
Nové poznatky v diagnostice a léčbě amyotrofické laterální sklerózy
New insights in the diagnosis and treatment of amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease of increased prevalence with age. The main findings are loss of peripheral and central motoneurons and their pathways with extraocular and sphincter muscle sparing. These are classical forms of ALS with loss of central and peripheral motoneurons, as well as progressive bulbar paralysis with impairment of bulbar muscles. Progressive muscle atrophy with only peripheral motoneuron lesions and primary lateral sclerosis with only central motoneuron involvement are rarely found. There are some forms of ALS associated with dementia (frontotemporal lobar dementia-motor neuron disease [FTLD-MND]) with behavioral changes, cognitive and executive dysfunction. The cause of ALS has not yet been elucidated. It is a chain of follow-up events, at the end of which is cell death in selective subpopulations of neurons. In the present paper, we describe in detail the neuropathological findings and molecular genetic analysis in familial forms of ALS. A specific drug for this disease is still unknown. Neuroprotective drugs (such as riluzole and recently edaravon) have an ambiguous effect. Symptomatic treatment is designed to manage concomitant manifestations. Different treatment options are discussed in detail.
Key words:
amyotrophic lateral sclerosis – ALS – neurophysiology – motoneuron lesion – riluzole – edaravone
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
I. Štětkářová 1; R. Matěj 2,3; E. Ehler 4
Authors‘ workplace:
Neurologická klinika 3. LF UK a FNKV, Praha
1; Oddělení patologie a molekulární medicíny, Thomayerova nemocnice, Praha
2; Ústav patologie, 3. LF UK a FNKV, Praha
3; Neurologická klinika FZS UP a Pardubické krajské nemocnice, a. s.
4
Published in:
Cesk Slov Neurol N 2018; 81(5): 546-554
Category:
Review Article
doi:
https://doi.org/10.14735/amcsnn2018546
Overview
Amyotrofická laterální skleróza (ALS) patří mezi progredující neurodegeneativní onemocnění, jehož prevalence s věkem stoupá. Dochází k pozvolnému zániku periferních a centrálních motoneuronů a jejich drah s ušetřením extraokulárních a sfinkterových svalů. Rozlišuje se klasická forma ALS s postižením centrálního a periferního motoneuronu, a dále progresivní bulbární paralýza s postižením bulbárních svalů. Vzácnější jsou progresivní svalová atrofie s lézí pouze periferního motoneuronu a primární laterální skleróza s postižením pouze centrálního motoneuronu. Existují formy ALS sdružené s demencí (frontotemporální lobární demence [frontotemporal lobar dementia-motor neuron disease; FTLD-MND]), u kterých jsou přítomny poruchy chování, kognitivní dysfunkce a postižení exekutivních funkcí. Příčina nemoci zatím není objasněna. Jde o řetězec následných dějů, na jehož konci je programově řízená smrt buňky v selektivních subpopulacích neuronů. V práci je detailně popsán neuropatologický nález vč. podrobného molekulárně genetického rozboru u familiárních forem ALS. V současné době neexistuje specifický lék na toto onemocnění. Používá se neuroprotektivní léčba (např. riluzol a nově edaravon) s nejednoznačným efektem a léčba symptomatická, která je určena k zvládání doprovodných projevů. Jednotlivé léčebné možnosti jsou podrobně rozebrány.
Klíčová slova:
amyotrofická laterální skleróza – ALS – neurofyziologie – léze motoneuronu – riluzol – edaravon
Úvod
Neurodegenerativní nemoci, mezi něž se řadí amyotrofická laterální skleróza (ALS), jsou sporadická onemocnění, některá s hereditárním podkladem, jejichž prevalence stoupá s věkem. Vznikají na podkladě excesivního odumírání neuronů apoptotickým mechanizmem, k němuž dochází pravděpodobně vlivem kumulace abnormních proteinových agregátů [1]. Apoptóza je programovaná buněčná smrt, která vede k úbytku neuronů globálně či v určitých neuronálních populacích, typických pro dané onemocnění, kde v rámci selektivní neuronální vulnerability dochází k jejich úbytku. Tento zánik koreluje s progresí klinických symptomů nemoci. Objevují se abnormální agregáty proteinů, které jsou specifické pro jednotlivá onemocnění. Jejich vznik, interakce a nemožnost fyziologické degradace jsou v úzkém vztahu s přítomností volných kyslíkových radikálů. Zásadní úlohu rovněž hrají poruchy degradačních mechanizmů, které potencují vznik depozit patologických proteinových agregátů v buňce, ať už v cytoplazmě nebo v jejím jádře, tak i extracelulárně. Důležitou roli hraje rovněž oxidativní stres [2]. Mitochondrie jsou nejen zdrojem buněčné energie, ale i producentem volných kyslíkových radikálů [3]. Další vliv mají různé genové polymorfizmy a postižení genomu patogenními mutacemi, které v konečném důsledku usnadňují či zrychlují vznik proteinových agregátů. Souhrou všech výše zmíněných patofyziologických mechanizmů dochází k poškození neuronu úbytkem funkčního proteinu, a tím k výpadku jeho funkce (tzv. loss of function). Neuron je dále toxicky poškozen nově vzniklými patologickými proteinovými agregáty (tzv. gain of function). V současné době je léčba neurodegenerativních nemocí pouze omezená; ve většině případů jde o léčbu symptomatickou [1].
Definice a klasifikace ALS
Amyotrofická laterální skleróza se řadí mezi neurodegenerativní onemocnění a je charakterizována progresivním úbytkem centrálního motoneuronu (pyramidové buňky v kůře, kortikospinální dráha) a míšního motoneuronu s ušetřením extraokulárních a sfinkterových svalů [4,5]. Poruchu čití lze klinicky detekovat vzácně, ale subklinická dysfunkce senzitivního systému již byla dokumentována pomocí neurofyziologických metod a difuzní traktografie (DTI) v rámci MR [6,7]. Existují formy sdružené s demencí (frontotemporální lobární demence [frontotemporal lobar dementia-motor neuron disease; FTLD-MND]), u kterých jsou přítomny poruchy chování, kognitivní dysfunkce a postižení exekutivních funkcí [8,9].
Amyotrofická laterální skleróza patří do skupiny onemocnění zvané „motor neuron disease (MND)“ [4,5,10,11]. Klasická forma ALS má postižení centrálního a periferního motoneuronu (bývá přítomna u 65 – 70 % případů). Další formou je progresivní bulbární paralýza s postižením bulbárních svalů, která se vyskytuje u 25 % osob. Vzácnější je progresivní svalová atrofie s lézí pouze periferního motoneuronu, která se vyskytuje v 5 – 8 %. Extrémně vzácnou formou je primární laterální skleróza, která je charakterizována lézí pouze centrálního motoneuronu a bývá u 1 – 2 % osob. V tomto případě jde o klinickou diagnózu per exclusionem; po vyloučení jiných možných příčin postižení. Vzácné jsou rovněž varianty s fokální svalovou atrofií – monomelická spinální amyotrofie. Tzv. ALS plus syndromy s kognitivním postižením převážně typu frontotemporální demence (FTD) tvoří skupinu FTLD-MND a v poslední době se diagnostikují relativně často [12]. Je to proto, že byl nalezen společný jmenovatel postižení a tím je přítomnost ubikvitinových inkluzí v jádrech či cytoplazmě motorických neuronů, ale i v dalších neuronech a gliích u ALS i FTD v histopatologickém nálezu [13,14]. Jde nejčastěji o agregáty proteinu Tar DNA vazebný protein 43 (TDP-43) ze skupiny RNA-/ DNA-vazebných proteinů. Tento protein se nejspíše může šířit per continuitatem pomocí vezikulární exocytózy mezi jednotlivými neurony nebo vzdáleně kortikospinální dráhou [9]. ALS a FTD mají i další společný patogenní protein zvaný FUS (fused-in-sarcoma protein), jehož přítomnost je však ve srovnání s TDP-43 významně méně častá [15].
Jednotlivé podtypy ALS mají různou prognózu. Do klasické formy mohou vyústit všechny výše jmenované podskupiny. Onemocnění je fatální, příčina není známa. Medián přežití je 2 – 4 roky [16]. Kolem 50 % osob umírá do 3 let od vzniku prvních příznaků, 90 % umírá do 5 let a jen kolem 5 – 10 % žije více než 10 let od počátku choroby. Relativně delší přežití se objevuje u mladších osob a u těch, u kterých stanovení diagnózy trvalo delší dobu. Příznivější z hlediska dlouhodobého přežití je také převaha postižení centrálního motorického neuronu nad periferním motorickým neuronem. Krátké přežití zaznamenáváme u starších osob, které mají časné příznaky z bulbární oblasti a u kterých se brzy objeví dechové potíže. Nepříznivou prognózu z hlediska rychlosti přežití má i brzké objevení se klinických příznaků z postižení v cervikální oblasti a přítomnost kognitivního deficitu.
Výskyt
Incidence v Evropě se uvádí kolem 1 – 2/ 100 000 obyvatel/ rok. Prevalence je 4 – 6/ 100 000 obyvatel, resp. medián prevalence je 5,4/ 100 000 obyvatel [17]. Obvykle jsou postiženy osoby mezi 60 a 70 lety, vzácně před 40. rokem života. Častěji se vyskytuje u mužů. Kolem 5 – 10 % jsou familiární formy ALS (FALS), ostatní případy jsou sporadické formy ALS (SALS). Nejvyšší prevalence ALS je hlášena z Japonska (11,3/ 100 000 obyvatel), naopak nejméně případů je uváděno v Íránu (0,4/ 100 000 obyvatel) [17].
Etiologie
Příčina této nemoci zatím není objasněna. Uvažuje se o působení virů, vlivu exotoxinů, vč. excitotoxicity zprostředkované glutamátem a homocysteinem, či o poruše imunitního systému ve smyslu indukce chronického zánětu, ale tyto hypotézy zatím nebyly prokázány. Jde o řetězec následných dějů, na jehož konci je programově řízená smrt buňky v selektivních subpopulacích neuronů. Nejčastější je SALS. V případě FALS jde obvykle o autozomálně dominantní dědičnost se známými mutacemi genů (např. superoxidová dismutáza 1 [SOD1], senataxin, mutace dynactinu, alsinu apod.). Nejčastěji se jedná o mutace v genech C9orf72, SOD1 a FUS [13,14]. ALS se také spojuje s četnými geny a lokusy s mutacemi genů regulujících DNA/ RNA, např. TARDBP [16,18]. Vzácně se může najít i paraneoplastická etiologie s pozitivitou protilátek anti-Hu. Bývá i asociace s karcinomem prsu nebo s lymfomem, ale bez průkazu onkoneurálních protilátek.
Nově byly publikovány práce, ve kterých se uvádí výskyt selektivní atrofie hypotalamu u SALS i FALS a u rozvinuté formy FALS i v asymptomatickém stadiu [19]. Zdá se, že nápadnější úbytek hmotnosti může předcházet začátek nemoci o 5 – 10 let. Atrofie hypotalamu nekoreluje s motorickým postižením. Vyskytuje se více u osob s časným začátkem nemoci.
Časné motorické projevy SALS s přítomností TDP-43 odrážejí selhání adaptivních komplexních motorických dovedností. Vývoj těchto dovedností koreluje s rozvojem motorického systému, který je jedinečný pro primáty a u lidí je výrazně zdokonalen. Porucha tohoto systému vede k rozštěpené prezentaci ruky (split hand), poruchám chůze, syndromu rozštěpené nohy (split leg) a bulbární symptomatologii, která souvisí s vokalizací [8].
Charakteristickým znakem proteinopatologie TDP-43 je její omezení na kortikální oblasti a podkorová jádra, která jsou pod přímou kontrolou kortikálních projekcí. Patologický protein TDP-43 se nachází v mozkové kůře, kortikofugálních vláknech a subkortikálních jádrech a motorických neuronech předních rohů míšních. O ALS se nyní uvažuje jako o primární neurodegenerativní poruše, která v sobě zahrnuje koncept prionu podobného (prion-like) rozšíření na synaptických zakončeních kortikofugálních axonů [9]. Tento koncept teoreticky vysvětluje šíření do neokortexu a vztah mezi ALS a FTD.
Klinický obraz
U rozvinuté nemoci je klinický obraz poměrně charakteristický, ale diagnostické obtíže mohou nastat na začátku nemoci. Mohou se objevit bulbární příznaky s poruchou artikulace a obtížným polykáním. Lze pozorovat zřetelné atrofie a fascikulace jazyka. Často nemoc začíná asymetrickou slabostí ohraničené svalové skupiny, nejčastěji na horní končetině, např. jako neobratnost při odemykání, oslabení dorzální flexe ruky, slabost plantární flexe nohy apod. Klinický nález může připomínat mononeuropatii či radikulopatii. Méně často svalové oslabení postihuje šíjové svaly. Nápadná je výrazná hrudní kyfóza. Častá je svalová únava. Dochází k úbytku hmotnosti v důsledku svalové atrofie, ale i v důsledku vlastní nemoci. V pletencových svalech horních a dolních končetin (m. deltoideus, m. quadriceps femoris) se objevují fascikulace či krampy. V klinickém obraze lze nalézt atrofie drobných svalů ruky a nohy (zejména interoseálních svalů) a generalizované fascikulace v pletencových svalech, které někteří pacienti vnímají subjektivně nepříznivě. Vzácně lze fascikulace nalézt i na trupovém svalstvu. V objektivním nálezu nacházíme u rozvinuté formy smíšený obraz centrální a periferní kvadruparézy, bulbární příznaky, kvadruhyperreflexii, pozitivní pyramidové jevy, atrofie svalů horních a dolních končetin a jazyka, masivně fascikulace, zejména na končetinách a v jazyku. Nejsou přítomny sfinkterové potíže a bývá normální funkce čití. U formy s demencí (FTD-ALS) jsou porušeny exekutivní a rozpoznávací funkce a jsou přítomny poruchy chování. V pokročilých stadiích nemoci není pacient sebeobslužný a stává se imobilním. Není schopen přijímat tekutou ani tuhou stravu.
Diagnostika
Vychází z klinického obrazu, který ukazuje na postižení centrálního a periferního motoneuronu. Objektivní průkaz léze periferního motoneuronu v předních rozích míšních přinese EMG, která má v diagnostice ALS stále klíčové místo. Kondukční studie prokazuje na počátku nemoci normální vedení senzitivními a motorickými vlákny, při progresi nemoci s atrofií svalů se snižují amplitudy motorických odpovědí a lehce se zpomaluje i rychlost vedení. Je to dáno zánikem nejrychleji vedoucích motorických vláken. Stanovení diagnózy ALS je závažná informace, a proto musí být náležitě podložena elektrofyziologicky, což vyžaduje vyšetření značného počtu svalů (tab. 1) [20,21]. Je třeba vyšetřovat ve čtyřech regionech – hlava, hrudní segmenty, horní a dolní končetiny. Na končetinách je nezbytné vždy vyšetřit dva svaly, a to jeden proximální a jeden distální. Tyto svaly nesmí být inervovány stejným nervem ani stejným míšním segmentem. V oblasti hlavových nervů a hrudních segmentů je dostačující průkaz změn v jednom svalu. Je doporučeno vyšetřit paravertebrální svaly (nejlépe v segmentech Th5 – 8) nebo přímý břišní sval. V postižených i nepostiženích svalech je nutno pátrat po známkách aktivního denervačního procesu (fibrilace, pozitivní ostré vlny), po starším denervačním postižení (neurogenní, regenerační, delší, vyšší, polyfazický a nestabilní potenciál motorické jednotky [motor unit potential; MUP]). U ALS se velmi často vyskytují fascikulace. Dle Awaji-Shima kritérií jsou pro ALS charakteristické nestabilní MUP v různé fázi neurogenní přestavby, s pomalejším náborem, vyšší únavností a frekvencí pálení > 10 Hz [21].
![Doporučený protokol vyšetření EMG při podezření na ALS/MND
dle Awaji-Shima [21].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/5a6c7fa754b6d6e1521c19540e4c5d5a.jpeg)
K diagnostice se používají revidovaná El Escorial kritéria, která rozdělují formy ALS na klinicky definitivní, klinicky pravděpodobné, klinicky pravděpodobné laboratorně podpořené (EMG) a možné. Od roku 2008 se užívají Awaji-Shima kritéria, která jsou senzitivnější pro pacienty s bulbární symptomatologií [22]. Na rozdíl od revidovaných EL Escorial kritérií byl u Awaji-Shima kritérií postaven elektrofyziologický průkaz léze periferního motoneuronu na stejnou úroveň jako jeho klinické příznaky. Byla zcela vynechána kategorie klinicky pravděpodobných laboratorně podporovaných ALS. Přítomnost fascikulací pak byla uznána jako projev léze periferního motoneuronu. Při použití těchto kritérií je diagnóza ALS stanovena u mnoha nemocných podstatně dříve a nevyskytuje se tolik falešně pozitivních diagnóz ALS [23].
Motorické evokované potenciály po transkraniální magnetické stimulaci (TMS) potvrdí lézi centrálního motoneuronu (resp. kortikospinální dráhy). Většinou jsou tyto kortikální odpovědi zcela nevýbavné. Pro ALS je typická přítomnost kortikální hyperexcitability se zvýšeným motorickým prahem [24], absencí intrakortikální inhibice (zejména short-interval intracortical inhibition; SICI) v rámci párové TMS, doprovázené zvýšenou facilitací.
Nutné je také doplnit vyšetření mozkomíšního moku k vyloučení jiné etiologie (např. zánětlivé – neuroboreliózy apod.). Zobrazení mozku a míchy pomocí MR vyloučí další afekce nervového systému. Úbytek kortikospinální nebo kortikobulbární dráhy lze prokázat v sekvenci DTI snížením frakční anizotropie [25]. V laboratorních nálezech se mohou vyskytnout zvýšené hodnoty svalových enzymů (kreatinkináza, laktátdehydrogenáza, myoglobin) jako důsledek rychlého rozpadu svalové tkáně.
Diferenciální diagnostika
V případě dysfagie a dysartrie je třeba vyloučit poruchu nervosvalového přenosu (zejména myastenii), pseudobulbární symptomatologii v rámci aterosklerotických změn mozku, infiltrativně rostoucí tumory, infekční a autoimunitní záněty apod. (tab. 2 [26]). Je nutné vyšetřit mozkomíšní mok a provést MR mozku a míchy. U převážně spinální formy se provádí MR krční míchy k vyloučení jiných léčitelných afekcí (např. cervikální spondylogenní myelopatie, syringomyelie apod.). Vzácně se může indikovat i svalová biopsie, např. k vyloučení polymyozitidy nebo myozitidy s inkluzními tělísky.
![Diferenciální diagnostika ALS [21].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/0c17705dbeb9c1ae4292a98fd834da7f.jpeg)
Neuropatologie ALS
Při pitvě těla dominuje různě vyjádřená kachexie s úbytkem podkožního tuku, povětšinou je už makroskopicky nápadná atrofie kosterního svalstva, která je zřetelná nejen na svalech končetin, ale i interkostálních svalů, jazyka a bránice. Atrofie postihuje míchu především v oblastech intumescencí, přední motorické kořeny jsou zmenšené, hnědé, mnohem více postižené atrofií ve srovnání se zadními senzorickými kořeny. Některé hlavové motorické nervy (zejména n. hypoglossus) mohou být také makroskopicky atrofické. Mozek ve většině případů prostých MND nejeví makroskopické abnormity, jen při dlouhodobém průběhu může být atrofovaný gyrus praecentralis. V případě MND s doprovodnou demencí však je různě výrazně patrná atrofie oblastí prefrontálního, frontálního a temporálního kortexu [27].
V histopatologickém vyšetření jsou degenerativní změny pozorovatelné většinou v motorické oblasti. Dominuje numerická atrofie velkých motorických neuronů předních rohů míšních a motorických neuronů jader hlavových nervů v mozkovém kmeni (obr. 1). Postiženy bývají i Betzovy pyramidy v primární motorické kůře. V důsledku úbytku motorických neuronů nacházíme známky chronické denervační neurogenní atrofie s tukovou pseudohypertrofií v příčně pruhovaných svalech končetin a respiračních svalů (obr. 2). Ve standardním barvení v přehledném zvětšení dominují změny předních rohů, kde je výrazná atrofie s reaktivní astrogliózou a atrofie předních kořenů. Ve specializovaném histochemickém průkazu myelinu jsou nejnápadnější změny patrné v anterolaterálních provazcích, kde bývá zřetelné projasnění a skleróza. Tyto změny ale můžeme najít při dlouhodobém průběhu onemocnění i v zadních provazcích. Klíčovým diagnostickým znakem je výrazná numerická atrofie velkých motorických neuronů předních rohů míšních, která je nejvíce vyjádřena v cervikální a lumbální intumescenci. Zbylé neurony jeví známky regresivních změn, bývají svraštělé, v cytoplazmě se nachází depozita lipofuscinu, v jádrech může být patrná centrální chromatolýza [28]. V cytoplazmě takto postižených neuronů je možné najít různé typy inkluzí. Za nejvíce diagnosticky specifická byla v éře před imunohistochemickými metodami považována tělíska Buninové, což jsou malé eozinofilní granulární inkluze (velikost 2 – 4 μm) v cytoplazmě a dendritech postižených motorických neuronů, v imunohistochemickém vyšetření pozitivní v reakci s protilátkou proti cystatinu C. Pomocí protilátky proti ubikvitinu či proteinu P62 lze ozřejmit další typy inkluzí. Jsou to jednak tzv. skein-like inkluze protaženého či vláknitého tvaru, které tvoří až bizarní sférické struktury v perikariu neuronů, jednak světle eozinofilní kulaté hyalinní inkluze [29]. Podstatou proteinových inkluzí jsou depozita proteinu TDP-43 [30] (obr. 3). U některých případů především FALS je možné v cytoplazmě pozorovat Lewyho tělískům podobné hyalinní inkluze, které v případech mutace genu SOD1 jsou silně imunoreaktivní v reakci s protilátkou proti superoxiddismutáze (SOD1) [31]. Vzácně se dají u FALS prokázat další charakteristické inkluze v cytoplazmě zbylých motorických neuronů či reaktivních astroglií.
Fig. 1. Scattered motor neurons of the anterior horns of the spinal cord reveal advanced
regressive changes (arrow). Standard hematoxylin and eosin staining with luxol blue,
original magnification 200×.
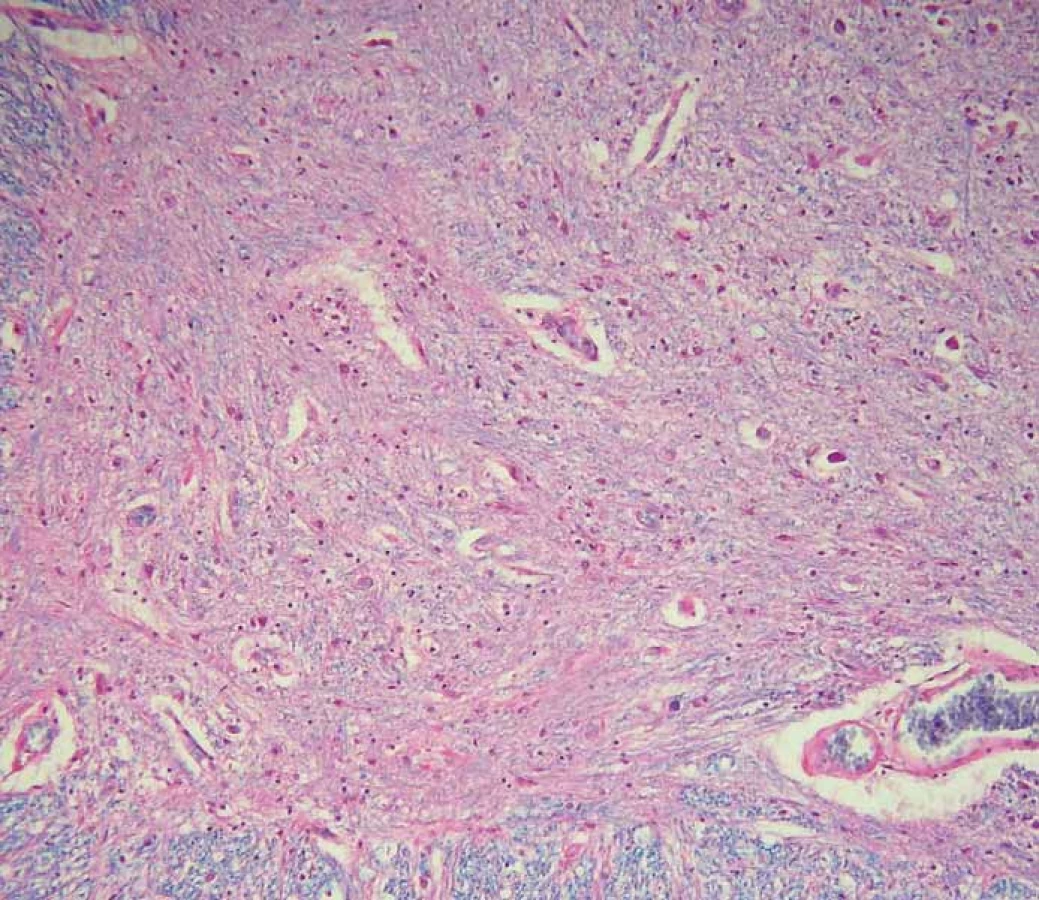
Fig. 2. Chronic neurogenic atrophy (arrow) of skeletal muscle of the diaphragm. Standard
hematoxylin and eosin staining, original magnification 200×.
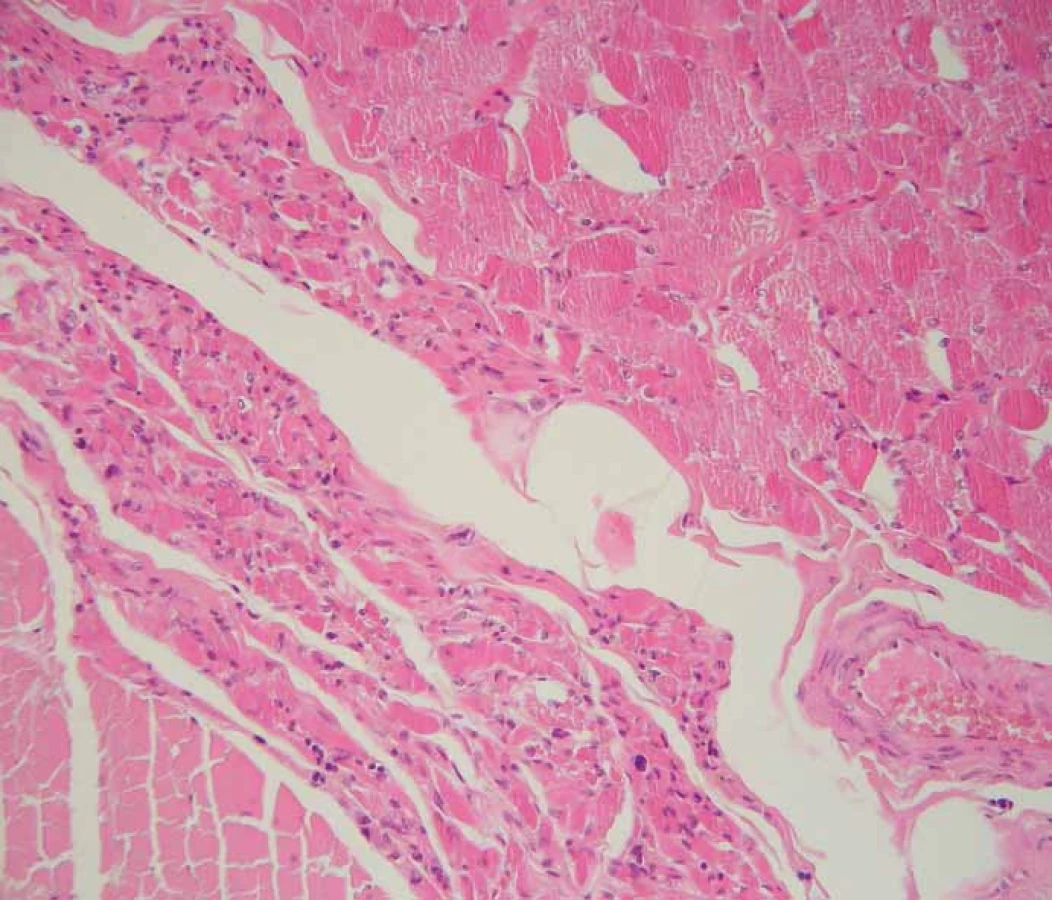
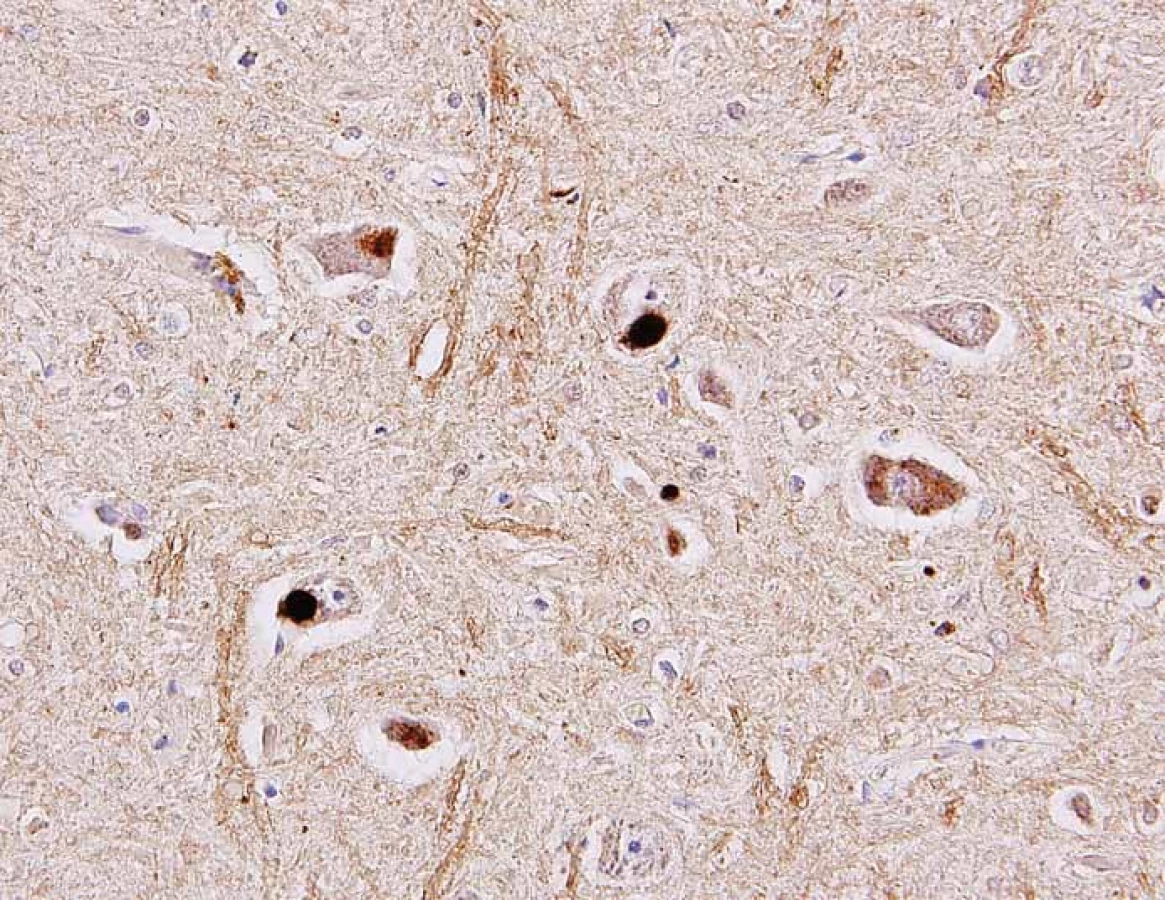
Fig. 3. Different types of inclusions in the cytoplasm of the spinal cord motor neurons
identified as positive in the immunohistochemical reaction using antibody against TDP-
43 protein (arrows). Original magnification 400×.
Molekulární genetika ALS
Familiární forma ALS se odhaduje na 5 – 10 %. Většinou jde o autozomálně dominantní dědičnost, avšak jsou známy i případy s X-vázaným typem dědičnosti. V současné době bylo identifikováno 16 genů, které jsou zodpovědné za FALS. U 10 – 20 % z těchto familiárních případů lze zjistit až 80 různých mutací. Na 21. chromozomu bylo zjištěno již přes 60 genových mutací, které způsobují tvorbu defektní SOD1 a vedou k FALS. V současné době jsou nejdůležitější geny spojeny s metabolizmem proteinu TDP-43, který hraje zásadní roli v patogenezi ALS. Kromě vlastního genu proteinu TAR DNA binding protein (TARDBP) jde o gen pro protein fus (fused in sarcoma; FUS), vesicle-associated membránový protein B (VAPB), valosin obsahující peptid (VCP), ubiquillin 2 (UBQLN2), optineurin (OPTN), progranulin (GRN) a především relativně recentně objevená významná asociace se 72. otevřeným čtecím rámcem na chromozomu 9 (C9orf72) [32]. V něm dochází k patologické expanzi „hexarepeatu“ (GGGGCC) [33] (obr. 4). Tato genetická aberace se zdá být zásadní nejen u řady pacientů s FALS, kde se odhaduje výskyt až u 20 %, ale je možné ji detekovat i u 10 % SALS. Navíc je tento genetický defekt úzce spjat s behaviorální variantou frontotemporálních demencí (bvFTD), které se vyskytují buď v přímé asociaci s ALS (FTLD-MND) nebo jako čistě kognitivní postižení bez MND. Spektrum postižení se v rodinách s expanzemi v genu C9orf72 výrazně liší od klasické formy ALS, přes kombinaci ALS a kognitivního postižení (FTLD-MND) až po čistě behaviorální symptomatologii (bvFTD) bez prokazatelného postižení motorické dráhy [34].
Fig. 4. Pathological hexarepeate (GGGGCC) expansion in the chromosome 9 open reading frame 72 (C9orf72). Full penetration represents
> 29 GGGGCC repeats (upper panel) compared to the normal repeat count (< 20 – lower panel).
Léčba
V současné době neexistuje specifický lék na toto onemocnění. Používá se neuroprotektivní léčba, jejíž účinky nejsou jednoznačné, a léčba symptomatická, která je určena k zvládání doprovodných projevů.
Farmakoterapie
Dlouhodobě se podává riluzol [35], což je benzothiazolový derivát, který inhibuje uvolňování glutamátu na presynaptických nervových zakončeních. Antagonizuje účinky excitačních aminokyselin na postsynaptických nervových zakončeních a tím snižuje koncentraci glutamátu na synapsích. Podává se perorálně v dávce 2 × 50 mg denně, obvykle 1,5 h před jídlem. Ve studiích bylo prokázáno, že tento glutamátový antagonista může významně prodloužit dobu přežití [36]. Nejsou důkazy o zlepšení motorických nebo plicních funkcí a neprokázala se účinnost v pozdních stadiích nemoci. Kvůli riziku postižení jater je třeba po nasazení zkontrolovat jaterní funkce (přípustná hodnota transamináz je do trojnásobku jejich normální hodnoty) a sledovat je v prvním roce léčby v 3měsíčních intervalech.
Dalším lékem, který se nyní používá v léčbě ALS, je edaravon [37], zametač volných radikálů. Je to další lék, do kterého se vkládalo plno nadějí, ale ani jeho klinický účinek není nijak převratný. Je uveden jako novinka, a to po více než 20 letech od zavedení léčby riluzolem. V Japonsku je registrován od roku 2015, dále je schválen v Jižní Koreji a od května 2017 je registrován v USA. Při 2letém užívání zpomaluje u vybrané subpopulace nemocných s ALS progresi ztráty hybných funkcí o 30 % [37].
Ve studiích na zvířatech se zkouší léčba ALS pomocí vysokých dávek metylcobalaminu (vitamin B12), což je koenzym syntázy methioninu. Prokazuje se významné zpomalení progrese motorických funkcí [38]. Jeho klinický efekt u lidí nebyl dosud prokázán.
Ve stadiu testování je i léčba pomocí perampanelu, selektivního antagonisty AMPA receptorů, který se používá k léčbě epilepsie. V animálních modelech na myších bylo zjištěno významné zmírnění progrese hybnosti [39].
Testují se také monoklonální protilátky, např. masitinib a bosutinib. Masitinib je selektivní inhibitor tyrozinkinázy, který má protizánětlivé působení, ovlivňuje funkci mikroglie, resp. u zvířat redukuje její aberantní expanzi [40]. Bosutinib u laboratorních zvířat snižuje množství aberantního proteinu SOD1 a potlačuje porušenou expresi mitochondriálních genů [41].
Mírné zlepšení bylo také pozorováno u osob, které užívaly dexpramipexol [42]. Jiné studie na zvířecích modelech tento příznivý efekt dexpramipexolu nepotvrdily [43].
Zkoušel se lék tirasemtiv, což je aktivátor troponinového komplexu u rychlých svalových vláken [44]. Zvyšuje jeho selektivitu k vápenatým iontům. Bohužel ani tento lék nesplnil primární cíl, tj. oddálení progrese poruchy motorických funkcí a zlepšení dechových funkcí. Jeho testování bylo ve fázi III ukončeno na podzim roku 2017.
Lze podávat i další látky, např. antioxidanty (vitamin E, koenzym Q10, betakaroten), u kterých ale nebyl prokázán jasný klinický účinek. V léčbě ALS se zkoušelo i lithium pro jeho prokázaný neuroprotektivní účinek na zvířatech, ale v humánních studiích nebyl prokázán jeho vliv na delší přežití [45].
Dalším možným léčebným cílem se stává nervosvalová ploténka a vlastní sval [46]. U osob se SALS s rychle progredujícím funkčním motorickým úbytkem byla zjištěna snížená exprese genů kódujících sarkomerický protein titin [47]. Modifikace exprese titinu by se tedy mohla stát dalším léčebnou možností.
Kmenové buňky
Použití kmenových buněk v léčbě neurodegenerativních nemocí vč. ALS [48] se zdálo být velkým příslibem, ale výsledky jsou nadále nejednoznačné. Většina experimentálních prací se opírá o hypotézu, že kmenové buňky, nejčastěji mezenchymální nebo neurální, jsou schopny podpořit přežití motoneuronů několika různými mechanizmy. Může to být prostřednictvím sekrece růstových faktorů, imunomodulačním působením na aktivované astrocyty a mikroglii, diferenciací do podoby funkční glie, respektive více zmiňovanými způsoby současně [49,50,51]. Kmenové buňky byly u nemocných s ALS aplikovány intravenózně, intratékálně, intraspinálně i intracerebrálně. I když experimentální práce dokládají pozitivní efekt buněčné terapie na průběh nemoci, dosud publikované klinické studie tento účinek nepodporují.
Symptomatická terapie
Klíčovým přístupem v léčbě zůstává symptomatická terapie, která zahrnuje širokou mezioborovou spolupráci. Pacient by si měl udržovat dostatečnou výživu a neměl by výrazně hubnout. Při ambulantních kontrolách doporučujeme pravidelně kontrolovat váhu. Pokud dochází ke zhoršení polykacích obtíží i s nemožností polykání, je třeba nečekat a včas doporučit zavedení nazogastrické sondy nebo řešit dlouhodobě tento problém pomocí perkutánní endoskopické gastrostomie (PEG). Jde o malý zákrok v lokální anestezii, který provádí gastroenterolog. Při zhoršení dechových obtíží je dobré konzultovat specialisty ze spánkové medicíny a diskutovat možnosti podpůrných dýchacích prostředků. Mezi ně řadíme domácí plicní ventilaci (biphasic positive airway pressure; BiPAP). Je-li již stav dechových funkcí neúnosný pro spontánní ventilaci, je nutné znát předem vyslovené přání nemocného, který má právo odmítnout dechovou podporu. V terminálním stadiu je možné podat morfin ke snížení úzkosti, myoskeletálních bolestí, deprese a dechové tísně.
U některých pacientů s bulbárním postižením bývá přítomná hypersalivace, kterou je možné ovlivnit podáním amitriptylinu v malé dávce nebo lokálně aplikovat botulotoxin do slinných žláz. Dávky botulotoxinu jsou relativně malé [52]. Například do podčelistní a příušní žlázy se podává celkově 250 U BoNT-A (Dysport [Galderma, Švýcarsko]) nebo 2500 U BoNT-B(Neurobloc [Eisai Inc., Woodcliff Lake, NJ, USA]) [53]. Doporučuje se aplikovat za pomocí sonografie. Klinický efekt trvá většinou 3 – 4 měsíce, ale u některých pacientů může být až 6 měsíců.
Další důležitou součástí péče o nemocné s ALS je ovlivnění spasticity, pokud je postižení centrálního motoneuronu dominantní lézí. Začíná se perorálním podáváním baklofenu a tizanidinu. Dobré výsledky jsou s použitím lokální léčby botulotoxinu A do spastických svalů u ALS s převážným postižením centrálního motoneuronu a u primární laterální sklerózy, kdy se může významně zlepšit zejména chůze [54]. V případě těžké generalizované spasticity je možné uvažovat o zavedení baklofenové pumpy, která uleví i od doprovodné bolesti při spastickém postižení [55].
Na zmírnění svalových krampů a fascikulací se v některých studiích zkoušel memantin, tetrahydrokanabinol, vitamin E, gabapentin, či baklofen, ale podle rozsáhlé metaanalýzy z databáze Cochrane nebyl účinek těchto léků potvrzen [56].
Nepřesvědčivé výsledky jsou podle této metaanalýzy i při použití repetitivní transkraniální magnetické stimulace TMS, která rovněž nepřinesla významné zlepšení disability u nemocných s ALS oproti významně pozitivnímu účinku intenzivního 3měsíčního cvičení [56].
Neměli bychom zapomenout na urologické a gastrointestinální obtíže a pacienty odeslat k příslušným specialistům. Urologické obtíže (záněty močového měchýře, inkontinence, apod.) jsou u ALS časté, a pokud se objeví na začátku onemocnění, zhoršují její celkový průběh [57]. Gastrointestinální obtíže jsou spojeny se zhoršeným polykáním a s nedostatečným příjmem potravy. V případě úzkosti podáváme nízké dávky antidepresiv nebo benzodiazepinů. Je-li přítomna deprese, zahájíme léčbu antidepresivy ze skupiny selektivních inhibitorů zpětného vychytávání serotoninu (SSRI) (citalopram, escitalopram, sertralin) nebo selektivních inhibitorů zpětného vychytávání serotoninu a noradrenalinu ( SNRI) (venlafaxin). Důležitá je cílená a dlouhodobá rehabilitace a včasné požádání o vhodné pomůcky (vozík, polohovací postel, sedák do vany nebo na WC apod.).
Pokud je onemocnění provázeno příznaky FTD, je možné využít trazodon nebo antipsychotika. Při kombinaci s Alzheimerovou nemocí lze podávat inhibitory acetylcholinesterázy.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
prof. MUDr. Ivana Štětkářová, CSc., MHA
Neurologická klinika 3. LF UK
Ruská 87
100 00 Praha 10
e-mail: ivana.stetkarova@fnkv.cz
Přijato k recenzi: 9. 5. 2018
Přijato do tisku: 29. 8. 2018
Sources
1. Mathis S, Couratier P, Julian A et al. Current view and perspectives in amyotrophic lateral sclerosis. Neural Regen Res 2017; 12(2): 181 – 184. doi: 10.4103/ 1673-5374.200794.
2. Jaiswal MK. Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: implications for motoneurons specific calcium dysregulation. Mol Cell Ther 2014; 2 : 26. doi: 10.1186/ 2052-8426-2-26.
3. Federico A, Cardaioli E, Da Pozzo P et al. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci 2012; 322(1 – 2): 254 – 262. doi: 10.1016/ j.jns.2012.05.030.
4. Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med 2013; 377(2): 162 – 172. doi: 10.1056/ NEJMra1603471.
5. Ambler Z, Bednařík J, Růžička E et al. Klinická neurologie – část speciální II. Praha: Triton 2010.
6. Vucic S, Kiernan MC. Utility of transcranial magnetic stimulation in delineating amyotrophic lateral sclerosis pathophysiology. Handb Clin Neurol 2013; 116 : 561 – 575. doi: 10.1016/ B978-0-444-53497-2.00045-0.
7. Iglesias C, Sangari S, E Mendili MM et al. Electrophysiological and spinal imaging evidences for sensory dysfunction in amyotrophic lateral sclerosis. BMJ Open 2015; 5(2): e007659. doi: 10.1136/ bmjopen-2015-007659.
8. Eisen A, Braak H, Del Tredici K et al. Cortical influences drive amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2017; 88(11): 917 – 924. doi: 10.1136/ jnnp-2017-315573.
9. Bräuer S, Zimyanin V, Hermann A. Prion-like properties of disease-relevant proteins in amyotrophic lateral sclerosis. J Neural Transm (Vienna) 2018; 125(4): 591 – 613. doi: 10.1007/ s00702-018-1851-y.
10. Al-Chalabi A, Hardiman O, Kiernan MC et al. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol 2016; 15(11): 1182 – 1194. doi: 10.1016/ S1474-4422(16)30199-5.
11. Vlčková E. Amyotrofická laterální skleróza. Neurol praxi 2016; 17(6): 362 – 365.
12. Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 2011; 7(11): 639 – 649. doi: 10.1038/ nrneurol.2011.153.
13. Geser F, Brandmeir NJ, Kwong LK. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol2008; 65(5): 636 – 641. doi: 10.1001/ archneur.65.5.636.
14. Geser F, Martinez-Lage M, Robinson J. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol 2009; 66(2): 180 – 189. doi: 10.1001/ archneurol.2008.558.
15. Aulas A, Vande Velde C. Alterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS? Front Cell Neurosci 2015; 9 : 423. doi: 10.3389/ fncel.2015.00423.
16. Vucic S, Rothstein JD, Kiernan MC. Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci 2014; 37(8): 433 – 442. doi: 10.1016/ j.tins.2014.05.006.
17. Chiò A, Logroscino G, Traynor BJ et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 2013; 41(2): 118 – 130. doi: 10.1159/ 000351153.
18. Wu CH, Fallini C, Ticozzi N et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012; 488(7412): 499 – 503. doi: 10.1038/ nature11280.
19. Gorges M, Vercruysse P, Müller HP et al. Hypothalamic atrophy is related to body mass index and age at onset in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2017; 88(12): 1033 – 1041. doi: 10.1136/ jnnp-2017-315795.
20. Katirji B, Kaminski HJ, Ruff RL (eds). Neuromuscular disorders in clinical practice. New York: Springer 2014.
21. Goutman SA. Diagnosis and clinical management of amyotrophic lateral sclerosis and other motor neuron disorders. Continuum 2017; 23(5): 1332 – 1359. doi: 10.1212/ CON.0000000000000535.
22. de Carvalho M, Swash M. Fasciculation potentials and earliest changes in motor unit physiology in ALS. J Neurol Neurosurg Psychiatry 2013; 84(9): 963 – 968. doi: 10.1136/ jnnp-2012-304545.
23. Schrooten M, Smetcoren C, Robberecht W et al. Benefit of the Awaji diagnostic algorithm for amyotrophic lateral sclerosis: a prospective study. Ann Neurol 2011; 70(1): 79 – 83. doi: 10.1002/ ana.22380.
24. Vucic S, Kiernan MC. Transcranial magnetic stimulation for the assessment of neurodegenerative disease. Neurotherapeutics 2017; 14(1): 91 – 106. doi: 10.1007/ s13311-016-0487-6.
25. Baldaranov D, Khomenko A, Kobor I et al. longitudinal diffusion tensor imaging-based assessment of tract alterations: an application to amyotrophic lateral sclerosis. Front Hum Neurosci 2017; 11 : 567. doi: 10.3389/ fnhum.2017.00567.
26. Mélé N, Berzero G, Maisonobe T et al. Motor neuron disease of paraneoplastic origin: a rare but treatable condition. J Neurol 2018; 265(7): 1590 – 1599. doi: 10.1007/ s00415-018-8881-0.
27. Ellison D, Love S, Cardao Chimelli LM et al. Neuropathology: a reference text of CNS pathology. 3rd ed. London: Mosby Elsevier 2013.
28. Dickson DW, Weller RO. Neurodegeneration. The molecular pathology of dementia and movement disorders. 2nd ed. Chichester: Wiley-Blackwell 2011.
29. Love S, Perry A, Ironside J et al. Greenfield‘s Neuropathology. 9th ed. Boca Raton: CRC Press 2015.
30. Mackenzie IR, Frick P, Neumann M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol 2014; 127(3): 347 – 357. doi: 10.1007/ s00401-013-1232-4.
31. Kato S, Hayashi H, Nakashima K et al. Pathological characterization of astrocytic hyaline inclusions in familial amyotrophic lateral sclerosis. Am J Pathol 1997; 151(2): 611 – 620.
32. Ji AL, Zhang X, Chen WW et al. Genetics insight into the amyotrophic lateral sclerosis/ frontotemporal dementia spectrum. J Med Genet 2017; 54(3): 145 – 154. doi: 10.1136/ jmedgenet-2016-104271.
33. DeJesus-Hernandez M, Mackenzie IR, Boeve BF et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72(2): 245 – 256. doi: 10.1016/ j.neuron.2011.09.011.
34. Millecamps S, Boillée S, Le Ber I et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J Med Genet 2012; 49(4): 258 – 263. doi: 10.1136/ jmedgenet-2011-100699.
35. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/ Riluzole Study Group. N Engl J Med 1994; 330(9): 585 – 591. doi: 10.1056/ NEJM199403033300901.
36. Fang T, Al Khleifat A, Meurgey JH et al. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose-ranging study. Lancet Neurol 2018; 17(5): 416 – 422. doi: 10.1016/ S1474-4422(18)30054-1.
37. Writing Group; Edaravone (MCI-186) ALS 19 StudyGroup. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2017; 16(7): 505 – 512. doi: 10.1016/ S1474-4422(17)30115-1.
38. Ikeda K, Iwasaki Y, Kaji R. Neuroprotective effect of ultra-high dose methylcobalamin in wobbler mouse model of amyotrophic lateral sclerosis. J Neurol Sci 2015; 354(1 – 2): 70 – 74. doi: 10.1016/ j.jns.2015.04.052.
39. Akamatsu M, Yamashita T, Hirose N et al. The AMPA receptor antagonist perampanel robustly rescues amyotrophic lateral sclerosis (ALS) pathology in sporadic ALS model mice. Sci Rep 2016; 6 : 28649. doi: 10.1038/ srep28649.
40. Trias E, Ibarburu S, Barreto-Núñez R et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflammation 2016; 13(1): 177. doi: 10.1186/ s12974-016-0620-9.
41. Imamura K, Izumi Y, Watanabe A et al. The Src/ c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci Transl Med 2017; 9(391): pii: eaaf3962. doi: 10.1126/ scitranslmed.aaf3962.
42. Bozik ME, Mitsumoto H, Brooks BR et al. A post hoc analysis of subgroup outcomes and creatinine in the phase III clinical trial (EMPOWER) of dexpramipexole in ALS. Amyotroph Lateral Scler Frontotemporal Degener 2014; 15(5 – 6): 406 – 413. doi: 10.3109/ 21678421.2014.943672.
43. Vieira FG, LaDow E, Moreno A et al. Dexpramipexole is ineffective in two models of ALS related neurodegeneration. PLoS One 2014; 9(12): e91608. doi: 10.1371/ journal.pone.0091608.
44. Shefner JM, Wolff AA, Meng L et al. A randomized, placebo-controlled, double-blind phase IIb trial evaluating the safety and efficacy of tirasemtiv in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2016; 17(5 – 6): 426 – 435. doi: 10.3109/ 21678421.2016.1148169.
45. Morrison KE, Dhariwal S, Hornabrook R et al. Lithium in patients with amyotrophic lateral sclerosis (LiCALS): a phase 3 multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2013; 12(4): 339 – 345. doi: 10.1016/ S1474-4422(13)70037-1.
46. Gotaas HT, Skeie GO, Gilhus NE. Myasthenia gravis and amyotrophic lateral sclerosis: A pathogenic overlap. Neuromuscul Disord 2016; 26(6): 337 – 341. doi: 10.1016/ j.nmd.2016.03.003.
47. Watanabe H, Atsuta N, Hirakawa A et al. A rapid functional decline type of amyotrophic lateral sclerosis is linked to low expression of TTN. J Neurol Neurosurg Psychiatry 2016; 87(8): 851 – 858. doi: 10.1136/ jnnp-2015-311541.
48. Thomsen GM, Gowing G, Svendsen S, Svendsen CN. The past, present and future of stem cell clinical trials for ALS. Exp Neurol 2014; 262 : 127 – 137. doi: 10.1016/ j.expneurol.2014.02.021.
49. Staff NP, Madigan NN, Morris J et al. Safety of intrathecal autologous adipose-derived mesenchymal stromal cells in patients with ALS. Neurology 2016; 87(21): 2230 – 2234. doi: 10.1212/ WNL.0000000000003359.
50. Petrou P, Gothelf Y, Argov Z et al. Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis: results of phase 1/ 2 and 2a clinical trials. JAMA Neurol 2016; 73(3): 337 – 344. doi: 10.1001/ jamaneurol.2015.4321.
51. Baumgartner D, Marusič P, Mazanec R. Kmenové buňky v léčbě amyotrofické laterální sklerózy – přehled současných klinických zkušeností. Cesk Slov Neurol N 2017; 80/ 113(1): 27 – 33. doi: 10.14735/ amcsnn201727.
52. Hosp C, Naumann MK, Hamm H. Botulinum ToxinTreatment of Autonomic Disorders: Focal Hyperhidrosis and Sialorrhea. Semin Neurol 2016; 36(1): 20 – 28. doi: 10.1055/ s-0035-1571214.
53. Guidubaldi A, Fasano A, Ialongo T et al. Botulinum toxin A versus B in sialorrhea: a prospective, randomized, double-blind, crossover pilot study in patients with amyotrophic lateral sclerosis or Parkinson‘s disease. Mov Disord 2011; 26(2): 313 – 319. doi: 10.1002/ mds.23473.
54. Vázquez-Costa JF, Máñez I, Alabajos A et al. Safety and efficacy of botulinum toxin A for the treatment of spasticity in amyotrophic lateral sclerosis: results of a pilot study. J Neurol 2016; 263(10): 1954 – 1960. doi: 10.1007/ s00415-016-8223-z.
55. McClelland S 3rd, Bethoux FA, Boulis NM et al. Intrathecal baclofen for spasticity-related pain in amyotrophic lateral sclerosis: efficacy and factors associated with pain relief. Muscle Nerve 2008; 37(3): 396 – 398. doi: 10.1002/ mus.20900
56. Ng L, Khan, Young CA et al. Symptomatic treatments for amyotrophic lateral sclerosis/ motor neuron disease. Cochrane Database Syst Rev 2017; 1: CD011776. doi: 10.1002/ 14651858.CD011776.pub2.
57. Vázquez-Costa JF, Arlandis S, Hervas D et al. Clinical profile of motor neuron disease patients with lower urinary tract symptoms and neurogenic bladder. J Neurol Sci 2017; 378 : 130 – 136. doi: 10.1016/ j.jns.2017.04.053.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2018 Issue 5
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Nové poznatky v diagnostice a léčbě amyotrofické laterální sklerózy
- Přehled onemocnění s obrazem restrikce difuze na magnetické rezonanci mozku
- Cervikální vertigo – fikce či realita?
- Anestezie a nervosvalová onemocnění