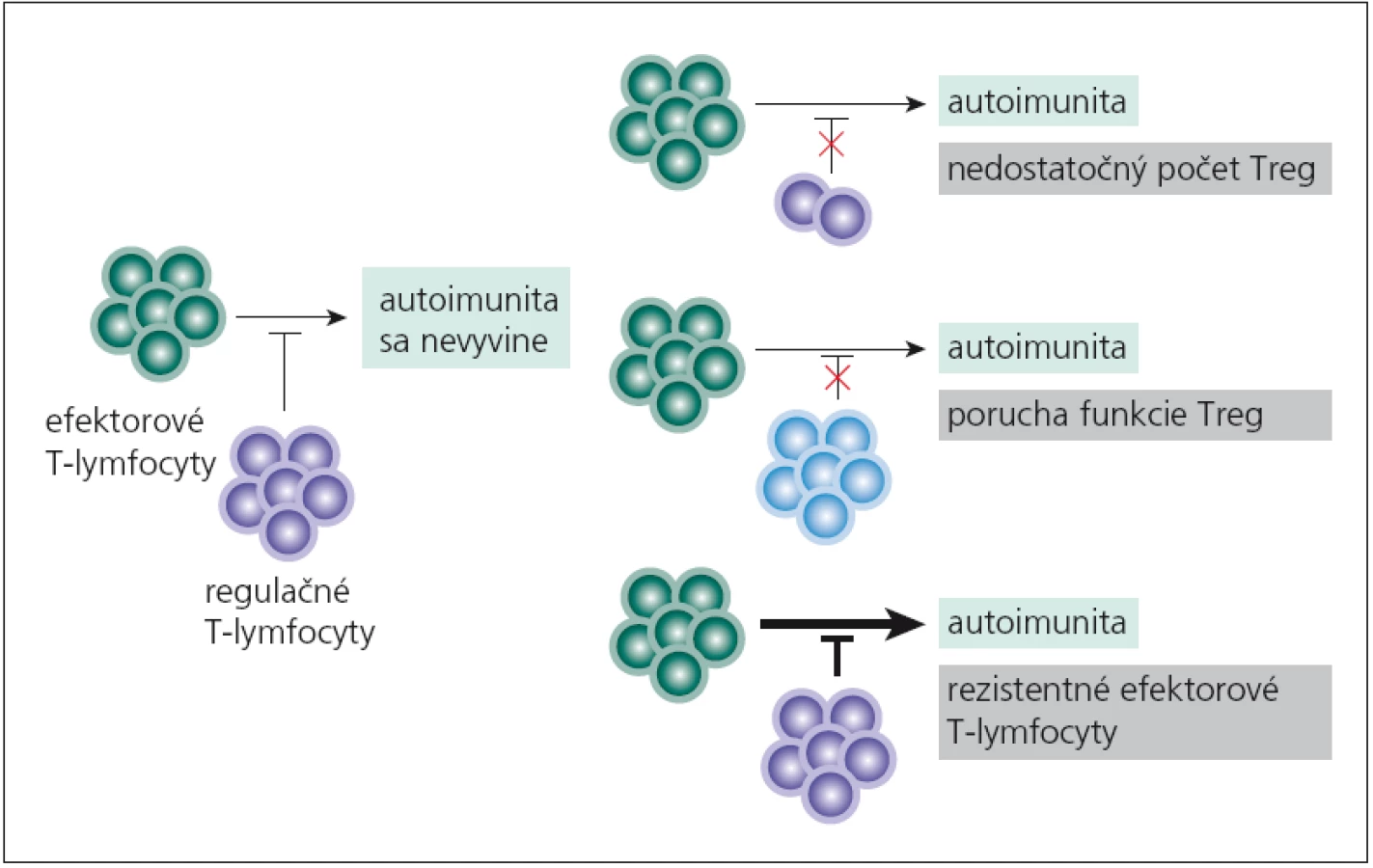
Vychádzajúc z indukcie autoimunitných procesov pre mutáciu vo FOXP3 - géne, predpokladalo sa, že porucha funkcie či nedostatok nTreg‑lymfocytov sa bude podieľať aj na imunopatologických procesoch „bežných“ autoimunitných chorôb. Tento predpoklad sa potvrdil. nTreg‑lymfocyty môžu mať svoj podiel na vývoji autoimunitných chorôb tým, že chýbajú, ich funkcia je znížená alebo že efektorové T‑lymfocyty na ich pôsobenie neodpovedajú (obr. 1). Aká je situácia pri sclerosis multiplex (SM)? Viaceré práce poukazujú na nedostatočnú funkciu nTreg‑lymfocytov [20 – 22], i keď sú aj práce, ktoré udávajú pokles počtu týchto buniek v periférnej krvi [23]. Aká je príčina ich nedostatočnej funkcie? Zrejme pôjde o komplexnú poruchu, ako je znížená expresia ko‑inhibičných molekúl (CLTA4, TIM ‑ 3, TIGIT) v membránach buniek, znížená syntéza imunosupresívnych cytokínov a pod. [22,24]. Nedostatočná tlmivá aktivita nTreg‑lymfocytov umožní na periférii aktivovaným autoreaktívnym T‑lymfocytom prechádzať do CNS a rozvíjať tu imunopatologický proces.
Sclerosis multiplex je typická autoimunitná choroba. Poznatky o jej imunopatogenéze v posledných rokoch podstatne vzrástli, čo umožnilo aj lepšiu, adresnejšiu biologickú liečbu. V stručnosti, pri SM dochádza k deštrukcii myelínových obalov v mozgu, periférny nervový systém ostáva neporušený. Príčinu choroby nepoznáme. Predpokladá sa interakcia faktorov vonkajšieho prostredia s genetickou predispozíciou k chorobe. Zo spúšťacích faktorov sa obviňujú vírusy, ktoré mechanizmom molekulového mimikri (tj. podobnosť vlastných a mikrobiálnych antigénov) indukujú rozvoj imunopatologických procesov. V skutočnosti, početné vírusy obsahujú proteíny, ktoré majú príbuznú štruktúru s principiálnym autoantigénom pri SM – myelínovým bázickým proteínom (MBP). Ide o vírus osýpok, CMV, EBV, HBV, HSV, humánny herpetický vírus 6 alebo 7, koronavírusy a iné. Vzťah medzi vírusmi a SM podporujú aj údaje, že vírusové infekcie často predchádzajú záchvaty choroby. Pravdepodobne IFN‑γ, ktorý sa produkuje počas vírusovej infekcie, spustí do pohybu imunopatologické procesy vrcholiace demyelinizáciou [25].
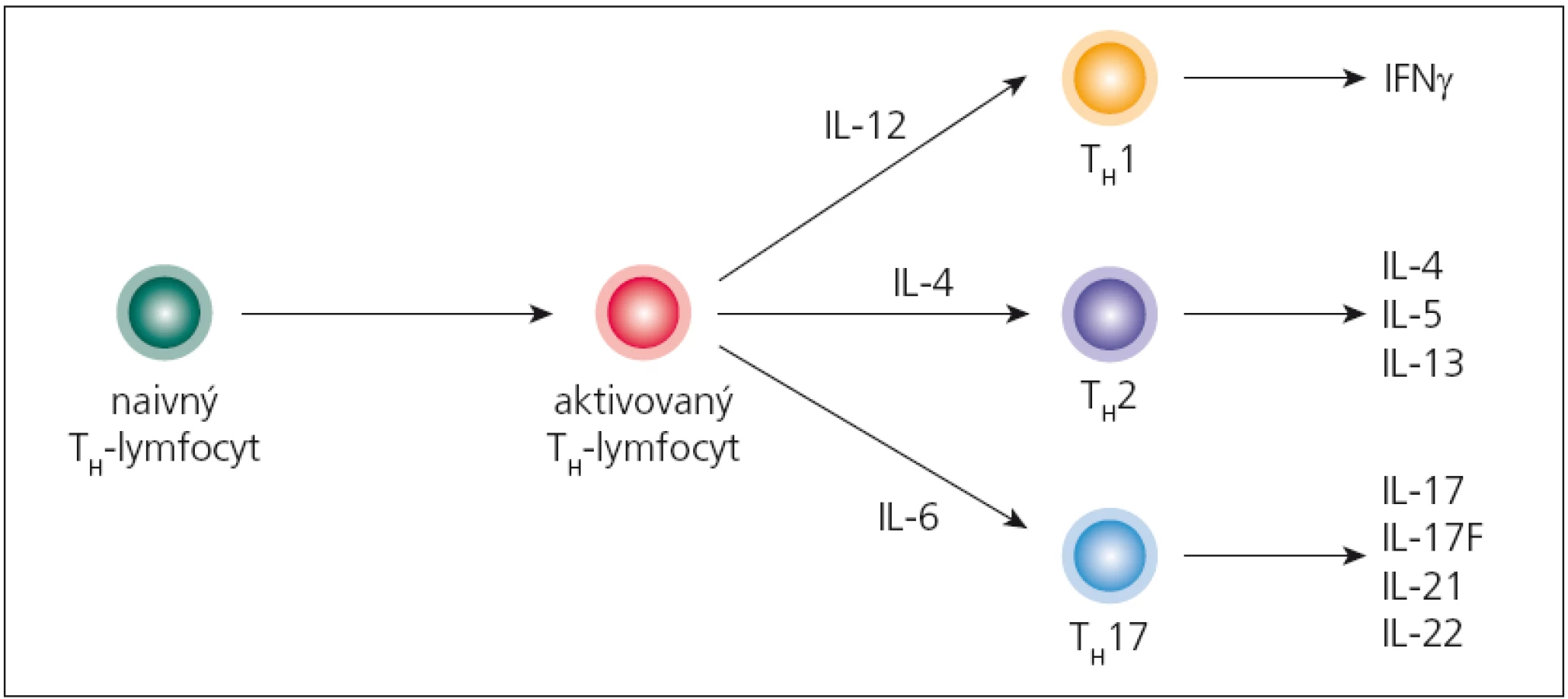
Imunopatologický proces sa začína aktiváciou autoreaktívnych T‑lymfocytov na periférii, ktoré patria TH1 a najmä TH17 - subpopuláciám (obr. 2). Aktivované T‑lymfocyty následne začnú do svojej bunkovej membrány zabudovávať adhezívne molekuly (najdôležitejšie sú α4β1 - integríny), čo im umožní viazať sa na adhezívne molekuly na endotelových bunkách CNS (najmä VCAM‑1, ktorý sa tu v malých množstvách konštitutívne exprimuje; po stimulácii cytokínmi sa jeho expresia zvyšuje) a následne prechádzať do parenchýmu mozgu. Tento mechanizmus prechodu využívajú TH1‑lymfocyty, kým TH17‑lymfocyty využívajú interakciu medzi ich chemokínovým receptorom CCR6 a jeho ligandom CCL20, ktorý sa tiež konštitutívne exprimuje na endotelových bunkách [26,27]. Po prechode autoreaktívnych T‑lymfocytov, v perivaskulárnom priestore, sa začnú odohrávať iniciálne imunopatologické procesy. Tu sa dostávajú do kontaktu s myeloidnými dendritovými bunkami, ktoré prostredníctvom svojich HLA‑molekúl druhej triedy prezentujú peptidy pochádzajúce z myelínu, čím sa reaktivujú, začnú invadovať do parenchýmu a produkovať prozápalové cytokíny (najmä IFN ‑ γ, IL‑17 a TNF). Tieto následne stimulujú parenchýmové mikrogliové bunky, ktoré produkciou svojich cytokínov (IL‑12, IL‑23, osteopontín) a toxických mediátorov (reaktívne intermediálne produkty dusíka a kyslíka) poškodzujú myelín; z neho sa uvoľňujú ďalšie autoantigény a „circulus vitiosus“ pokračuje. Na deštrukcii myelínu sa podieľajú aj cytotoxické T‑lymfocyty svojím perforínovo ‑ granzýmovým mechanizmom. Súčasne dochádza aj k aktivácii B‑lymfocytov; týchto je v plaku spočiatku málo, ich relatívny počet sa zvyšuje, až keď sa ložisko stáva chronickým. Aktivácia B‑lymfocytov sa prejaví intracerebrálnou syntézou IgG, ktoré sú typicky oligoklonové [25,28,29].
Neuromyelitis optica (NMO) sme až donedávna považovali za variant SM. Dnes predstavuje samostatnú chorobnú jednotku. Pri tejto chorobe ide o autoimunitný zápal spôsobujúci demyelinizáciu optického nervu a miechy. Hoci môže postihnúť aj mozog, lézie sú rozdielne od tých, ktoré sa pozorujú pri SM. Pre syndróm sú charakteristické protilátky proti aquaporínu 4 v membráne astrocytov (AQP4) (aquaporíny sú integrálne proteíny bunkových membrán, ktoré regulujú tok vody) [30,31]. Dominantnými bunkami infiltrujúcimi lézie sú neutrofily, bunky, ktoré pri SM prakticky chýbajú. Ďalšia odlišnosť od SM je, že v cerebrospinálnej tekutine sú vysoké hladiny IL‑17. Tie pravdepodobne zodpovedajú za indukciu produkcie chemokínov (IL‑8, G‑CSF a CXCL1), ktoré priťahujú a aktivujú granulocyty [32]. IFN‑β, ktorý sa používa v liečbe SM, pri NMO sa nesmie podávať. Nielenže sú pacienti, ktorí na túto liečbu neodpovedajú, ale u niektorých liečba indukuje ťažké relapsy a exacerbácie choroby [33,34]. V súčasnosti sa na liečbu NMO používa azatioprin, prednizon, rituximab, cyklofosfamid, metotrexát, mitoxantron, mykofenolát mofetil, intravenózne imunoglobulíny (IVIG) či výmenná plazmaferéza. Tu však sa tiež ukazuje, že biologická liečba by mohla tiež značne prispieť k liečbe choroby – vyvinuli sa monoklonové protilátky (aquaporumab), ktoré sa viažu na AQP4, čím sa zabráni patogenetickým anti‑AQP4 protilátkam, aby uplatnili svoju aktivitu [35,36]. Aquapuromab neaktivuje ani komplement ani K-bunky, takže k poškodeniu buniek, na ktoré sa viaže, nedochádza.
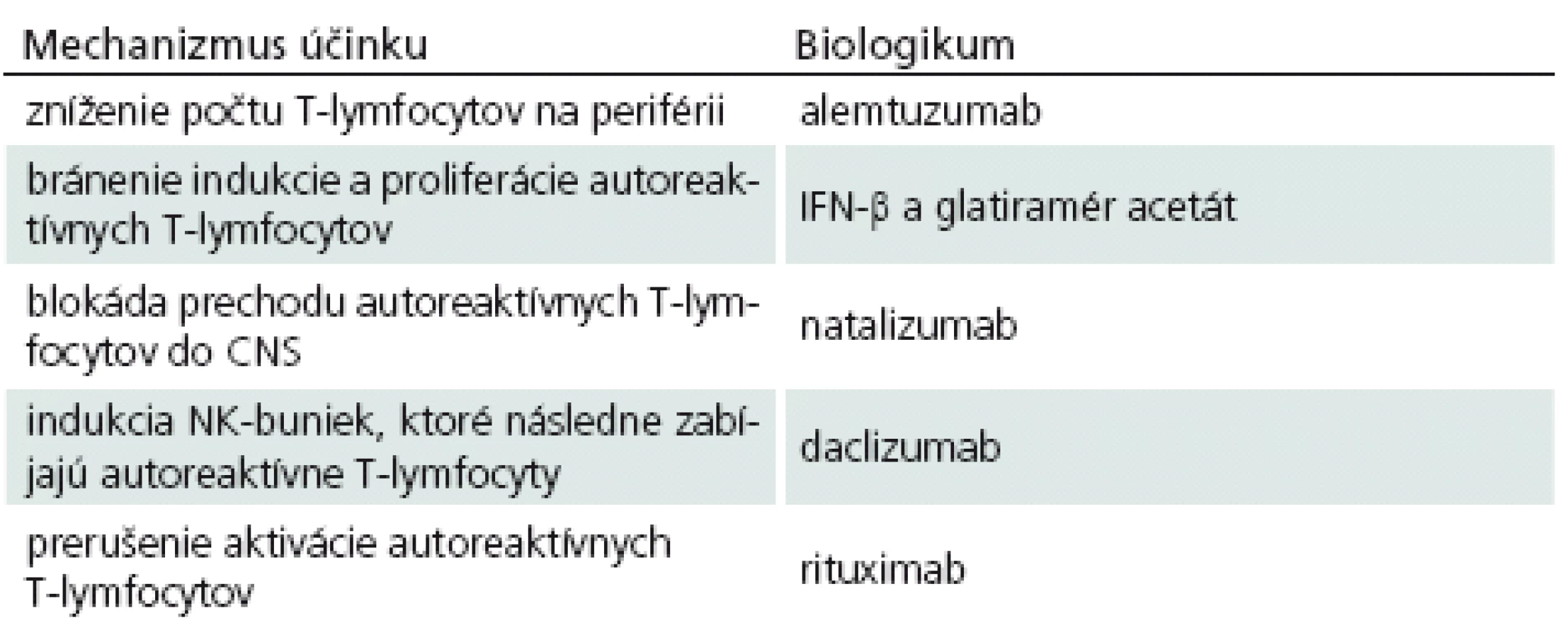
Ako sa zmieňujeme vyššie, spoznanie imunopatogenézy SM sa odrazilo na rozmachu vývoja rôznych imunoterapeutických látok. Ako prvé biologikum, ktoré sa pri liečbe SM začalo podávať, bol IFN‑β, neskôr do hry vstúpil glatiramér acetát, monoklonové protilátky, FTY ‑ 720 a iné. Každé z uvedených imunoterapeutík má svoje vlastné mechanizmy, ktorými ovplyvňuje prebiehajúce imunopatologické procesy a pokúša sa o návrat do fyziologických podmienok (tab. 1). Treba však hneď povedať, že ani jednému v súčasnosti používaných biologík sa to doteraz nepodarilo, vždy ide iba o úpravu – zlepšenie klinického stavu pacientov, ale nie o vyliečenie.
Interferón beta (IFN‑β) svoje imunosupresívne pôsobenie uplatňuje viacerými spôsobmi. Pôsobí na bunky prezentujúce antigén, t.j. makrofágy a mikrogliu, tým, že znižuje v ich membránach množstvo HLA‑molekúl druhej triedy a indukuje objavenie sa inhibičných molekúl PDL‑2, ktoré po interakcii s PD1 - receptormi na T‑lymfocytoch navodia v nich apoptotický proces. IFN‑β znižuje aj proliferáciu makrofágov, čím zmenšuje ich počet, a tým aj schopnosť aktivovať ďalšie a ďalšie autoreaktívne T‑lymfocyty. Ďalej IFN‑β znižuje ich vstup do mozgu tým, že znižuje množstvo VLA4 - adhezívnych molekúl, na ktoré sa T‑lymfocyty musia prostredníctvom svojich VCAM‑1 molekúl pred vstupom do parenchýmu viazať [36].
Významnú úlohu má podávanie IFN‑β aj na pôsobenie Treg‑lymfocytov. IFN‑β v membránach dendritových buniek zvyšuje množstvo ligandov pre GITR ‑ receptory (GITRL). Interakcia medzi GITR na Treg‑lymfocytoch s GITRL na dendritových bunkách podmieni v Treg‑lymfocytoch prenos stimulačného signálu, ktorý spôsobí, že tieto začnú proliferovať, a tým zvyšovať svoje počty a následne aj svoju tlmivú aktivitu. Tejto proliferácii napomáha aj druhý efekt IFN‑β pôsobenia na Treg ‑ bunky, a to v podobe zníženia množstva ich CTLA4 - molekúl, ktoré potláčajú aktivitu Treg‑lymfocytov; stávajú sa takto vnímavejšie na pôsobenie stimulačných podnetov, najmä na IL‑2, ktorý je ich základným homeostatickým cytokínom [37].
Glatiramer acetát (GA) je druhým imunopreparátom prvolíniovej liečby. Ide o zmes syntetických peptidov, ktoré sa skladajú z 50 až 90 aminokyselinových jednotiek. Konkrétne ide o L ‑ glutámovú kyselinu, L ‑ lyzín, L ‑ alanín a L ‑ tyrozín, ktoré sú v peptide zastúpené náhodne. Tieto aminokyseliny sa na tvorbu peptidov nevybrali náhodne, ale cielene, lebo sa v myelíne vyskytujú najčastejšie. Mechanizmus účinku GA môže byť v blokáde žliabku HLA‑molekúl, ale ukazuje sa, že GA má aj imunomodulačné vlastnosti. Dokázalo sa, že GA indukuje produkciu sIL‑1Ra, prirodzeného inhibítora IL‑1, čím znižuje jeho pôsobenie a aj jeho syntézu. Navyše monocyty, resp. makrofágy pod jeho vplyvom produkovali aj menej IL‑1, TNF, teda najdôležitejších mediátorov zápalu a IL‑12, ktorý podporuje TH1 - imunitnú odpoveď; naopak, GA zvyšoval ich syntézu imunosupresívneho IL‑10 [38]. Predpokladá sa, že GA ‑ aktivované T‑lymfocyty vstupujú aj do CNS a tu presadzujú svoj protizápalový a neuroprotektívny účinok [39]. GA zlepšuje aj imunosupresívne pôsobenie regulačných T‑lymfocytov, a to zvyšovaním expresie ko‑inhibičných molekúl TIGIT a TIM ‑ 3, ktoré sú u pacientov so SM v membránach ich Treg‑lymfocytov znížené [22,24,40].
K druholíniovej biologickej liečbe pacientov so SM patrí natalizumab a FTY720.
Natalizumab sú monoklonové protilátky (mAb) proti α4 - reťazcu adhezívnej molekuly VLA4 (α4β1 - integrín), ktorý sa exprimuje v membránach T‑lymfocytov; jeho partnerská molekula na endotelových bunkách ciev v CNS je VCAM‑1. Podávaním týchto mAb sa zabraňuje prechodu autoreaktívnych T‑lymfocytov do CNS, pretože T‑lymfocyty nemôžu priľnúť na endotelovú výstelku ciev, a tak sa následne preplaziť do parenchýmu mozgu, čo makroskopicky vnímame ako lymfocytózu. Natalizumab redukuje tiež počet dendritových buniek v perivaskulárnom priestore mozgu poukazujúc na to, že sa jeho pôsobenie neobmedzuje iba na T‑lymfocyty [36].
Po prerušení liečby sa u niektorých pacientov po troch až šiestich mesiacoch pozoruje relaps choroby. U jedincov, ktorí relabovali, alebo mali zhoršené prejavy MR, sa pozorovala menšia lymfocytóza oproti tým, u ktorých sa relaps nevyskytol. Jedna z možných príčin tohto javu je aj v genetickom polymorfizme génu pre antiapoptotický proteín Akt. Jedna jeho forma, rs2498804T (jedná sa o polymorfizmus na úrovni jednotlivého nukleotidu – SNP, Single Nucleotide Polymorphism), sa spája z jeho zvýšenou aktivitou a teda zvýšenou apoptózou aktivovaných T‑lymfocytov, a teda aj nižšou lymfocytózou [42,42].
Jedna z komplikácii liečby SM natalizumabom je aj indukcia progresívnej multifokálnej leukoencefalopatie (PML) vyvolanej JC ‑ vírusom [43]. Ďalšia z komplikácií liečby pacientov natalizumabom je indukcia IRIS ‑ syndrómu (Immune Reconstitution Inflammatory Syndrome), ktorý poznáme aj ako syndróm z imunitnej obnovy, zotavenia. Pozoruje u niektorých pacientov po imunosupresii, keď imunitný systém sa pomaly vracia do pôvodného stavu a začína reagovať na predchádzajúce oportunistické infekcie, a to prehnanou hyperinflamačnou odpoveďou, čo paradoxne celú situáciu ešte zhoršuje. Na indukcii IRIS ‑ syndrómu po prerušení liečby natalizumambom sa snáď podieľajú aj Treg‑lymfocyty, skôr indukované ako prirodzené, pravdepodobne pre nedostatočné podmienky ich indukcie. Napriek tomu, natalizumab má svoje miesto v liečbe SM, keď lekár zvažuje riziko pacienta na rozvoj PML s ohľadom na predchádzajúcu imunosupresívnu liečbu v minulosti a pozitivitu anti‑JCV protilátok v jeho plazme.
V druholíniovej liečbe SM máme ešte k dispozícii FTY720 (fingolimod). Je to derivát myriocínu, ktorý sa izoluje z čínskej rastliny Iscaria sinclarii. FTY720 je štruktúrnym analógom sfingozínu, ktorý sa fosforyluje sfingozínovou kinázou a následne pôsobí ako veľmi silný agonista receptorov pre sfingozín 1 - fosfát (ide o receptory S1P1 až S1P5, na S1P2 sa neviaže. T‑lymfocyty využívajú najmä S1P1 a B‑lymfocyty zase S1P3) [44,45]. Zníženie, resp. vymiznutie S1P1/ S1P3 - receptorov na T ‑ a B‑lymfocytoch spôsobí poruchu ich vycestovania zo sleziny, lymfatických uzlín a z Peyerových plakov. Reverzibilná sekvestrácia sa týka až 80 % lymfocytov za 3 – 5 hod po aplikácii [46 – 48].
Pôsobenie FTY720 na regulačné T‑lymfocyty nie je ešte úplne objasnené. Existujú literárne údaje, ktoré poukazujú, že FTY720 podporuje ich proliferáciu a imunosupresívnu aktivitu, i keď mechanizmus, akým to dosahujú, autori neuvádzajú [46]. Tejto vlastnosti FTY720 by zodpovedali aj klinické údaje, ktoré dokumentujú, že po prerušení liečby týmto preparátom dochádza k indukcii príznakov, ktoré sa podobajú vyššie uvedenému IRIS ‑ syndrómu [49,50]. Naznačovalo by to, že okrem iného došlo aj k poklesu aktivity regulačných a zvýšeniu aktivity efektorových T‑lymfocytov. Naopak, iné práce dokazujú, že FTY720 znižuje aktivitu regulačných T‑lymfocytov, a to aj ich proliferačnú aj funkčnú aktivitu. Spôsob, ktorým to FTY720 dosahuje, sa zakladá na jeho interferencii s fosforyláciou, a tým aktiváciou transkripčného faktora STAT5, ktorý je potrebný na transkripciu FOXP3 - génu a vznik nTreg‑lymfocytov [51]. Klinické skúsenosti v súvislosti s vyššie uvedeným IRIS ‑ syndrómom po prerušení liečby však tomu nenasvedčujú, resp. získané poznatky v predklinických štúdiách nie vždy musia korelovať s účinkom preparátu pri samotnej liečbe pacientov. Pripomína to udalosti z r. 2006, kedy sa použila super ‑ agonistická monoklonová protilátka anti‑CD28, ktorá v klinickom použití viedla k cytokínovej búrke a k ťažkým klinickým príznakom, ktoré hraničili s prežitím dobrovoľníkov. Bolo to neočakávané, lebo v predklinických štúdiách táto mAb podporovala expanziu práve nTreg‑lymfocytov [52].
V poslednej dobe sa do klinických skúšok dostávajú monoklonové protilátky, ktoré rozpoznávajú CD20 na povrchu B‑lymfocytov, a pomocou komplementu alebo aktiváciou K ‑ buniek ich likvidujú. Vychádza sa tu z poznatkov o inej biologickej funkcii B‑lymfocytov ako len buniek, ktoré zodpovedajú za syntézu protilátok. Už vyššie sme písali, že existuje subpopulácia B‑lymfocytov, ktorá má imunosupresívne vlastnosti. Podobne začína sa zisťovať, že B‑lymfocyty majú významnú úlohu aj pri prezentácii antigénov. B‑lymfocyty totiž majú v svojich membránach HLA‑molekuly druhej triedy a pohltené antigény, ktoré predtým rozpoznali a viazali svojim antigénovým receptorom, dokážu spracovať, nadviazať na HLA‑molekuly a prezentovať T‑lymfocytom. Monoklonové protilátky anti‑CD20 znižujú počet B‑lymfocytov, čím sa znižuje možnosť ich interakcie autoreaktívnymi T‑lymfocytmi, a tým aj k útlmu imunitných procesov. Súčasne dochádza k zmene cytokínového mikroprostredia, ktoré podmieni vznik a expanziu Treg‑lymfocytov, čím sa autoimunitný proces ďalej tlmí [17,53,54]. Prečo sa vybral práve CD20 - antigén? Odpoveď je pomerne jednoduchá – nachádza sa v membránach prakticky všetkých vývojových štádií B‑lymfocytov, od pre‑B‑lymfocytov až po plazmablasty [55].Anti‑CD20 mAb existujú v troch prevedeniach – rituximab, ocrelizumab a ofatimumab. Rituximab a ofatimumab ničia B‑lymfocyty prostredníctvom aktivácie komplementu, ocrelizumab aktiváciou K ‑ buniek, čo je výhodnejšie, lebo nevznikajú prozápalové fragmenty komplementového systému, ktoré vyvolávajú prozápalové komplikácie, ale apoptotické telieska, ktoré likvidujú makrofágy bez akejkoľvek indukcie zápalových procesov [56].Rituximab sa osvedčil aj pri zvládaní neuromyelitis optica (NMO) ako sa o tom píše vyššie [31,57].
Do štádií klinických skúšok sa dostávajú aj ďalšie dve monoklonové protilátky – alemtuzumab a daclizumab. Alemtuzumab sú monoklonové protilátky proti CD52. Táto molekula sa nachádza v membránach lymfocytov, monocytov a granulocytov. Biologickú úlohu CD52 zatiaľ ešte nepoznáme. Alemtuzumab aktiváciou komplementu vedie k rýchlej lýze T ‑ , B‑lymfocytov, NK ‑ buniek a monocytov. Hoci má biologický polčas len 6 dní a neovplyvňuje prekurzorové bunky, aj pomocné (CD4+) aj cytotoxické (CD8+) lymfocyty sa vracajú k východiskovým hodnotám až za 61, resp. 30 mesiacov, čo môže vysvetliť jeho pozitívne účinky v liečbe SM. V protiklade s tým B‑lymfocyty sa dostávajú na pôvodnú úroveň už za 3 mesiace a potom ich počty dokonca aj stúpajú [56]. Táto skutočnosť (t.j. zvýšená aktivita B‑lymfocytov a nedostatočná tlmivá funkcia regulačných T‑lymfocytov) môže vysvetliť aj sklon takto liečených pacientov vyvinúť autoimunitné komplikácie [58,59].
Daclizumab sú monoklonové protilátky proti alfa‑reťazcu pre IL‑2 (CD25). Mechanizmus ich účinku je zaujímavý – indukujú vznik NK ‑ buniek, ktoré prechádzajú do intratékálneho priestoru a tu zabíjajú autoreaktívne T‑lymfocyty [60].
Napokon, v súvislosti s úlohou Treg‑lymfocytov v rozvoji a terapii SM sa treba zmieniť aj o možnosti indukcie a expanzie vlastných Treg‑lymfocytov pacienta in vitro a následné ich spätné podanie pacientovi. Tento spôsob zvládania imunitných procesov sa úspešne odskúšal pri zvládaní GvHD u ľudí [61] a pri liečbe experimentálnej autoimunitnej encefalitídy (EAE) indukovanej podaním MOG [62].
V poslednej dobe pribudlo množstvo poznatkov o jednotlivých imunopatologických procesoch pri SM. Našli si odraz aj v lepšej terapii, ktorú sme v stručnosti vyššie načrtli, ale stále nemáme odpoveď, ktoré (laboratórne) ukazovatele by nám mali naznačiť, aký typ liečby máme použiť, resp. kedy zameniť jeden typ liečby za druhý. Určovanie jednotlivých subpopulácií T‑lymfocytov v periférnej krvi na základe ich rozdielnych membránových znakov informuje o odpovedi pacienta na daný preparát, ale vzhľadom na najnovšie poznatky o ich veľkej plasticite a schopnosti rediferenciácie z jednej subpopulácie na druhú (napr. TH2 na TH1 a pod.) [63] sa zrejme v budúcnosti presunieme na určovanie cytokínového profilu – ak je posunutý v smere prozápalových cytokínov (IL‑17, IFN ‑ γ, TNF-α i.) a v neprospech protizápalových (IL‑10, IL‑4, TGF‑β a i.), zrejme bude patologický proces intenzívnejší a liečba asi menej úspešná. Napr. pomer hladín imunosupresívneho IL‑10 k prozápalovému IL‑12 koreluje s aktivitou choroby a u pacientov, ktorí odpovedajú na IFN‑β, sa zlepšuje, t.j. stúpa. Podobný zmysel má aj sledovanie hladín adhezívnych a ko‑inhibičných molekúl (VLA ‑ 4, LFA ‑ 1, VCAM‑1, CTLA ‑ 4, TIM ‑ 3) a niektorých chemokínov (RANTES, MIP‑1α). Napr. ak úspešne liečime pacienta IFN‑β, hladiny jeho adhezívnych molekúl v periférnej krvi stúpajú a v membránach lymfocytov klesajú; hladiny uvedených chemokínov tiež klesajú [64]. Ako liečiť, kedy terapiu meniť, či použiť kombináciu liečiv, zostáva teda zatiaľ vždy na rozhodnutí lekára, a to už aj vzhľadom k tomu, že SM tiež nie je jednotná choroba, ale má svoje odtiene, podmienené prevahou jedných alebo druhých imunopatologických procesov, ktoré u každého pacienta rozdielne dominujú. Samozrejme teoretický výskum bude lekárom v ich rozhodovaní pomáhať, a to tým viac, čím budeme lepšie poznať podstatu týchto procesov a ich vzájomnú prepojenosť.
V článku sa uvádzajú základné údaje o lymfocytoch, ich subpopuláciách, cytokínoch, povrchových molekulách buniek imunitného systému. Samozrejme charakter článku neumožňuje podrobnejšie sa týmito poznatkami zaoberať, vysvetľovať ich, resp. poukazovať na ich súvislosti. Rád by som preto odporučil čitateľov, ktorí by sa radi hlbšie ponorili do tejto oblasti imunológie na knižné publikácie, ktoré vyšli v ČR [65], SR [66] alebo v zahraničí [67]. Problematika autoimunitných chorôb je tiež fascinujúca a opäť možno o nej nájsť podrobnejšie informácie v monografiách, ktoré vyšli v ČR [65,68], v SR [69]alebo v zahraničí [70]. Napokon pre neurológov, ktorí by radi hlbšie prenikli do tajov ako sa imunitné mechanizmy uplatňujú na rozvoji autoimunitných procesoch postihujúcich nervový systém, možno odporučiť monografiu neuroimunológie [71].
Autor deklaruje, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Článok vznikol v rámci riešenia grantovej úlohy MŠ SR VEGA 1/0810/12.
prof. MUDr. Milan Buc, DrSc.
Imunologický ústav LF UK
Odborárske nám. 14
813 72 Bratislava
e-mail: milan.buc@fmed.uniba.sk
Prijato k recenzii: 11. 12. 2012
Prijato do tlače: 15. 1. 2013
Sources
1. Liu YJ. A unified theory of central tolerance in the thymus. Trends Immunol 2006; 27(5): 215 – 221.
2. Lan RY, Ansari AA, Lian ZX, Gershwin ME. Regulatory T cells: development, function and role in autoimmunity. Autoimmun Rev 2005; 4(6): 351 – 363.
3. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 2010; 10(7): 490 – 500.
4. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012; 30 : 531 – 564.
5. Mizoguchi A, Bhan AK. A case for regulatory B cells. J. Immunol 2006; 176(2): 705 – 710.
6. Park SG, Mathur R, Long M, Hosh N, Hao L, Hayden MS et al. T regulatory cells maintain intestinal homeostasis by suppressing gammadelta T cells. Immunity 2010; 33(5): 791 – 803.
7. Ohkura N, Sakaguchi S. Maturation of effector regulatory T cells. Nat Immunol 2011; 12(4): 283 – 284.
8. Ouyang W, Li MO. Foxo: in command of T lymphocyte homeostasis and tolerance. Trends Immunol 2011; 32(1): 26 – 33.
9. Langier S, Sade K, Kivity S. Regulatory T cells: the suppressor arm of the immune system. Autoimmun Rev 2010; 10(2): 112 – 115.
10. Shevach EM. Mechanisms of foxp3+ T regulatory cell ‑ mediated suppression. Immunity 2009; 30(5): 636 – 645.
11. Cvetanovich GL, Hafler DA. Human regulatory T cells in autoimmune diseases. Curr Opin Immunol 2010; 22(6): 753 – 760.
12. Tejera ‑ Alhambra M, Alonso B, Teijeiro R, Ramos ‑ Medina R, Aristimuno C, Valor L et al. Perforin expression by CD4+ regulatory T cells increases at multiple sclerosis relapse: sex differences. Int J Mol Sci 2012; 13(6): 6698 – 6710.
13. Hori S. Developmental plasticity of Foxp3+ regulatory T cells. Curr Opin Immunol 2010; 22(5): 575 – 582.
14. Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012; 482(7385): 395 – 399.
15. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol 2012; 30 : 733 – 758.
16. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN et al. Neuropilin 1 is expressed on thymus ‑ derived natural regulatory T cells, but not mucosa ‑ generated induced Foxp3+ T reg cells. J Exp Med 2012; 209(10): 1723 – 1742.
17. Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4(+) T cell immunity. Nat Rev Immunol 2010; 10(4): 236 – 247.
18. Vitale G, Mion F, Pucillo C. Regulatory B cells: evidence, developmental origin and population diversity. Mol Immunol 2010; 48(1 – 3): 1 – 8.
19. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI et al. Regulatory B cells control T ‑ cell autoimmunity through IL‑21 – dependent cognate interactions. Nature 2012; 491(7423): 264 – 268.
20. Haas J, Hug A, Viehöver A, Fritzsching B, Falk CS,Filser A et al. Reduced suppressive effect of CD4+CD25 high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol 2005; 35(11): 3343 – 3352.
21. Kumar M, Putzki N, Limmroth V, Remus R, Lindemann M, Knop D et al. CD4+CD25+FoxP3+ T lymphocytes fail to suppress myelin basic protein‑induced proliferation in patients with multiple sclerosis. J Neuroimmunol 2006; 180(1 – 2): 178 – 184.
22. Lowther DE, Hafler DA. Regulatory T cells in the central nervous system. Immunol Rev 2012; 248(1): 156 – 169.
23. Praksova P, Stourac P, Bednarik J, Vlckova E, Mikulkova Z, Michalek J. Immunoregulatory T cells in multiple sclerosis and the effect of interferon beta and glatiramer acetate treatment on T cell subpopulations. J Neurol Sci 2012; 319(1 – 2): 18 – 23.
24. Joller N, Peters A, Anderson AC, Kuchroo VK. Immune checkpoints in central nervous system autoimmunity. Immunol Rev 2012; 248(1): 122 – 139.
25. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol 2005; 23 : 683 – 747.
26. Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood ‑ brain barriers. Trends Immunol 2012; 33(12): 579 – 589.
27. Krumbholz M, Theil D, Steinmeyer F, Cepok S, Hemmer B, Hofbauer M et al. CCL19 is constitutively expressed in the CNS, up ‑ regulated in neuroinflammation, active and also inactive multiple sclerosis lesions. J Neuroimmunol 2007; 190(1 – 2): 72 – 79.
28. Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest 2012; 122(4): 1180 – 1188.
29. Peelen E, Damoiseaux J, Smolders J, Knippenberg S,Menheere P, Tervaert JW et al. Th17 expansion in MS patients is counterbalanced by an expanded CD39+ regulatory T cell population during remission but not during relapse. J Neuroimmunol 2011; 240 – 241 : 97 – 103.
30. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364(9451): 2106 – 2112.
31. Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R et al. Mechanisms of disease: aquaporin‑4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol 2008; 4(4): 202 – 214.
32. Wang HH, Dai YQ, Qiu W, Lu ZQ, Peng FH, Wang YGet al. Interleukin‑17 - secreting T cells in neuromyelitis optica and multiple sclerosis during relapse. J Clin Neurosci 2011; 18(10): 1313 – 1317.
33. Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol 2012; 11(6): 535 – 544.
34. Palace J, Leite MI, Nairne A, Vincent A. Interferon Beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol 2010; 67(8): 1016 – 1017.
35. Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC et al. Anti‑aquaporin‑4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol 2012; 71(3): 314 – 232.
36. Graber JJ, McGraw CA, Kimbrough D, Dhib ‑ Jalbut S. Overlapping and distinct mechanisms of action of multiple sclerosis therapies. Clin Neurol Neurosurg 2010; 112(7): 583 – 591.
37. Chen M, Chen G, Deng S, Liu X, Hutton GJ, Hong J. IFN‑beta induces the proliferation of CD4+CD25+Foxp3+ regulatory T cells through upregulation of GITRL on dendritic cells in the treatment of multiple sclerosis. J Neuroimmunol 2012; 242(1 – 2): 39 – 46.
38. Burger D, Molnarfi N, Weber MS, Brandt KJ, Benkhoucha M, Gruaz L et al. Glatiramer acetate increases IL‑1 receptor antagonist but decreases T cell‑induced IL‑1beta in human monocytes and multiple sclerosis. Proc Natl Acad Sci USA 2009; 106(11): 4355 – 4359.
39. Lalive PH, Neuhaus O, Benkhoucha M, Burger D, Hohlfeld R, Zamvil SS et al. Glatiramer acetate in the treatment of multiple sclerosis: emerging concepts regarding its mechanism of action. CNS Drugs 2011; 25(5): 401 – 414.
40. Haas J, Korporal M, Balint B, Fritzsching B, Schwarz A, Wildemann B. Glatiramer acetate improves regulatory T ‑ cell function by expansion of naive CD4(+)CD25(+) FOXP3(+) CD31(+) T ‑ cells in patients with multiple sclerosis. J Neuroimmunol 2009; 216(1 – 2): 113 – 117.
41. Martelli AM, Tabellini G, Bressanin D, Ognibene A,Goto K, Cocco L et al. The emerging multiple roles of nuclear Akt. Biochim Biophys Acta 2012; 1823(12): 2168 – 2178.
42. Rossi S, Motta C, Studer V, Monteleone F, De Chiara V, Buttari F et al. A genetic variant of the anti‑apoptotic protein Akt predicts natalizumab‑induced lymphocytosis and post‑natalizumab multiple sclerosis reactivation. Mult Scler 2013; 19(1): 59 – 68.
43. Mancini N, Clementi M, Burioni R. Natalizumab‑associated progressive multifocal leukoencephalopathy. N Engl J Med 2012; 367(9): 871 – 872.
44. Girkontaite I, Sakk V, Wagner M, Borggrefe T, Tedford K, Chun J et al. The sphingosine ‑ 1 - phosphate (S1P) lysophospholipid receptor S1P3 regulates MAdCAM ‑ 1+ endothelial cells in splenic marginal sinus organization. J Exp Med 2004; 200(11): 1491 – 1501.
45. Cinamon G, Matloubian M, Lesneski MJ, Xu Y, Low C, Lu T et al. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol 2004; 5(7): 713 – 720.
46. Sawicka E, Dubois G, Jarai G, Edwards M, Thomas M, Nicholls A et al. The sphingosine 1 - phosphate receptor agonist FTY720 differentially affects the sequestration of CD4+/ CD25+ T ‑ regulatory cells and enhances their functional activity. J Immunol 2005; 175(12): 7973 – 7980.
47. Havla JB, Pellkofer HL, Meinl L, Gerdes LA, Hohlfeld R, Kümpfel T. Rebound of disease activity after withdrawal of fingolimod (FTY720) treatment. Arch Neurol 2012; 69(2): 262 – 264.
48. Gross CM, Baumgartner A, Rauer S, Stich O. Multiple sclerosis rebound following herpes zoster infection and suspension of fingolimod. Neurology 2012; 79(19): 2006 – 2007.
49. Rigau V, Mania A, Béfort P, Carlander B, Jonquet O,Lassmann H et al. Lethal multiple sclerosis relapse after natalizumab withdrawal. Neurology 2012; 79(22): 2214 – 2216.
50. Marousi S, Travasarou M, Karageorgiou CE, Gheuens S, Koralnik IJ. Simultaneous PML‑IRIS after discontinuation of natalizumab in a patient with MS. Neurology 2012; 79(21): 2160.
51. Wolf AM, Eller K, Zeiser R, Dürr C, Gerlach UV, Sixt M et al. The sphingosine 1 - phosphate receptor agonist FTY720 potently inhibits regulatory T cell proliferation in vitro and in vivo. J Immunol 2009; 183(6): 3751 – 3760.
52. Suntharalingam G, Perry MR, Ward S, Brett SJ,Castello ‑ Cortes A, Brunner MD et al. Cytokine storm in a phase 1 trial of the anti‑CD28 monoclonal antibody TGN1412. N Engl J Med 2006; 355(10): 1018 – 1028.
53. Barun B, Bar ‑ Or A. Treatment of multiple sclerosis with anti‑CD20 antibodies. Clin Immunol 2012; 142(1): 31 – 37.
54. Ray A, Basu S, Williams CB, Salzman NH, Dittel BN.A novel IL‑10 – independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol 2012; 188(7): 3188 – 3198.
55. Stashenko P, Nadler LM, Hardy R, Schlossman SF. Characterization of a human B lymphocyte ‑ specific antigen. J Immunol 1980; 125(4): 1678 – 1685.
56. Saidha S, Eckstein C, Calabresi PA. New and emerging disease modifying therapies for multiple sclerosis. Ann N Y Acad Sci 2012; 1247 : 117 – 1137.
57. Yamamura T, Miyake S. B ‑ cell ‑ directed therapy: which B cells should be targeted and how? Immunotherapy 2012; 4(5): 455 – 457.
58. Thompson SA, Jones JL, Cox AL, Compston DA, Coles AJ. B ‑ cell reconstitution and BAFF after alemtuzumab (Campath ‑ 1H) treatment of multiple sclerosis. J Clin Immunol 2010; 30(1): 99 – 105.
59. Coles AJ, Wing M, Smith S, Coraddu F, Greer S, Taylor C et al. Pulsed monoclonal antibody treatment and autoimmune thyroid disease in multiple sclerosis. Lancet 1999; 354(9191): 1691 – 1695.
60. Bielekova B. Daclizumab therapy for multiple sclerosis. Neurotherapeutics 2013; 10(1): 55 – 67.
61. Brusko T, Bluestone J. Clinical application of regulatory T cells for treatment of type 1 diabetes and transplantation. Eur J Immunol 2008; 38(4): 931 – 934.
62. Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen ‑ specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol 2002; 169(9): 4712 – 4716.
63. Nakayamada S, Takahashi H, Kanno Y, O’Shea JJ. Helper T cell diversity and plasticity. Curr Opin Immunol 2012; 24(3): 297 – 302.
64. Graber JJ, Dhib ‑ Jalbut S. Biomarkers of disease activity in multiple sclerosis. J Neurol Sci 2011; 305(1 – 2): 1 – 10.
65. Krejsek J, Kopecký O. Klinická imunologie. Nucleus: Hradec Králové 2004.
66. Buc M. Základná a klinická imunológia. Veda: Bratislava 2012.
67. Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. 7th ed. Philadelphia: Saunders, Elsevier 2012.
68. Schoenfeld Y, Fučíková T, Bartůňková J. Autoimunita – vnitřní nepřítel. Grada Publishing: Praha 2007.
69. Buc M. Autoimunita a autoimunitné choroby. Veda: Bratislava 2007.
70. Rich RR. Clinical Immunology. Principles and Practice. 3rd ed. Philadelphia: Mosby, Elsevier 2008.
71. Havrdová E. Neuroimunologie. Praha: Maxdorf 2001.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2013 Issue 3
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Mechanizmy spasticity a její hodnocení
- Lidské prionové nemoci v České republice – 10 let zkušeností s diagnostikou
- Myozitida s inkluzními tělísky se slabostí šíjových svalů a pozitivním efektem imunoglobulinu – kazuistika
- Extrakraniálně metastazující meningeomy