
Úskalí diagnostiky atypické formy kongenitální svalové dystrofie – parciálního deficitu merosinu – kazuistiky
Diagnostic Pitfalls of an Atypical Form of Congenital Muscular Dystrophy – Partial Merosin Deficiency – Case Reports
Congenital muscular dystrophies are a group of inherited muscle disorders characterized by early onset of muscle weakness and dystrophic changes in muscle biopsy. Mutation of the laminin α2 gene resulting in total or partial merosin deficiency is the most common cause in European population. Total loss of merosin manifests as congenital muscular dystrophy type 1A, partial deficiency results in limb girdle weakness of variable severity. Diagnosis of partial deficiency is based on clinical manifestation, brain magnetic resonance imaging (MRI) with apparent leucodystrophy, electromyography with documented peripheral neuropathy and genetic examination, muscle MRI pattern of pathological changes may be beneficial in differential diagnosis. Muscle biopsy shows dystrofic changes but immunohistochemistry may not reveal merosin deficiency and so it can be misleading. Treatment is symptomatic, correct diagnosis can help in predicting the course of the disease and associated risks and facilitates genetic counselling. It is likely that, due to the difficult diagnosis, partial merosin deficiency occurs more frequently than currently estimated. The authors present three illustrative case reports.
Key words:
congenital muscular dystrophy – partial merosin deficiency – laminin α2 – leucodystrophy – peripheral demyelinating neuropathy – magnetic resonance imaging
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Chinese summary - 摘要
诊断困难先天性肌的非典型 营养不良 - 偏阴虚Merosin's - 病例报告摘要
先天性肌营养不良症是一组罕见的遗传性肌一个疾病的特点是困难和形象改变营养不良发病初期
肌肉活检。欧洲人口是最常见的原因基因的突变层粘连蛋白α2,从而导致降低或不存在的生产Merosin's。临床区分这两种表现型:完全赤字Merosin's,即先天性经典的表型。肌营养不良1A型,并与Merosin有条件局部不足的表型可变严重性带肌肉无力。诊断依据不足部分在临床情况,脑白质病变的磁共振成像发现脑肌电变化与周围神经病变的证据,遗传检查过程中检查,辅助技术可能会被削弱肌纹磁共振。肌肉活检是很明显的营养不良的变化免疫组化可能成为常态,使诊断常复杂。治疗主要是对症诊断允许课程的预测和风险和遗传咨询。由于诊断陷阱可能
Merosin's 的偏阴虚较成功的展会更常见。作为一个例子作者目前三个案例报告。
关键词:
先天性肌营养不良 - 偏阴虚Merosin - 层粘连蛋白α2 -脑白质营养不良 - 主要是外周脱髓鞘病变 - 磁共振成像
Authors:
A. Slabá 1; L. Fajkusová 2; K. Stehlíková 2; R. Barresi 3; M. Kynčl 4; J. Zámečník 5; J. Haberlová 1
Authors‘ workplace:
Klinika dětské neurologie 2. LF UK a FN Motol, Praha
1; Centrum molekulární biologie a genové terapie, FN Brno
2; University of Newcastle, Institute of Human Genetics, International Centre for Life, Newcastle upon Tyne, United Kingdom
3; Klinika zobrazovacích metod 2. LF UK a FN Motol, Praha
4; Ústav patologie a molekulární medicíny, 2. LF UK a FN Motol, Praha
5
Published in:
Cesk Slov Neurol N 2017; 80/113(1): 101-106
Category:
Case Report
doi:
https://doi.org/10.14735/amcsnn2017101
Tento článek vznikl za podpory nadace Pohyb bez pomoci.
Overview
Kongenitální svalové dystrofie jsou skupina vzácných vrozených svalových onemocnění charakterizovaná časným počátkem obtíží a obrazem dystrofických změn ve svalové biopsii. V evropské populaci je nejčastější příčinou mutace v genu pro laminin α2, jejímž důsledkem je snížená či chybějící produkce merosinu. Klinicky odlišujeme dva fenotypy: úplný deficit merosinu, tzv. klasický fenotyp kongenitální svalové dystrofie typ 1A, a fenotyp podmíněný parciálním deficitem merosinu s variabilní tíží pletencové svalové slabosti. Diagnostika parciálního deficitu je založena na klinickém obraze, nálezu leukodystrofických změn na magnetické rezonanci mozku, elektromyografickém vyšetření s průkazem periferní neuropatie a genetickém vyšetření, pomocnou metodou může být vzorec postižení svalů při vyšetření magnetickou rezonancí. Ve svalové biopsii jsou patrné dystrofické změny, imunohistochemické vyšetření může být v normě, což diagnostiku mnohdy komplikuje. Léčba je symptomatická, stanovení diagnózy umožní predikci průběhu a rizik a genetické poradenství. Vzhledem k diagnostickým úskalím je pravděpodobné, že parciální deficit merosinu je častější, než se daří prokázat. Jako příklad autoři prezentují tři kazuistiky.
Klíčová slova:
kongenitální svalová dystrofie – parciální deficit merosinu – laminin α2 – leukodystrofie – periferní primárně demyelinizační neuropatie – magnetická rezonance
Úvod
Kongenitální svalové dystrofie (Congenital Muscular Dystrophy; CMD) jsou klinicky a geneticky heterogenní skupina vzácných vrozených onemocnění svalů. Společnou charakteristikou je časný nástup obtíží (typicky do 6. měsíce věku) a obraz dystrofických změn ve svalové biopsii [1]. Dědičnost je převážně autozomálně recesivní. Pro nedostatek dostupných informací v současné době nelze uvést přesná epidemiologická data, odhadovaná incidence těchto onemocnění je 1 : 20 000; prevalence je udávána 0,68– 2,5 na 100 000 a je pravděpodobně podhodnocena [2,3]. Je identifikováno nejméně 13 kauzálních genů [3]. Popis celé této heterogení skupiny přesahuje rámec tohoto sdělení.
V evropské populaci je jednou z nejčastějších jednotek z této skupiny onemocnění merosin deficientní kongenitální svalová dystrofie typ 1A (MDC1A), dle odhadů tvoří asi 30 % všech CMD [3– 6]. Je způsobena mutacemi v genu pro laminin α2 (LAMA2), který se nachází na dlouhém raménku chromozomu 6 v lokusu 6q22– 23. Lamininy jsou jedny z nejrozšířenějších glykoproteinů bazální membrány. Jedná se o heterotrimery, složené z řetězců označovných α, β a γ. Těchto podjednotek existuje několik typů a jejich výskyt a kombinace jsou tkáňově specifické. Merosin (neboli laminin-2 či podle novější nomenklatury laminin-211), jehož deficit je prokazován u MDC1A, je tvořen podřetězci α2, β1 a γ1. Jednotlivé molekuly jsou vzájemně propojeny v síťovitou strukturu a váží se jednak prostřednictvím nidogenu ke kolagenu IV, jednak na receptory α-dystroglykanů a integrinů v membráně příčně pruhovaných svalů. Dochází tak k propojení extracelulární matrix s cytoskeletem příčně pruhovaných svalů [7]. Typické klinické projevy úplného deficitu merosinu zahrnují těžkou kongenitální hypotonii periferního původu a svalovou slabost s dechovými a polykacími obtížemi. Pacienti nikdy nejsou schopni samostatné chůze, od počátku vývoje jsou odkázáni na mechanický vozík. Postupem času dochází k rozvoji sekundárních deformit – četných kontraktur a skoliózy. Průběh je velmi pomalu progredující, typickým příznakem druhé dekády věku je respirační insuficience s nutností dechové podpory. Až u třetiny pacientů jsou v adolescenci a dospělosti popisovány epileptické záchvaty. Intelekt pacientů nebývá postižen. V séru lze prokázat zvýšenou hladinu svalových enzymů kreatinkinázy (CK), alaninaminotransferázy, aspartátaminotransferázy a laktátdehydrogenázy. CK přesahuje normu obvykle 4–20násobně. Při magnetické rezonanci (MR) mozku nacházíme známky signálových změn v bílé hmotě (obr. 1). Elektromyografickým vyšetřením (EMG) lze prokázat myopatické změny a lehkou, obvykle asymptomatickou, periferní primárně demyelinizační motorickosenzitivní neuropatii. Ve svalové biopsii jsou patrné nespecifické dystrofické změny zahrnující degeneraci svalových vláken, kolísání jejich velikosti s převahou atrofie a zvýšené množství pojivové tkáně. Imunohistochemie (IHC) ukáže úplné chybění LAMA2. Výjimečně bývají popisovány případy s přidruženou mentální retardací, poruchami migrace neuronů či subklinickým kardiálními příznaky [8,9].
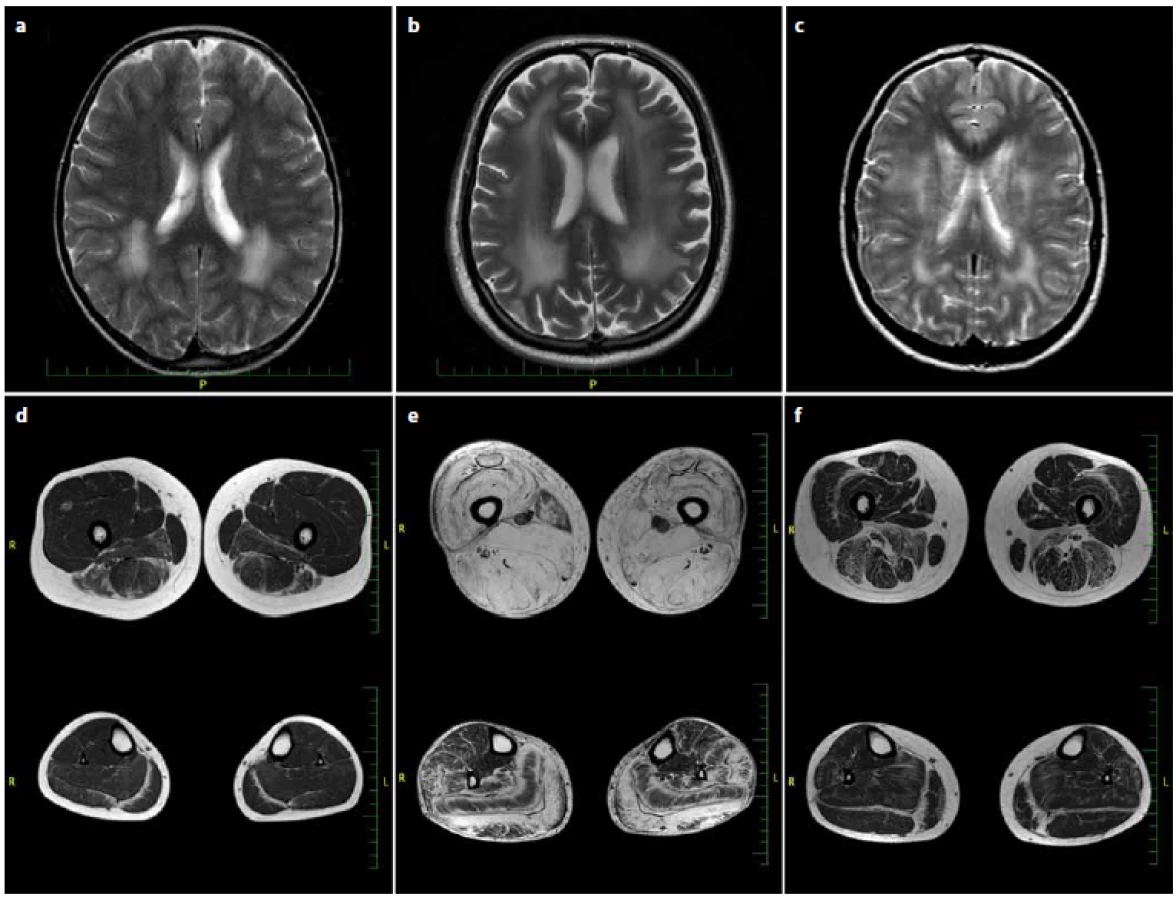
Pacienti se zachovanou reziduální expresí merosinu mají variabilnější fenotyp. Projevy onemocnění jsou mírnější, pacienti jsou alespoň dočasně schopni samostatné chůze [8]. Typická je lehká generalizovaná svalová slabost s pletencovým maximem, nástup obtíží bývá velmi časný, během prvních let života [10]. Progrese je velmi mírná. Brzy dochází k rozvoji sekundárních kontraktur zejména v oblasti Achillových šlach. V pozdní dospělosti je popisována ztráta schopnosti samostatné chůze a v některých případech i výskyt epileptických záchvatů. Svalové enzymy bývají mírně zvýšené či zcela v normě. Z hlediska diferenciální diagnostiky myopatií a svalových dystrofií je významná skutečnost, že i u parciálního deficitu merosinu jsou konstantně na MR mozku přítomny signálové změny v bílé hmotě a nález primárně demyelinizační periferní neuropatie. Ve svalové biopsii je nespecifický obraz dystrofických změn, imunohistochemie (IHC) může odhalit sníženou expresi merosinu, nicméně při použití pouze jedné protilátky se částečné chybění merosinu nemusí prokázat. Při podezření na parciální deficit merosinu se proto v literatuře doporučuje průkaz LAMA2 protilátkami proti dvěma fragmentům (80 kDa i 300 kDa) [11]. Potvrzením klinického podezření na diagnózu parciálního či úplného deficitu merosinu je genetické vyšetření, které je v České republice dostupné metodou masivně paralelního sekvenování panelu genů kauzálních pro nervosvalové nemoci s ověřením patologických variant Sangerovým sekvenováním v Centru molekulární biologie a genové terapie ve FN Brno.
Kauzální léčba v současnosti není možná, stanovení přesné diagnózy slouží k predikci průběhu onemocnění a z něj plynoucích rizik a je klíčové pro cílené genetické poradenství v rodině a případnou prenatální či preimplantační diagnostiku.
Pacienti
Pacient M.F.
První pacient je t.č. 13letý chlapec, syn nepříbuzných rodičů. Matka pacienta je zdráva, otec byl od dětství sledován pro nejasnou myopatii a bude podrobně popsán níže. Perinatální i kojenecké období pacienta M.F. bylo bez nápadností, první obtíže rodiče pozorovali ve 3 letech věku, kdy při běhu nestačil vrstevníkům.
Na Klinice dětské neurologie 2. LF UK a FN Motol byl poprvé vyšetřen ve věku 8 let. V této době zvládal zcela bez obtíží chůzi na běžné vzdálenosti i do schodů. Při objektivním neurologickém vyšetření byla patrná lehká ptóza oboustranně, další nález na hlavových nervech byl bez patologie, chlapec měl mírně oslabené šíjové svaly, svalová síla na horních končetinách i dolních končetinách byla dle svalového testu nižší v oblasti pletence (4/ 5 MRC) a rovněž na horních i dolních končetinách byly nižší, ale výbavné šlachookosticové reflexy. Oboustranně byly patrné lehké kontraktury Achillových šlach. Čití vibrační i taktilní bylo v normě. Intelekt chlapce není postižen.
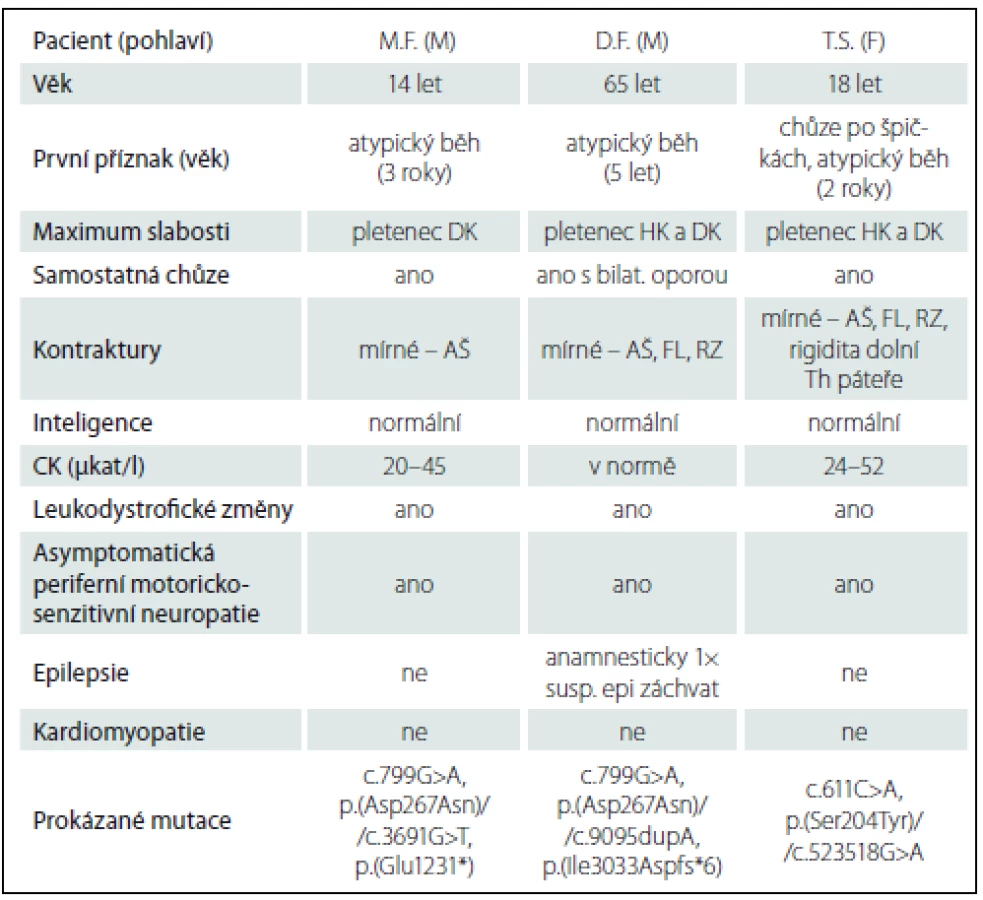
Při biochemickém vyšetření séra byla opakovaně zachycena zvýšená hladina CK (v rozmezí 20– 45 μkat/ l, norma do 2,27 μkat/ l) a myoglobinu (180– 230 μg/ l, norma do 50 μg/ l). Při EMG vyšetření byla kromě očekávaného myogenního vzorce v jehlové EMG zachycena také diskrétní primárně demyelinzační motorickosenzitivní periferní neuropatie. Vyšetření evokovaných potenciálů BAEP a pVEP bylo abnormální pro prodlouženou latenci vln III i V, resp. vlny p 100. Náhodně pro úraz hlavy bylo v 9 letech bylo provedeno CT a následně MR mozku s nálezem signálových změn v bílé hmotě (obr. 1). MR svalů dolních končetin zobrazila atrofizaci a tukové přestavby zejména v posteriorním a mediálním kompartmentu stehna a zadním kompartmentu lýtka (obr. 1). Kardiologické vyšetření bylo bez patologického nálezu.
Klinický obraz odpovídal obrazu kongenitální myopatie či svalové dystrofie. Vzhledem k obdobným obtížím u otce byly zvažovány klinické jednotky s autozomálně dominantním typem dědičnosti. Překvapením byl nález při vyšetření panelu genů v Centru molekulární biologie a genové terapie ve FN Brno metodou masivně paralelního sekvenování („next generation sequencing“), kde byly prokázány mutace v obou alelách genu LAMA2 (NM_000426): c.799G>A, p.(Asp267Asn)/c.3691G>T, p.(Glu1231*). Varianta c.799G>A zatím nebyla popsána, ale je přítomna i u pacientova otce, u kterého diagnóza parciálního deficitu merosinu byla potvrzena i imunohistochemií svalové biopsie (viz níže). Varianta c.3691G>T je rovněž zatím nepopsána, vede však ke vzniku stop kodonu, a lze tudíž předpokládat její poškozující charakter. Vzhledem ke skutečnostem, že obě nalezené varianty v LAMA2 genu jsou s vysokou pravděpodobností kauzální a klinický obraz je zcela ve shodě se zatím popsanými kazuistikami pacientů s parciálním deficitem merosinu, další vyšetření již nebyla indikována [8,10,11]. Po stanovení diagnózy bylo doplněno EEG s normálním nálezem, epileptický záchvat u chlapce nikdy nebyl pozorován. Matka pacienta je heterozygot pro c.3691G>T v LAMA2 genu.
Pacient D.F.
Pacient D.F., otec pacienta M.F., je 64letý muž. I u něj se obtíže začaly projevovat v předškolním věku. Od 4 let je sledován neurologem pro atypický stereotyp běhu. V klinickém obraze od počátku dominuje lehká pletencová svalová slabost, z tohoto důvodu byl v dětství sledován pod diagnózou Beckerova svalová dystrofie, později pro blíže neurčenou pletencovou svalovou dystrofii. V 17 letech prodělal operační prolongaci Achillových šlach. Od 18 let věku zvládal chůzi do schodů jen s oporou jedné horní končetiny (HK). Svalová slabost s věkem pomalu progredovala, od 60 let potřebuje oboustrannou oporu při chůzi, je neschopen vyjít schody či vstát ze země bez pomoci. V 61 letech byl hospitalizován pro nejasný stav poruchy vědomí, tato událost nebyla etiologicky uzavřena, pravděpodobná se jeví etiologie epileptická.
V objektivním neurologickém nálezu ve věku 64 let byla patrná ptóza a hypomimie, na končetinách dominovala proximální svalová slabost. HK pacient nezvedl nad horizontálu (flexe paže 4/ 5, extenze paže 4/ 5 MRC), pohyb v kyčli a koleni byl možný jen s vyloučením gravitace (extenze v kyčli 3/ 5, flexe v kyčli a flexe a extenze v koleni 2+/ 5 MRC). Distálně byla svalová síla v normě či jen mírně snížena. Šlachookosticové reflexy na končetinách byly povšechně nevýbavné. U pacienta byly mírně zkrácené rotátory zápěstí. Byla patrná hypotrofie svalstva dolních končetin s výjimkou lýtkových svalů, které byly hypertrofické. Patrné byly kontraktury Achillových šlach a perimaleolární otoky. Citlivost končetin nebyla postižena. Intelekt pacienta je zcela v normě, celý život pracoval jako vysoce postavený manažer.
Svalové enzymy jsou t.č. u pacienta D.F. při opakovaném vyšetření séra nezvýšeny. EMG je abnormální, s nálezem primárně demyelinizační motorickosenzitivní periferní neuropatie, s nálezem myogenních změn při jehlové EMG. Na MR mozku (doplněno ve věku 61 let) byly popsány signálové změny bílé hmoty. Ve svalové biopsii z oblasti m. deltoideus byl ve věku 50 let obraz dystrofických změn se zvýšenou intersticiální fibrózou, enzymatické a IHC a nález v elektronové mikroskopii byl v normě. Kardiologické vyšetření bylo u tohoto pacienta opakovaně bez patologického nálezu.
S novými možnostmi vyšetření genů kauzálních pro kongenitální myopatie jsme v roce 2012 spolu s výše uvedeným vyšetřením u syna doplnili vyšetření panelu neuromuskulárních genů metodou masivně paralelního sekvenování v Centru molekulární biologie a genové terapie ve FN Brno. Byly rovněž nalezeny dvě mutace v LAMA2 genu (NM_000426): c.799G>A, p.(Asp267Asn)// c.9095dupA, p.(Ile3033Aspfs*6). Varianta c.9095dupA již byla publikována jako kauzální, varianta c.799G>A dosud popsána nebyla, ale vyskytuje se i u syna s obdobným klinickým obrazem (viz výše). K ověření diagnózy bylo díky zahraniční spolupráci doplněno v ČR nedostupné imunohistochemické vyšetření vzorku svalové biopsie dvěma protilátkami proti LAMA2 (proti 80 kDa a 300 kDa fragmentům), průkaz parciálního deficitu merosinu byl tímto vyšetřením potvrzen. Vyšetření bylo provedeno v imunohistochemické laboratoři John Walton Muscular Dystrophy Research Centre v Newcastlu v UK dr. Ritou Barresi. Byla doplněna magnetická rezonance svalů dolních končetin, kde je obraz tukové přestavby všech svalů stehna se zbytky svalové tkáně v m. sartorius. Z lýtkových svalů jsou nejvíce postiženy m. gastrocnemius a m. soleus. Konečná diagnóza pacienta D.F. byla stanovena až 60 let po objevení prvních obtíží.
Pacientka T.S.
Třetí pacientka, nyní 17letá dívka, dcera nepříbuzných zdravých rodičů, byla od 2 let věku sledována neurologem pro lehký pletencový myopatický syndrom. Prvními příznaky byly atypický stereotyp chůze a běhu, tendence k chůzi po špičkách, obtíže při chůzi do schodů a zvýšená unavitelnost. Pre- i perinatální anamnéza byla bez abnormit. Motorický vývoj byl dle dokumentace v širší normě, chodila od 18. měsíce. Mentálně vždy odpovídala věku. Během první dekády života se její obtíže zásadně neměnily. Pro přidružený nález signálových změn v bílé hmotě na MR mozku byla ve věku 13 let detailně vyšetřována v Ústavu dědičných metabolických poruch 1. LF UK a VFN v Praze. Zde bylo mimo jiné provedeno i EMG s nálezem primárně demyelinizační periferní neuropatie. Pro nález na EMG byla odeslána ke konzultaci na naše pracoviště.
Při objektivním neurologickém vyšetření byla zřejmá mírná hypomimie a lehká bilaterální ptóza. Na horních končetinách byly patrné mírné (do 10°) kontraktury flexorů paží a rotátorů zápěstí. Svalová síla končetin byla lehce oslabena s maximem proximálně (v oblasti extenzorů paže 5– / 5 MRC, flexe kyčle 4– / 5 MRC, extenze 3+/ 5 MRC), distálně byla svalová síla v normě. Šlachookostcové reflexy byly povšechně nižší, ale výbavné, čití v normě, jemná motorika rovněž nepostižena. Na dolních končetinách byla viditelná atrofie svalstva stehen a kontraktury Achillových šlach bilaterálně. Páteř byla s lehkou rigiditou v bederní oblasti, bez známek skoliózy. Nápadná byla chůze po špičkách a vstávání se známkami šplhu.
Při biochemickém vyšetření séra byla opakovaně zvýšená hladina CK (v rozmezí 24– 52 μkat/ l, norma do 2,27 μkat/ l) a zvýšená hladina myoglobinu (250 μg/ l, norma do 50 μg/ l). EMG vyšetření potvrdilo symetrickou primárně demyelinzační motorickosenzitivní periferní neuropatii v kombinaci s myogenní lézí při jehlové EMG. MR mozku byla se známkami signálových změn v bílé hmotě (obr. 1). Ve věku 13 let pacientka podstoupila svalovou biopsii z m. gastrocnemius. Nález byl popsán jako necharakteristický, s kombinací lehké myogenní a neurogenní léze, imunohistochemické vyšetření základních strukturálních proteinů vč. merosinu bylo v normě. Byla doplněna MR svalů dolních končetin (obr. 1) s nálezem stranově symetrické tukové degenerace v oblasti svalů stehenních i lýtkových, s maximem změn v dorzálním kompartmentu stehna (m. biceps femoris a m. adductor magnus) a zadní skupině svalů lýtka (m. gastrocnemius a m. soleus). Kardiologické vyšetření bylo v normě.
Na základě klinického obrazu a pomocných vyšetření bylo indikováno vyšetření genu LAMA2 s nálezem dosud nepopsaných, dle predikčního programu Human Splicing Finder potenciálně patologických změn v obou alelách genu LAMA2 (NM_000426): c.611C>A, p.(Ser204Tyr)/ c.523518G>A, potenciálně ovlivňující sestřih mRNA. Otec pacientky nese mutaci c.5235-18G>A, matka analyzována nebyla. Vzhledem k nejasnostem ohledně kauzality zachycených sekvenčních změn byla revidována svalová biopsie, z výše zmíněných důvodů v imunohistochemické laboratoři John Walton Muscular Dystrophy Research Centre v Newcastlu v UK, kde při použití dvou protilátek byl imunohistochemicky parciální deficit merosinu potvrzen.
Po ověření diagnózy bylo pro možnost přidružené epilepsie doplněno EEG vyšetření, které bylo s normálním nálezem, žádný epileptický záchvat u pacientky pozorovám nebyl.
Data týkající se pacientů přehledně shrnuje tab. 1.
Diskuze
Na příkladech pacientů autoři ilustrovali možné klinické projevy parciálního deficitu merosinu a obtíže při diagnostice tohoto onemocnění. Diferenciálně diagnosticky jsou u této jednotky zvažovány nemoci ze skupiny pletencových svalových dystrofií a skupiny kongenitálních myopatií. Klinicky se jedná o velmi pomalu progredující, zejména pletencovou svalovou slabost. Ztráta schopnosti chůze je velmi individuální, dle literárních údajů i našich tří pacientů je samostatná chůze většinou zachována do pozdní dospělosti. Svalové enzymy v periferní krvi mohou, ale nemusí být zvýšené. S normálními hodnotami svalových enzymů se setkáváme zejména u dospělých pacientů, u kterých je již patrná výrazná atrofizace svalstva. Přidružené změny charakteru signálových změn v bílé hmotě na MR mozku a subklinická, elektromyograficky prokazatelná periferní demyelinizační neuropatie mohou na jednu stranu pomoci odhalit správnou diagnózu, na stranu druhou mohou být zdrojem diagnostických rozpaků a vést k úvahám o metabolické či primárně neuropatické příčině obtíží. V případě našich pacientů M.F. a D.F. byl neobvyklý i výskyt ve dvou generacích, který vzhledem k výskytu tří patologických alel v rodině imitoval autozomálně dominantní dědičnost.
Diagnóza parciálního deficitu merosinu byla u všech případů stanovena na základě anamnézy, klinického obrazu, nálezu změn na MR mozku, na základě genetického vyšetření a ve dvou případech na nálezu ve svalové biopsii. Byla u všech případů taktéž podpořena charakterem změn na MR svalů (jedná se pouze o podpůrné diagnostické kritérium, neboť u této klinické jednotky zatím není dostatek literárních údajů týkajících se vzorce postižení svalů při MR vyšetření) [12]. U našich pacientů byly v oblasti stehen nejdříve postiženy svaly zadní skupiny, následovaly mm. vastii. a m. gracilis. Na lýtku byl u všech pacientů charakteristický obraz tukové infiltrace mezi m. soleus a m. gatrocnemius, postupující z periferie do centra m. soleus a m. gastrocnemius medialis a lateralis, což může připomínat deficit kolagenu VI(tj. kongenitální svalovou dystrofii typ Bethlem) [12,13]. Přední a laterální skupina svalů byla postižena méně. Nejdéle zachovány byly v našem případě m. sartorius, m. tibialis posterior a m. flexor digit. long. (obr. 1).
Závěr
Parciální deficit merosinu má typický klinický obraz pomalu progredující svalové slabosti s pletencovým maximem. Vykazuje charakteristické abnormity při pomocných vyšetřeních:
- obraz signálových změn na MR mozku,
- EMG s nálezem periferní primárně demyelinizační motorickosenzitivní periferní neuropatie v kombinaci s myogenní lézí v jehlové EMG,
- obraz dystrofických změn ve svalové biopsii (IHC nemusí být deficit merosinu zachycen),
- charakteristický obraz dystrofických změn na MR svalů.
I přes tento charakteristický obraz bývá parciální deficit často neobjasněn, v uvedených kazuistikách diagnostika trvala i několik desítek let. Doporučujeme zvážit tuto diagnózu ve všech případech kombinace svalové dystrofie, periferní primárně demyelinizační motorickosenzitivní neuropatie a signálových změn bílé hmoty na MR mozku. V diagnostickém algoritmu doporučujeme klinické vyšetření, stanovení hladiny CK, EMG vyšetření a v případě podezření na parciální deficit merosinu MR mozku a MR svalů. K potvrzení klinického podezření je indikováno genetické vyšetření metodou masivně paralelního sekvenování panelu neuromuskulárních genů ve FN Brno. Svalovou biopsii doporučujeme až při nejasných nálezech, např. v případě nových, zatím nepopsaných variant LAMA2 genu.
S narůstajícími možnostmi a rozvojem genetiky významně přibývá etiologicky objasněných případů dříve nezařazených či nespecifikovaných myopatií. Zvyšuje se dostupnost a snižuje nákladnost genetických vyšetření, která se tak stávají rutinní součástí vyšetřovacích postupů. I přes nemožnost kauzální léčby většiny dedičných svalových onemocnění je stanovení diagnózy podstatné pro predikci průběhu a rizik (v našem případě se jedná o velmi pomalou progresi s riziky vzniku kontraktur a rozvoje sekundární epilepsie). Dále umožní genetické poradenství a cílenou prenatální či preimplantační diagnostiku v postižených rodinách. Obecným přínosem určení kauzální mutace je potenciál pro další vědecké zkoumání s rozšiřováním možností budoucí terapie vč. terapie genové.
Seznam použitých zkratek
BAEP – kmenové sluchové evokované potenciály
CK – kreatinkináza
CMD – kongenitální svalové dystrofie
CT – počítačová tomografie
EMG – elektromyografie
IHC – imunohistochemie
LAMA2 – laminin α2
MDC1A – kongenitální svalová dystrofie typ 1A
MR – magnetická rezonance
MRC – Medical Research Council
pVEP – zrakové evokované potenciály, typ „pattern reversal“
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Alžběta Slabá
Klinika dětské neurologie
2. LF UK a FN Motol
V Úvalu 84
150 06 Praha
e-mail: alzbeta.slaba@fnmotol.cz
Přijato k recenzi: 14. 6. 2016
Přijato do tisku: 23. 8. 2016
Sources
1. Reed UC. Congenital muscular dystrophy. Part I: a review of phenotypical and diagnostic aspects. Arq Neuropsiquiatr 2009;67(1):144– 68.
2. Sparks S, Quijano-Roy S, Amy Harper A. Congenital muscular dystrophy overview. [accessed 2016 May 20]. Available from URL: http://www.ncbi.nlm.nih.gov/books/NBK1291.
3. Ciafaloni E, Chinnery PF, Griggs RC. Evaluation and treatment of myopathies. 2nd ed. Oxford, GB: Oxford University Press 2014.
4. Vajsar J, Kraus J. Kongenitální svalové dystrofie. Neurol Praxi 2012;13(4):195– 7.
5. Graziano A, Bianco F, D’Amico A, et al. Prevalence of congenital muscular dystrophy in Italy: a population study. Neurology 2015;84(9):904– 11. doi: 10.1212/ WNL.0000000000001303.
6. Clement EM, Feng L, Mein R, et al. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001– 2008. Neuromuscul Disord 2012;22:522– 7. doi: 10.1016/ j.nmd.2012.01.010.
7. Miyagoe-Suzuki Y, Nakagawa M, Takeda S. Merosin and congenital muscular dystrophy. Microsc Res Tech 2000;48:181– 91.
8. Kim HJ, Choi YC, Park HJ, et al. Congenital muscular dystrophy type 1A with residual merosin expression. Korean J Pediatr 2014;57(3):149– 52. doi: 10.3345/ kjp. 2014.57.3.149.
9. Seeman P, Šišková D, Perníková I, et al. Kongenitální svalová dystrofie s úplným defektem merosinu – první dva případy prokázané v České republice – typický fenotyp a průkaz pomocí biopsie kůže. Cesk Slov Neurol N 2002;65/ 98(1):37– 44.
10. Chan SH, Foley AR, Phadke R, et al. Limb girdle muscular dystrophy due to LAMA2 mutations: diagnostic difficulties due to associated peripheral neuropathy. Neuromuscul Disord 2014;24(8):677– 83. doi: 10.1016/ j.nmd.2014.05.008.
11. Sewry CA, Naom I, D‘Alessandro M, et al. Variable clinical phenotype in merosin-deficient congenital muscular dystrophy associated with differential immunolabelling of two fragments of the laminin alpha 2 chain. Neuromuscul Disord 1997;7(3):169– 75.
12. Straub V, Carlier PG, Mercuri E. TREAT-NMD workshop: pattern recognition in genetic muscle diseases using muscle MRI: 25– 26 February 2011, Rome, Italy. Neuromuscul Disord 2012;22(Suppl 2):S42– 53. doi: 10.1016/ j.nmd.2012.08.002.
13. Nelson I, Stojkovic T, Allamand V, et al. Laminin α2 Deficiency-Related Muscular Dystrophy Mimicking Emery-Dreifuss and Collagen VI related Diseases. J Neuromuscul Dis 2015;2(3):229– 40. doi: 10.3233/ JND-150093.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2017 Issue 1
- Metamizole vs. Tramadol in Postoperative Analgesia
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Současný pohled na kontraindikace a komplikace elektromyografie
- Základní neurologické vyšetření – nastal čas pro změny?
- Endoskopická exstirpace koloidní cysty III. mozkové komory
- Periodické pohyby končetinami ve spánku jsou závažnější u narkolepsie s kataplexií než u narkolepsie bez kataplexie