
Akutní myelitida u dětí – soubor 20 pacientů
Acute pediatric myelitis – cohort of 20 patients
Aim:
In this study, we focused on the current issue of pediatric myelitis and shared our clinical experience.
Patients and methods:
20 patients (10 girls, 10 boys; age 4– 17 years, median 14 years; follow-up period 1– 73 months, median 15 months) with acute spinal syndrome and without encephalopathy; acute spinal compression was excluded and inflammatory etiology was considered. Analysis of clinical and para-clinical data (magnetic resonance imaging [MRI] of the brain and spinal cord, cerebrospinal fluid examination, microbiological and immunological testing, serum aquaporin 4 antibodies). We evaluated the clinical course, response to acute therapy and the final diagnosis; whether the current international diagnostic criteria were fulfilled for clinically isolated syndrome (CIS), pediatric multiple sclerosis (MS) or neuromyelitis optica spectrum disorders (NMOSD).
Results:
One patient had infectious Borrelia-related myelitis. Acute myelitis as a symptom of CIS/ MS was in eight cases and as a symptom of NMOSD in three cases. Idiopathic myelitis was diagnosed in eight children. CIS/ MS patients in comparison to NMOSD patients had different clinical courses, laboratory findings and responses to acute therapy. The most serious were patients with idiopathic myelitis – with rapid progression of spinal symptoms and extensive spinal lesions detected using MRI of spinal cord. They had poor response to corticotherapy. Some of these patients were successfully treated with combined immunosuppression.
Conclusions:
Acute myelitis is a complicated diagnosis, which requires a comprehensive approach and clinical experience.
Key words:
pediatric myelitis – neuromyelitis optica – multiple sclerosis – acute therapy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
H. Nohejlová 1,2; P. Nytrová 3; Z. Libá 2
Authors‘ workplace:
Neurologická klinika 2. LF UK a FN Motol, Praha
1; Klinika dětské neurologie 2. LF UK a FN Motol, Praha
2; Neurologická klinika a Centrum klinických neurověd, 1. LF UK a VFN v Praze
3
Published in:
Cesk Slov Neurol N 2018; 81(2): 213-219
Category:
Short Communication
doi:
https://doi.org/10.14735/amcsnn2018213
Overview
Cíl:
Naše práce upozorňuje na současnou problematiku akutní myelitidy u dětí a sdílí naše klinické zkušenosti.
Soubor a metodika:
20 pacientů (10 chlapců a 10 dívek; věk 4– 17 let, medián 14 let; doba sledování 1– 73 měsíců, medián 15 měsíců) s akutním míšním syndromem, bez encefalopatie a po vyloučení míšní komprese, u kterých byla zvažována zánětlivá etiologie. Analýza klinických dat a paraklinických vyšetření (MR mozku a míchy, likvor, mikrobiologický a imunologický skríning, protilátky proti akvaporinu 4 v séru). Hodnotili jsme: klinický průběh, odpověď na akutní terapii a výslednou diagnózu; zda byla naplněna diagnostická kritéria pro klinicky izolovaný syndrom (clinically isolated syndrome; CIS), RS nebo onemocnění ze spektra neuromyelitis optica (neuromyelitis optica spectrum disorders; NMOSD).
Výsledky:
Jeden pacient měl myelitidu boreliové etiologie. Akutní myelitida byla projevem/ součástí CIS/ RS u osmi a NMOSD u tří pacientů. Idiopatická myelitida byla u osmi dětí. Pacienti s CIS/ RS měli oproti pacientům s NMOSD rozdílné klinické i laboratorní projevy a odpověď na akutní terapii. Nejzávažnější byli pacienti s idiopatickou myelitidou, u kterých došlo k rychlému rozvoji neurologických příznaků s rozsáhlým nálezem na MR míchy. Odpověď na kortikoterapii byla u této skupiny nedostatečná, někteří profitovali z kombinované imunosuprese.
Závěr:
Akutní myelitida je komplikovaná diagnóza vyžadující komplexní přístup a klinické zkušenosti.
Klíčová slova:
myelitida u dětí – neuromyelitis optica – roztroušená skleróza – akutní terapie
Úvod
Akutní myelitida je u dětí vzácné a závažné onemocnění, které se projeví míšní dysfunkcí s typickým rozvojem příznaků v rozmezí 4 h až 21 dní [1]. Zánětlivé postižení bílé hmoty (s postižením míšní šedi nebo bez něj) vede k různě vyjádřenému senzomotorickému deficitu vč. možné autonomní dysfunkce se sfinkterovou poruchou. Oboustranný, i asymetrický, neurologický deficit spolu se zánětlivým nálezem v likvoru nebo ložiskovým sycením na MR míchy po podání kontrastní látky definují kompletní transverzální myelitidu (TM) [2,3].
Přesná incidence myelitid není známa, incidence TM se odhaduje na 1–3 případy/ milion dětí/ rok [4]. Infekční i autoimunitní zánět míchy je možný a jejich klinické a laboratorní projevy jsou si podobné. Pro infekční etiologii je průkazný nález patogenu v mozkomíšním moku nebo pozitivní protilátkový index při vyšetření protilátek v séru a likvoru [5]. Klinicky lze očekávat systémové projevy infekce a důležitá je epidemiologická souvislost [3]. Oproti tomu autoimunitní etiologie bývá často stanovena per exclusionem a v případě TM převažuje nad infekční [4]. Myelitida na autoimunitním podkladě může být projevem klinicky izolovaného syndromu (clinically isolated syndrome; CIS) nebo být součástí akutní demyelinizační encefalomyelopatie (ADEM). Pro ADEM je typické polyfokální postižení s dominující encefalopatií, zatímco CIS definuje monofokální nebo polyfokální postižení bez encefalopatie [5]. Už v době první ataky akutní myelitidy mohou být naplněna diagnostická kritéria RS (schéma 1) nebo onemocnění neuromyelitis optica (NMO) a jejího širšího spektra (neuromyelitis optica spectrum disorders; NMOSD) (schéma 2) [6–8]. Vzácněji je myelitida součástí systémových autoimunitních onemocnění, jako je např. systémový lupus erythematodes. Někdy zůstává příčina myelitidy neobjasněna (tab. 1) [5]. Mimo infekční a autoimunitní etiologii zánětu musíme vyloučit nezánětlivá onemocnění, jakými jsou především míšní komprese nebo ischemie. Na vaskulární etiologii pomýšlíme u náhlého a rychlého rozvoje transverzální míšní léze (< 4 h), zvlášť pokud se v anamnéze vyskytují rizikové faktory, jakými jsou trauma, koagulopatie, operace aorty apod. [9].
![Schéma 1. Zjednodušené diagnostické schéma pro RS s ohledem na pediatrickou populaci. Diagnostické schéma pro RS zahrnuje revizi McDonaldových kritérií z roku 2010, která zjednodušuje radiologická kritéria diseminace demyelinizačních ložisek v čase (lze odlišit podáním kontrastní látky a/nebo opakovaným MR vyšetřením) a prostoru (charakterizující počet a lokalizaci ložisek). Diseminace ložisek v čase a prostoru spolu s klinickými projevy definují RS [6,8]. Klinicky izolovaný syndrom je v našem případě reprezentován první atakou akutní myelitidy.](https://pl-master.mdcdn.cz/media/image/29035b730977efe0fd221a55d50b0cf9.png?version=1537793141)
![Schéma 2. Zjednodušené diagnostické schéma pro NMOSD s ohledem na pediatrickou populaci. Diagnostické schéma pro NMOSD zahrnuje poslední revizi diagnostických kritérií pro NMOSD z roku 2015, která nově definuje NMOSD u AQP4-IgG séronegativních pacientů a zahrnuje MR nálezy v oblasti mozku a míchy [12].](https://pl-master.mdcdn.cz/media/image/ad7b92055f78cc6836f48b3cb09d4623.png?version=1537795903)
V diagnostice myelitid se neobejdeme bez akutní MR míchy a mozku s podáním kontrastní látky. Zánětlivé míšní léze mohou mít charakter izolovaného ložiska nebo difuzního víceložiskového postižení [10]. Rozsáhlá ložiska na MR míchy přesahující tři a více obratlových segmentů splňují radiologická kritéria longitudinální extenzivní transverzální myelitidy (LETM) [2,11]. Asymptomatické léze v oblasti mozku se v době první ataky akutní myelitidy popisují až u 40 % pacientů a mohou vést k diagnóze CIS nebo RS [10]. Standardem je vyšetření likvoru, přítomnost pleiocytózy a/ nebo oligoklonální produkce IgG definuje v kontextu dalších vyšetření zánětlivé postižení [2]. Vhodné je vyšetření autoprotilátek asociovaných se systémovým onemocněním. Novinkou posledních let je stanovení protilátek proti vodnímu kanálu akvaporinu 4 (AQP4-IgG) v séru, které představují vysoce specifický biomarker pro NMO [12].
V léčbě akutního stadia je v případě autoimunitní etiologie první volbou vysoká dávka intravenózních kortikoidů [1]. Za dostatečnou je u dětí považována dávka intravenózního methylprednisolonu 20–30 mg/ kg/ den (max. 1 g/ den) v průběhu 5 dní [10,12,13]. Nebylo prokázáno, že by podání kortikoidů zhoršilo výsledný klinický obraz u pacientů s ischemií míchy nebo infekcí [10]. Pokud nenastane zlepšení stavu, přichází do úvahy série plazmaferéz (obvykle 5–7 cyklů), podání intravenózních imunoglobulinů (IVIG) v imunosupresivní dávce (obvykle celkově 2 g/ kg) nebo výjimečně při přetrvávajícím závažném stavu cyklofosfamid (obvykle 500–1 000 mg/ m2 intravenózně). Jednoznačné terapeutické standardy pro pediatrickou populaci však chybí [4,10,12]. Další léčba se odvíjí dle stanovené diagnózy.
Naše práce má za cíl upozornit na současnou problematiku akutní myelitidy v dětském věku a sdílet naše zkušenosti s touto diagnózou na ilustrativním souboru pacientů.
Metodika
Provedli jsme analýzu klinických dat a paraklinických vyšetření u pacientů s akutním míšním syndromem, bez encefalopatie a po vyloučení míšní komprese, u kterých byla zvažována zánětlivá etiologie. Zajímala nás výsledná diagnóza, zda se jednalo o infekční, autoimunitní či idiopatický zánět míchy nebo neobjasněné míšní onemocnění s diferenciálně diagnostickou možností zánětlivé etiologie. Případně zda byla naplněna diagnostická kritéria pro CIS/ RS, nebo NMO/ NMOSD dle aktuálních mezinárodních doporučení pro pediatrickou populaci (schéma 1, 2) [6,7].
Soubor zahrnoval 20 pacientů, kteří byli léčeni a sledováni nebo referováni na Klinice dětské neurologie 2. LF UK a FN Motol v letech 2010–2016. Jednalo se o 10 chlapců a 10 dívek ve věku 4–17 let (medián 14 let). Medián sledování pacientů byl 15 měsíců (rozmezí 1–73 měsíců). Všichni pacienti byli rasy indoevropské, pouze jeden pacient byl rasy mongoloidní. Z klinických dat jsme hodnotili neurologický nález a použitou terapii v akutním stadiu.
Z paraklinických dat jsme posuzovali: 1. MR míchy a mozku (sagitální a transverzální rovina, T2 vážené sekvence, FLAIR [fluid-attenuated inversion recovery] a postkontrastní T1 vážené sekvence) – zajímaly nás přítomnost a charakter míšních lézí a současná přítomnost zánětlivých změn v oblasti mozkového parenchymu a/ nebo optického nervu, která by byla v souladu s diagnózou RS nebo by podpořila diagnózu NMOSD (schéma 1, 2) [7–8]; 2. likvorový nález (přítomnost pleiocytózy > 5 buněk/μL, oligoklonální produkce IgG izoelektrickou fokusací); 3. mikrobiologický skríning (protilátky proti Borrelia burgdorferi, Mycoplasma pneumoniae a neurotropním virům, přímý průkaz Borrelia burgdorferi, herpetických virů a enterovirů v séru a likvoru pomocí polymerázové řetězové reakce); 4. imunologický skríning (IgM, IgA a IgG, C3 a C4 složka komplementu a cirkulující imunokomplexy, anti-nukleární protilátky, protilátky proti extrahovatelným nukleárním antigenům, protilátky proti cytoplazmě neutrofilů, protilátky proti dvouvláknové DNA a protilátky proti štítné žláze). U pacientů s obrazem LETM na MR míchy bylo dále doplněno vyšetření AQP4-IgG v séru.
Výsledky
Infekční etiologie akutní myelitidy byla jednoznačně prokázána u jednoho pacienta (tab. 2). Byl to 7letý chlapec s febrilními špičkami, bolestmi hlavy a noční bolestí zad s následným rozvojem senzorického deficitu, bez poruchy hybnosti. MR míchy ukázala difuzní zánětlivé postižení s postkontrastním sycením. MR mozku byla v normě. V likvoru byla výrazná pleiocytóza (1 106 buněk/ µL), bez oligoklonální produkce IgG a byla prokázána přítomnost boreliové DNA metodou PCR (polymerase chain reaction) a přítomnost protilátek proti Borrelia burgdorferi s pozitivním protilátkovým indexem. Po intravenózní léčbě antibiotiky (ceftriaxon 70 mg/ kg/ den, 21 dní) došlo k úpravě stavu ad integrum.

Autoimunitní etiologie akutní myelitidy byla jednoznačně stanovena u 11 pacientů (tab. 2). Systémové autoimunitní onemocnění se u žádného z nich neprokázalo. V okamžiku manifestace myelitidy splnilo 6 pacientů diagnostická kritéria pro RS a 2 pacienti byli diagnostikováni jako CIS. U pacientů s CIS/ RS (7 dívek, 1 chlapce, věk 12–17 let, medián 15 let) se rozvíjely příznaky lehkého až středně těžkého senzomotorického deficitu a u žádného se nevyskytla sfinkterová porucha. Na MR míchy měli pacienti s RS zánětlivé léze charakteru solitárního ložiska (n = 2) nebo víceložiskového postižení (n = 4) (obr. 1A), které se sytily po podání kontrastní látky u pěti pacientů. K diagnóze definitivní RS vedla současná přítomnost sytících se i nesytících se lézí na MR mozku. Pacienti s CIS měli na MR míchy solitární ložisko (n = 1) a víceložiskové postižení (n = 1), zánětlivé změny na MR mozku byly jen u jednoho. Oligoklonální produkce IgG byla přítomna u všech pacientů s CIS/ RS, u čtyř byla zachycena i pleiocytóza (8– 17 buněk/ µL, medián 13 buněk/ µL). V akutní terapii byly použity intravenózní kortikoidy s dobrým efektem a úpravou ad integrum u všech pacientů. U obou pacientů s CIS došlo při kontrolní MR mozku za 3 měsíce k radiologické progresi a naplnili tak diagnostická kritéria definitivní RS. Pacienti s RS byli předáni k další léčbě do příslušných center.
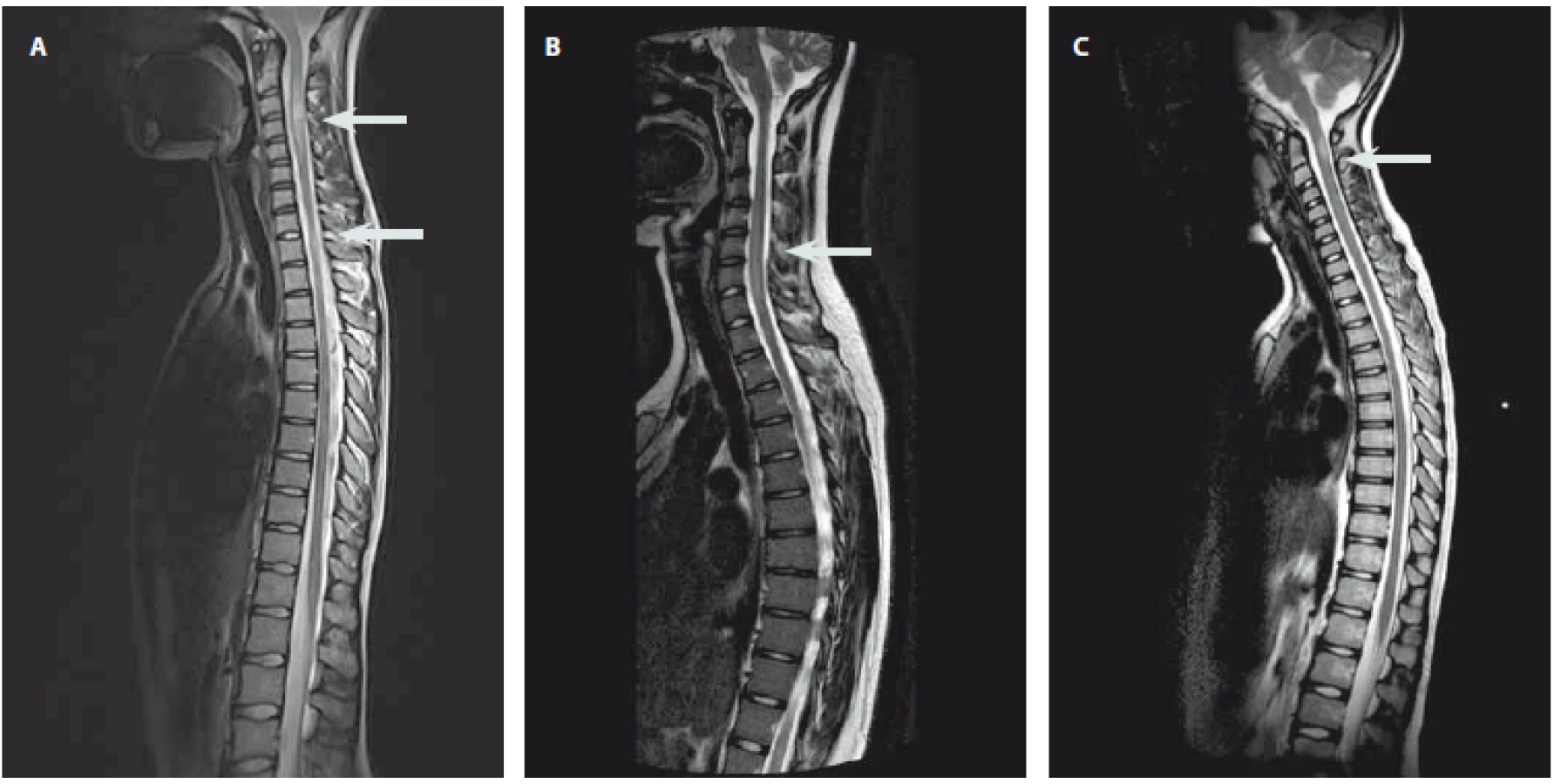
Diagnostická kritéria pro NMOSD splnili dva pacienti v okamžiku manifestace myelitidy a jeden v průběhu sledování (1 dívka, 2 chlapci, věk 6– 15 let, medián 14 let). První vyvinul obraz akutní transverzální míšní léze se sfinkterovou poruchou a současně poruchou zraku, druhý udával sfinkterové obtíže a poruchu zraku a třetí pacient se manifestoval pouze transverzální míšní lézí se sfinkterovou poruchou. Bez ohledu na klinické projevy měli všichni tito pacienti na MR míchy obraz LETM, u dvou s postkontrastním sycením (obr. 1B). Oba pacienti s poruchou vizu měli zánětlivé změny na MR mozku v oblasti optického nervu a jeden z nich také v oblasti mozkového kmene. V likvoru byla u všech pleiocytóza (60–254 buněk/ µL, medián 230 buněk/ µL), bez přítomnosti oligoklonální produkce IgG. Žádný z pacientů neměl pozitivní AQP4-IgG v séru. V akutní terapii byly u všech podány intravenózní kortikoidy, s dobrým efektem u dvou z nich. U pacienta s kompletní transverzální míšní lézí a poruchou zraku jsme terapii eskalovali za použití série plazmaferéz (10 cyklů) a jednorázového podání cyklofosfamidu (750 mg/ m2). Do vyloučení infekční příčiny obtíží dostali všichni pacienti antibiotika, jeden v kombinaci s virostatiky. U všech došlo po první atace k úpravě ad integrum, ale v dalším průběhu se u všech v souladu s diagnózou objevila recidiva neurologických obtíží (myelitida a/ nebo optická neuritida) a pacienti zůstali na dlouhodobé imunosupresivní terapii.
Diagnózu idiopatické myelitidy jsme stanovili u pěti chlapců (věk 4– 17 let, medián 14 let). Čtyři vyvinuli náhlý obraz kompletní transverzální míšní léze a jeden se manifestoval hemiparézou bez sfinkterové poruchy. U všech byl obraz LETM na MR míchy, u dvou se změny objevily až v opakovaném vyšetření do 48 h. Postkontrastní sycení bylo přítomno ve třech případech, u zbylých nebyla kontrastní látka podána. MR mozku byla u všech s normálním nálezem a vstupní likvorový nález také. Pouze u jednoho pacienta se objevila mírná pleiocytóza (7 buněk/ µL) s odstupem 48 h. AQP4-IgG v séru byly u všech negativní. Podání intravenózních kortikoidů mělo u všech jen částečný nebo nemělo žádný efekt, proto jsme na našem pracovišti léčbu eskalovali. U dvou pacientů, kteří byli po podání kortikoidů již bez sfinkterové poruchy, jsme podali IVIG (celkově 2 g/ kg) s úpravou ad integrum u jednoho, u druhého zůstala jen velmi lehká paraparéza s poruchou čití. U tří chlapců s přetrvávající sfinkterovou poruchou jsme provedli plazmaferézy (5–7 cyklů) a podali IVIG (celkově 2 g/ kg, max. 70 g). U jednoho z nich se podařilo obnovit schopnost chůze s oporou a získat kontrolu nad sfinktery, u druhého došlo ke zlepšení hybnosti končetin avšak bez možnosti samostatné chůze, u třetího zůstal neurologický nález beze změny a ani jednorázové podání cyklofosfamidu (500 mg/ m2) nevedlo k žádnému zlepšení. Jednalo se o pacienta, který prodělal v předchorobí epididymitidu a měl pozitivní IgM protilátky proti Mycoplasma pneumoniae v séru, k rozvoji kompletní TM došlo náhle (< 4 h) a abnormální nález v likvoru a na MR míchy se objevil v odstupu 48 h (viz výše). Do vyloučení infekční souvislosti byli tři pacienti léčeni virostatikem, u dvou v kombinaci s antibiotikem. Antibiotická terapie byla dokončena v plné dávce jen u pacienta s pozitivními protilátkami proti Mycoplasma pneumoniae. U žádného z pacientů nedošlo ve sledovaném období k recidivě neurologických potíží.
Neobjasněná diagnóza s možnou zánětlivou etiologií zůstala také u jednoho chlapce a dvou dívek (tab. 2). Desetiletý chlapec v anamnéze udával nezávažný pád na kostrč 6 týdnů a respirační infekt 4 týdny před rozvojem kompletní transverzální míšní léze. Neurologický deficit se u něj vyvinul rychle < 4 h) a na MR míchy byl obraz LETM v oblasti míšního konu. Zvažovala se především míšní ischemie, která však nebyla jednoznačně prokázána. Intravenózní podání kortikoidů bylo bez efektu. Na naše pracoviště se dostal s přetrvávajícím kompletním transverzálním míšním syndromem po několika měsících a léčbu jsme u něj již neeskalovali. U 13leté dívky předcházela rozvoji senzorického deficitu paréza lícního nervu boreliové etiologie. Na MR míchy měla víceložiskové postižení a v likvoru pozitivní oligoklonální produkci IgG. Přestože se borreliovou etiologii myelitidy nepodařilo prokázat, byla léčena pouze intravenózním ceftriaxonem s dobrou odezvou. Poslední, 10letá pacientka měla mírný senzorický deficit, nález solitárního ložiska na MR míchy (obr. 1C) a normální nález na MR mozku i v likvoru. Ložisko bylo podezřelé z tumoru, a tak byla pouze sledována, s odstupem 4 let se objevily nespecifické systémové potíže a nové demyelinizační změny na MR mozku v atypické lokalizaci pro RS. U této pacientky je zvažováno systémové autoimunitní onemocnění, které však dosud nebylo diagnostikováno.
Diskuze
Na ilustrativním souboru dětí jsme ukázali, že akutní myelitida může být jak samostatným onemocněním, tak jedním ze symptomů komplexních neurologických onemocnění a že je provázena různou tíží neurologického postižení s rozdílnými laboratorními a magneticko-rezonančními nálezy. Přestože jsme do souboru nezahrnuli pacienty s ADEM, dominovali pacienti s autoimunitním postižením míchy.
Diagnóza CIS, definitivní RS nebo NMO/ NMOSD v okamžiku manifestace akutní myelitidy umožní včasné zahájení specifické terapie a zlepší dlouhodobou prognózu. V našem souboru se jednalo o nadpoloviční většinu pacientů (55 %). Rozlišení mezi RS a NMO/ NMOSD může být náročné a ještě donedávna byla NMO vnímána jako agresivní podtyp RS. Samostatnou klinickou jednotkou se NMO stala až po objevení AQP4-IgG protilátek v roce 2006 a pojem NMOSD byl zaveden v roce 2007. Poslední revize diagnostických kritérií z roku 2015 umožňuje diagnostikovat NMOSD také u AQP4-IgG séronegativních pacientů (schéma 2) [7]. Likvorový nález není ke stanovení diagnózy NMOSD podmínkou, ale pomůže v diferenciální diagnostice, přítomnost oligoklonální produkce IgG je u NMOSD méně častá než u CIS/ RS, kolem 16– 40 % [14]. V našem souboru byli všichni pacienti s NMOSD AQP4-IgG séronegativní. Oproti pacientům s CIS nebo již definitivní RS měli pacienti s NMOSD výraznější klinické projevy, rozdílný nález na MR míchy i mozku a v likvoru výraznější pleiocytózu bez oligoklonální produkce IgG.
Nejvíce kontroverzní je diagnóza idiopatické myelitidy a přístup k pacientům s akutně vzniklou transverzální míšní lézí a normálním likvorovým nebo MR nálezem. Pokud je vstupní vyšetření likvoru a/ nebo MR míchy negativní, doporučuje se opakovat v odstupu 2–7 dní [15]. Normální likvorový nález je popisován až u 50 % a MR nález míchy u 6 % pacientů s TM, u některých z nich se později vyvine atrofie míchy [10]. Navíc náhlý rozvoj kompletní transverzální míšní léze (< 4 h) v kombinaci s normálním nálezem v likvoru a na MR mozku vždy vzbudí podezření na míšní ischemii a vede k nejistotě stran eskalace imunosupresivní terapie [2]. Míšní ischemie je však u dětí bez rizikových faktorů extrémně vzácná a její průkaz je velmi náročný, pomoci v diagnostice může CT angiografie [9]. Diagnóza idiopatické myelitidy ani rychlý rozvoj transverzální míšní léze nevylučují autoimunitní postižení [4]. V našem souboru bylo sedm pacientů (35 %) s normálním vstupním nálezem v mozkomíšním moku (bez pleiocytózy a oligoklonální produkce IgG), z toho šest chlapců s těžkým neurologickým deficitem a obrazem LETM na MR míchy. Jejich odpověď na kortikoterapii byla nedostatečná. Na našem pracovišti jsme se u pěti z nich rozhodli eskalovat imunosupresi a u čtyř jsme zaznamenali zlepšení, u tří se podařilo obnovit samostatnou chůzi a získat kontrolu nad sfinktery. Při eskalaci imunosuprese jsme obecně vycházeli z našich zkušeností a literárních údajů [4,10]. Pokud to bylo možné, snažili jsme se bezprostředně navázat na kortikoterapii. U pacientů s přetrvávající sfinkterovou poruchou jsme upřednostnili plazmaferézu a následně podávali IVIG, případně cyklofosfamid. U pacientů bez sfinkterové poruchy jsme podávali pouze IVIG, a to i přesto, že podávání IVIG u akutní myelitidy nebylo zatím ověřeno na větších studiích [12].
Prognóza myelitidy byla závažná, u tří pacientů zůstala neschopnost chůze se sfinkterovou poruchou. V souladu s literaturou byla nepříznivá prognóza spojena s rychlým rozvojem klinických příznaků, rozsáhlým postižením míchy a absencí pleiocytózy v likvoru [4].
Závěr
Myelitida je akutní stav se závažnou prognózou. Správná diagnóza se opírá o komplex vyšetření a klinickou zkušenost. Dostatečně razantní terapie v akutní fázi onemocnění a včasné zahájení specifické terapie v případě diagnózy RS nebo NMOSD může zlepšit prognózu pacientů. Vzhledem k riziku recidivy neurologických obtíží by děti po prodělaném onemocnění měly zůstat v dlouhodobém sledování.
Práce byla podpořena Nadačním fondem Neuron na podporu vědy a projektem Progres Q27/ LF1 Ministerstva školství ČR.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 26. 5. 2017
Přijato do tisku: 11. 1. 2018
MUDr. Hana Nohejlová
Neurologická klinika
2. LF UK a FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: hana.nohejlova@fnmotol.cz
Sources
1. Wolf VL, Lupo PJ, Lotze TE. Pediatric acute transverse myelitis overview and differential diagnosis. J Child Neurol 2012; 27(11): 1426– 1436. doi: 10.1177/ 088307381245 2916.
2. Transverse Myelitis Consortium Working Group. Proposed diagnostic criteria and nosology of acute transverse myelitis. Neurology 2002; 59(4): 499– 505.
3. Irani DN. Aseptic meningitis and viral myelitis. Neurologic Clinics 2008; 26(3): 635– 655. doi: 10.1016/ j.ncl.2008.03.003.
4. Deiva K, Absoud M, Hemingway C et al. Acute idiopathic transverse myelitis in children: early predictors of relapse and disability. Neurology 2015; 84(4): 341– 349. doi: 10.1212/ WNL.0000000000001179.
5. Rostasy K, Bajer-Kornek B, Venkateswaran S et al. Differential diagnosis and evaluation in pediatric inflammatory demyelinating disorders. Neurology 2016; 87(9): S28– S37. doi: 10.1212/ WNL.0000000000002878.
6. Tardieu M, Banwell B, Wolinsky JS et al. Consensus definitions for pediatric MS and other demyelinating disorders in childhood. Neurology 2016; 87(9): S8– S11. doi: 10.1212/ WNL.0000000000002877.
7. Winderchuk DM, Banwell B, Bennett JL et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85(2): 177– 189. doi: 10.1212/ WNL.0000000000001729.
8. Polman CH, Reingold SC, Banwell B et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011; 69(2): 292– 302. doi: 10.1002/ ana.22366.
9. Vargas MI, Gariani J, Sztajzel R et al. Spinal cord ischemia: practical imaging tips, pearls, and pitfalls AJNR Am J Neuroradiol 2015; 36(5): 825– 830. doi:10.3174/ ajnr.A4118.
10. Absoud M, Greenberg BM, Lim M et al. Pediatric transverse myelitis. Neurology 2016; 87(9): S46– S52. doi: 10.1212/ WNL.0000000000002820.
11. Tobin WO, Weinshenker BG, Lucchinetti CF. Longitudinally extensive transverse myelitis. Curr Opin Neurol 2014; 27(3): 279– 289. doi: 10.1097/ WCO.0000000000000093.
12. Tenembaum S, Chitnis S, Nakashima I et al. Neuromyelitis optica spectrum disorders in children and adolescents. Neurology 2016; 87(9): S59– S66. doi: 10.1212/ WNL.0000000000002824.
13. Havrdová E, Piťha J. Klinický standard pro diagnostiku a léčbu roztroušené sklerózy a neuromyelitis optica. Praha: Národní referenční centrum 2012. Dostupné z URL: http:/ / www.czech-neuro.cz/ archiv/ data/ q/ O/ V/ 0031-rs-odborna.pdf.
14. Flanagan EP, Weinshenker BG, Krecke KN et al. Short myelitis lesions in aquaporin-4-IgG– positive neuromyelitis optica spectrum disorders. JAMA Neurol 2015; 72(1): 81– 87. doi: 10.1001/ jamaneurol.2014.2137.
15. Sorte DE, Poretti A, Newsome SD et al. Longitudinally extensive myelopathy in children. Pediatr Radiol 2015; 45(2): 244– 257. doi: 10.1007/ s00247-014-3225-4.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2018 Issue 2
- Metamizole vs. Tramadol in Postoperative Analgesia
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Ataxie
- Biopsie mozku v deseti bodech – co může neurolog očekávat od neurochirurga a neuropatologa?
- Fabryho choroba, přehled problematiky a nejčastější neurologické projevy
- Syndrom GLUT-1 deficience – expandující klinické spektrum léčitelného onemocnění