
Poruchy cirkadiánního systému u Huntingtonovy choroby – implikace pro terapii světlem
Circadian system disturbances in Huntington’s disease – implications for light therapy
Huntington disease (HD) is an autosomal-dominant, hereditary neurodegenerative disease with a fatal prognosis. Besides the typical progressive deterioration of motor functions, cognitive and behavioral disorders can also be observed in patients with HD. The most common symptoms also include sleep disorders that seriously affect the quality of life of the patients but also of their relatives and which are being associated with a disrupted circadian system. Stabilization of sleep lenght and quality by strengthening the circadian system could mitigate or suppress many HD symptoms, which, although being a direct result of the disease etiology, can secondarily be heightened by long-term insufficient sleep or circadian system disturbances. Such interventions could lead to slower especially cognitive symptom progression or onset in pre-manifesting patients. Synchronizing bright light therapy, which has already proven useful as a complementary tool for the treatment of affective disorders, as well as some neurodegenerative diseases, could lead to radical improvement of the patients’ quality of life, at least in the early stages of disease development.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Keywords:
Huntington disease – circadian system – clock genes – sleep – light therapy
Authors:
D. Pačesová 1,2; S. Moravcová 1,2; J. Kopřivová 1; Z. Bendová 1,2
Authors‘ workplace:
Národní ústav duševního zdraví, Klecany
1; Přírodovědecká fakulta, UK Praha
2
Published in:
Cesk Slov Neurol N 2019; 82(3): 289-294
Category:
Review Article
doi:
https://doi.org/10.14735/amcsnn2019289
Overview
Huntingtonova choroba (Huntington disease; HD) je autozomálně dominantní, dědičné neurodegenerativní onemocnění s fatální prognózou. Kromě typicky progresivně se zhoršujících motorických funkcí lze u pacientů s HD pozorovat i kognitivní a behaviorální poruchy. Mezi nejčastější symptomy patří také poruchy spánku, které mají velmi závažný dopad na kvalitu života jak pacientů, tak jejich blízkých a které bývají asociovány s narušeným cirkadiánním systémem. Stabilizace délky i kvality spánku posílením cirkadiánního systému by mohla zmírnit či potlačit mnohé symptomy HD, jež mají sice přímou příčinu v etiologii HD, sekundárně mohou být ale zesíleny dlouhodobým nedostatečným spánkem či právě poruchami cirkadiánního systému. U premanifestujících pacientů by takové zásahy mohly vést k pomalejšímu rozvoji či nástupu zejména kognitivních symptomů nemoci. Terapie synchronizujícím jasným světlem, která se již osvědčila jako doplňkový nástroj k léčbě afektivních i některých neurodegenerativních nemocí, by mohla vést k radikálnímu zlepšení života pacientů alespoň v počátečních stadiích
onemocnění.
Klíčová slova:
Huntingtonova choroba – cirkadiánní systém – hodinové geny – spánek – terapie světlem
Úvod
Huntingtonova choroba (Huntington disease; HD) je zapříčiněna abnormálním zmnožením repetic trinukleotidu CAG v exonu 1 genu Huntingtinu na 4. chromozomu [1]. Tento genetický defekt vede k formování abnormálního mutantního proteinu se strukturálními a funkčními změnami. Huntingtin (HTT) je multifunkční protein, který se podílí na několika buněčných procesech, jejichž výčet je skvěle shrnut v review Saudoua a Humberta [2]. V souhrnu lze říct, že HTT je nosný, tzv. scaffold, protein, který svazuje proteiny do komplexů, a tím koordinuje buněčné procesy, jako jsou transport organel v buňce, endocytóza či transkripce. Gen pro HTT byl detekován ve většině tělních tkání. V nervovém systému není jeho exprese omezena pouze na oblasti, které degenerují v důsledku mutace HD, ale vyskytuje se hojně v celém mozku, vč. mozečku [3].
Huntingtonova choroba se nejčastěji manifestuje mezi 35. a 45. rokem života, ovšem doba nástupu nemoci se může lišit podle závažnosti mutace [4]. HD je charakterizována především progresivním zhoršováním motorických funkcí, neboť degenerace primárně zasahuje neurony striata a motorického kortexu [5]. Nemoc se však také projevuje významnými deficity v kognitivních procesech a změnami chování, např. iritabilitou a psychiatrickými symptomy [6]. Velmi často se u pacientů s HD objevuje deprese, která koreluje s poruchami spánku [7]. Poruchy spánku jsou detekovány až u 80 % pacientů s HD [8,9]. Pomocí dotazníkových šetření, aktigrafických a polysomnografických záznamů bylo zjištěno, že u pacientů s HD je ovlivněna jak kvalita, tak kvantita spánku a dochází také k poruše jeho časování, což značí změny v cirkadiánním systému. Změny spánku a cirkadiánní rytmicity jsou mnohdy detekovatelné již ve velmi časných stadiích nemoci, ještě dříve, než se objevují první motorické symptomy [9,10]. Jejich včasná korekce by tak mohla výrazně přispívat k oddálení manifestace HD.
Cirkadiánní systém
Mnohé fyziologické procesy i mnohé formy chování člověka se periodicky opakují v průběhu 24h cyklů, tj. probíhají s tzv. cirkadiánní rytmicitou. Cirkadiánní signál vzniká na úrovni jednotlivých buněk a jeho molekulární podstatou jsou autoregulační transkripčně-translační zpětnovazebné smyčky mezi hodinovými geny a jejich proteinovými produkty [11]. Molekulární smyčky zahrnují transkripční represory, tj. proteiny CRY1, CRY2, PER1, PER2 a REV-ERB α, a transkripční aktivátory BMAL1 a CLOCK a RORA. Základní princip vzájemných interakcí je uveden na obr. 1.
Aby byly molekulární oscilace v jednotlivých buňkách tkání vzájemně synchronizovány a aby byly synchronizovány cirkadiánní oscilace jednotlivých tkání mezi sebou, musí buňky dostávat synchronizační časové signály z centrálního cirkadiánního pacemakeru, který leží v suprachiazmatických jádrech hypotalamu (suprachiasmatic nucleus; SCN). Jednotlivé neurony SCN jsou podobně jako jiné buňky těla autonomními cirkadiánními oscilátory s vlastním hodinovým mechanizmem. Významné práce ze začátku tohoto století ukázaly, že pokud tyto neurony přežívají v disociované kultuře, oscilují každý s vlastní fází i periodou [12]. Jak tedy dokáže SCN jako celek vydávat silný jednotný výstupní signál synchronizující periferní oscilátory? Celá řada současných studií ukázala, že oscilace jednotlivých neuronů jsou vzájemně synchronizovány několika neurotransmitery a neuropeptidy, z nichž nejdůležitější jsou vazoaktivní intestinální peptid (VIP), arginin vazopresin (AVP) a GABA [13–18]. Neuropeptid VIP se váže na receptory VPAC2 a spouští intracelulární kaskádu s G-proteinem jako druhým poslem. Ta vede k fosforylaci transkripčního faktoru CREB (cAMP response element binding) a k aktivaci transkripce genů s promotorovou sekvencí CRE (cAMP response element). Sekvenci CRE mají ve svém promotoru i geny Per1 a Per2, které tak mají dvojí regulaci transkripce; jednak vazbou dimeru CLOCK/ BMAL1 na E-box element, jednak vazbou aktivovaného CREB na CRE element ve svém promotoru (obr. 1) [19]. Tento princip zajišťuje sjednocení fází transkripčních oscilací a zvyšuje amplitudu cirkadiánních rytmů [14,17].
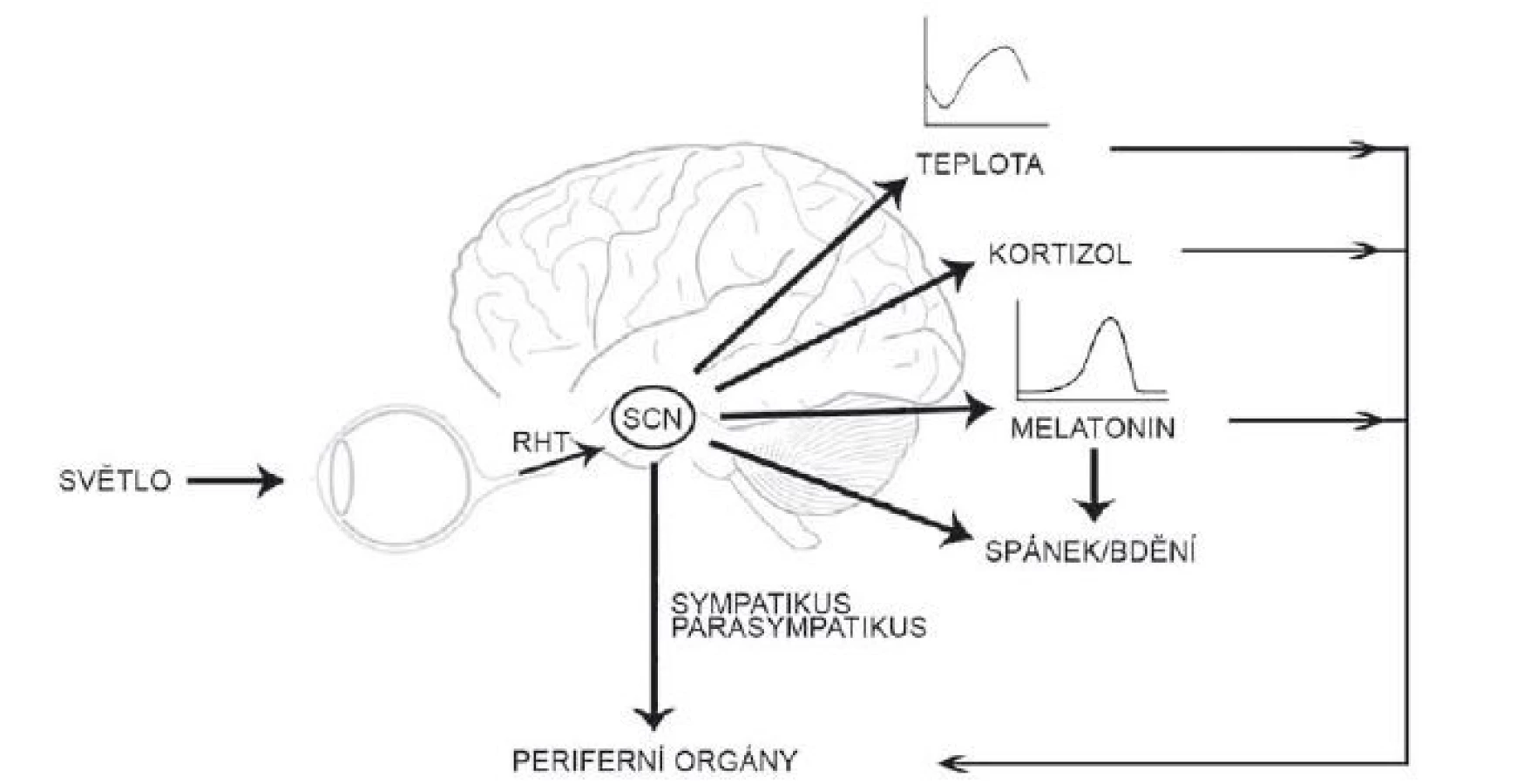
Suprachiasmatická jádra hypotalamu generují a udržují cirkadiánní signál s periodou blízkou 24 h, a to i v neperiodickém prostředí stálé tmy. Tato vnitřní perioda je adaptována k přesnému geofyzikálnímu času pomocí světla [20]. K přenastavení hodin světlem dochází jen během noci či subjektivní noci jedince; světlo vyvolá fázové zpoždění při působení zvečera či předběhnutí hodin při působení zrána. Informace o světelných podmínkách okolí jsou do SCN vedeny přímou drahou z retiny, tzv. retinohypotalamickým traktem (RHT), který je tvořen axony tzv. vnitřně senzitivních gangliových buněk (intrinsically photosensitive retinal ganglion cells; ipRGC) obsahujících fotopigment melanopsin [21]. Glutamát uvolněný z RHT aktivuje receptory kyseliny N-methyl-D-asparagové s GluN2B podjednotkou a vzestup intracelulárního Ca aktivuje signální dráhu, která, podobně jako signalizace od receptoru VPAC2, vede k transkripční aktivaci Per1 vazbou CREB na CRE sekvenci v jeho promotoru [22–24]. Tato světlem indukovaná změna v expresi hodinového genu je považována za hlavní mechanizmus, kterým světlo synchronizuje cirkadiánní systém [21]. Cirkadiánní pacemaker v SCN tedy integruje endogenní časové signály s časem okolí a o výsledku informuje neuronálními i humorálními drahami celý organizmus. Synchronizuje tak rytmy v jednotlivých tkáních a umožňuje jejich adaptaci na časové změny v okolním prostředí. Udává organizmu vnitřní časový řád (obr. 2).
Změny cirkadiánního systému u pacientů s Huntingtonovou chorobou
Ztráta pravidelného střídání denní aktivity a spánku je nejzřetelnější pozorovanou poruchou cirkadiánního systému u pacientů s HD. Bývá popisována fragmentace spánku a zvýšená noční aktivita [9,10], je změněna architektura spánku a též zvýšena latence usnutí [25]. Ve dne jsou pacienti ospalí, často podřimují během dne. Toto chování bývá spojováno se zhoršením kognitivních schopností a také s rozvojem deprese [7,26]. Jednou z možných příčin tohoto stavu mohou být úbytek orexigenních neuronů laterálního hypotalamu a snížená amplituda cirkadiánního rytmu v aktivitě orexigenních neuronů, které významně přispívají k udržování stavu bdělosti [27,28]. Nedávná studie také ukázala, že až 63 % pacientů s HD má narušený cirkadiánní rytmus v krevním tlaku, což významně koreluje právě se sníženou kvalitou spánku u těchto pacientů [29].
Analýza cirkadiánního rytmu melatoninu v plazmě v průběhu 24 h u kontrolních osob, u premanifestujících a u pacientů v pokročilých stadiích HD ukázala, že amplituda rytmu sekrece melatoninu je u pacientů snížena a noční koncentrace melatoninu v plazmě je výrazně nižší. Sekrece melatoninu klesá s rozvojem a postupem nemoci, ale výrazně nižší koncentrace melatoninu než u zdravých osob je patrna již v preklinických stadiích nemoci [25,30]. Vedle melatoninu jsou u pacientů s HD pozorovány také změny v produkci kortizolu. Celková produkce kortizolu za 24 h je v časné fázi choroby signifikantně vyšší a denní profil rytmu v sekreci kortizolu má zvýšenou amplitudu [31]. Pacienti s HD vykazují zejména zvýšenou ranní produkci kortizolu, kterou lze obecně pozorovat při spánkových deprivacích [31,32]. Přestože spánkové poruchy spolu s abnormální sekrecí kortizolu mohou mít příčinu také ve změnách HPA osy u pacientů s HD [31,33], abnormální sekrece melatoninu naznačuje, že změny by mohly pramenit i z narušené signalizace z SCN.
O změnách v SCN u pacientů s HD není známo mnoho. Jediné dostupné informace pocházejí z post mortem studií. V jedné takové studii bylo pomocí imunocytochemie v SCN zjištěno signifikantní snížení počtu neuronů produkujících VIP, a v menší míře také redukce neuronů produkujících AVP [34]. Změny v hladinách VIP a AVP se zdají být specifické pro SCN, v jiných hypotalamických strukturách, jako je paraventrikulární jádro, nebyly detekovány [35,36].
Animální modely Huntingtonovy choroby a cirkadiánní systém
Identifikace genetické mutace HD vedla k vytvoření několika animálních modelů, které exprimují HTT se zvýšeným množstvím repetic CAG tripletů [37]. Zdaleka nejpoužívanější model je linie transgenních myší R6/ 2, do jejichž genomu byl vpraven fragment lidského genu nesoucího prodlouženou repetici CAG tripletů [38]. Tento model vykazuje mnoho symptomů analogických symptomům pacientů s HD [9,39–43]. Cirkadiánní fenotyp HD mutace se u tohoto modelu objevuje po 16. týdnu života. Zvířata pozvolna ztrácí rytmicitu v pohybové aktivitě i v podmínkách střídání světla a tmy a kolem 18. týdne jsou již zcela arytmická. Kromě úbytku orexigenních neuronů v laterálním hypotalamu a snížené amplitudy rytmu jejich elektrické aktivity [28] byly u těchto myší zjištěny také snížené hladiny VIP a VPAC2 v SCN [44]. Behaviorální fenotyp myší R6/ 2 se výrazně podobá fenotypu myší Vipr2-/ - a Vip-/ -, tj. myší s delecí těchto genů. Tato zvířata mají narušený cirkadiánní rytmus ve střídání aktivity a spánku; vykazují zvýšenou denní a sníženou noční aktivitu v režimu střídání světla a tmy (light/ dark) nebo vykazují úplně arytmické chování v režimu stálé tmy (dark/ dark), mají narušený rytmus tělesné teploty a změny v EEG [41–45]. Je možné, že narušená signalizace VIP v SCN je jednou z příčin patologických změn cirkadiánních rytmů u pacientů s HD. Jednotlivé neurony SCN nejsou mezi sebou dobře synchronizovány, každý osciluje s odlišnou periodou a fází a SCN jako celek není schopné generovat jednotný časový signál, který předává k periferním tkáním organizmu. Tato úvaha je podpořena nálezem desynchronizovaných periferních hodin v játrech u jednoho z myších modelů HD [46].
Alternativní vysvětlení či paralelní fenotyp nabízí dvě nedávné studie, které ukázaly postupnou degeneraci sítnice oka, zejména čípků a také gangliových ipRGC s melanopsinem [43,47]. Jejich výsledky naznačují, že cirkadiánní fenotyp 20týdenních myší R6/ 2 je podobný fenotypu myší s genetickou delecí melanopsinu [48]. To pravděpodobně vede ke snížení fotorecepce sítnice, a tím i významu světelné informace jako synchronizátoru cirkadiánního pacemakeru v SCN. Ačkoliv úbytek melanopsinu nebyl studován u pacientů s HD, degenerace čípků a změny v barevném vidění potvrzují degeneraci sítnice i u těchto pacientů [49,50].
Světelná synchronizace u modelu R6/ 2
Z experimentů na animálních modelech HD vyplývá, že mutovaný HTT v buňkách SCN pracuje v neprospěch intercelulární synchronizace, která podmiňuje sebeudržující chod cirkadiánních hodin, a to minimálně potlačením syntézy VIP ve světločivných buňkách ventrolaterálního SCN. Z pokusů na myších Vipr2-/ - vyplývá, že absence VIP nemusí měnit intracelulární signální kaskádu aktivovanou světelnými podněty, která vede k transkripci hodinových genů, mění ale vzájemnou komunikaci mezi buňkami, a tím i celkové nastavení cirkadiánního pacemakeru. To má za následek jeho změněnou reaktivitu na synchronizační světelné stimuly. Nedostatek VIP může být bezpochyby podpořen také změněnou GABAergní signalizací, která je podstatnou součástí intercelulární synchronizace v SCN regulované VIP, či chronickou hyperpolarizací neuronů SCN bez VIP [50,51].
K patologickým změnám v synchronizaci hodin přispívá jistě také postupná degenerace sítnice oka a zejména ipRGC. Protože ipRGC inervují kromě SCN také olivární pretektální jádro, kromě histologické detekce úbytku melanopsinu se v experimentech s R6/ 2 myšmi testovala též jejich funkce – měřením pupilárního reflexu [43,47]. Při použití nízké a střední intenzity světla byl pupilární reflex redukován již u myší ve věku 12 a 15 týdnů a zcela selhával ve věku 20 týdnů i při použití jasného silného světla. Tyto změny ve fotorecepci sítnice mohou přispět k progresivní deregulaci cirkadiánního systému pozorované u pacientů s HD. Jak onemocnění progreduje, cirkadiánní systém se stává čím dál necitlivějším k vnějšímu cyklu světla a tmy, zvláště při nízké intenzitě osvětlení. V souladu s těmito nálezy další experimenty ukázaly, že zvýšení intenzity synchronizačního světla může zpomalit progresi dysfunkce cirkadiánního systému u myší R6/ 2. Pokud byl těmto myším aplikován hodinový světelný pulz o vysoké intenzitě 1 500 luxů, rytmus jejich pohybové aktivity se lépe synchronizoval s 24h režimem. Ještě lepších výsledků bylo dosaženo kombinací silného světla a pravidelné pohybové aktivity načasované na stejnou denní dobu [53].
Ačkoliv cirkadiánní systém animálních modelů funguje v principu podobně jako cirkadiánní systém člověka, myši jako noční živočichové jsou výrazně citlivější na intenzitu světla než denní živočichové a 1 500 lx použitých pro synchronizaci nočních hlodavců nemusí být lidským cirkadiánním systémem vnímáno jako silné světlo. Je známo, že nesynchronizovaný, tzv. volný běh cirkadiánních hodin podle molekulárního mechanizmu (obr. 1) může u člověka nastat již při nízké intenzitě denního světla v kombinaci s nedostatečnou tmou v noci. Stejně jako u všech organizmů je totiž cirkadiánní systém člověka závislý na vysokém kontrastu mezi světlem ve dne a tmou v noci [54,55]. Je možné, že narušená signalizace VIP v SCN může projevit svůj patologický dopad na amplitudu a fázi cirkadiánních rytmů zejména u těch pacientů s HD, kteří žijí ve špatně osvětleném prostředí denním a přisvětlovaném prostředí nočním. Zvýšení kontrastu mezi dnem a nocí či cílená terapie jasným světlem by mohly tento deficit částečně kompenzovat, alespoň do té doby, dokud ipRGC buňky sítnice produkují dostatečné množství melanopsinu. Synchronizačního účinku světla na cirkadiánní systém, jehož narušení bývá průvodním jevem řady dalších neurodegenerativních onemocnění, se již využívá při terapii Alzheimerovy i Parkinsonovy nemoci, které jsou v pozdějších stadiích také provázeny degenerací sítnice a ztrátou fotosenzitivity. Obvykle se terapie provádí ráno nebo 2× denně silným světlem o intenzitě 5 000–10 000 l lx (měřeno ve výšce očí) [56–60].
Fig. 1. Transcriptional activators (BMAL1 and CLOCK) induce transcription of repressors (CRY1, CRY2, PER1, PER2) by binding to the E-box
element in their promoter sequences. The repressors inhibit the activity of CLOCK/BMAL1 after translocation to the nucleus and they
are simultaneously a substrate for post-translational modifications and controlled protein degradation. Upon significant decrease in the
repressors levels, the activators CLOCK/BMAL1 are released and the cycle commences again. The levels of transcripts and protein products
of clock genes oscillate with circadian period in the cells. The dynamics of these oscillations are changed by expression of Per1
and Per2 triggered by activation of CRE via VIP signalization and/or photic stimulation. The transcriptional activators induce transcription
of their own repressors as well as large groups of clock-controlled genes. The proteins of clock-controlled genes do not have direct
feedback to the circadian mechanism but exhibit their own tissue-specific functions. Genome studies have shown that up to 10% of the
whole gene transcription may be regulated by the circadian mechanism [19].
VIP – vasoactive intestinal peptide
![Transkripční aktivátory (BMAL1 a CLOCK) indukují transkripci represorů (CRY1, CRY2, PER1, PER2) vazbou k E-boxu v jejich promotorových
sekvencích. Ty po vstupu do jádra inhibují aktivitu aktivátorů CLOCK/BMAL1 a zároveň jsou substrátem pro posttranslační
modifi kace a řízenou proteinovou degradaci. Když jejich hladina klesne dostatečně, aktivátory CLOCK/BMAL1 se uvolní a cyklus začíná
znovu. Hladiny transkriptů i proteinů hodinových genů takto oscilují v buňkách s cirkadiánní periodou. Dynamiku těchto oscilací mění
exprese Per1 a Per2 spouštěná aktivací CRE místa v promotoru těchto genů signalizací VIP nebo/a světelnými stimuly. Transkripční aktivátory
však neindukují transkripci pouze vlastních represorů, ale také velké skupiny tzv. hodinami kontrolovaných genů, jejichž proteiny
nemají přímou zpětnou vazbu v cirkadiánním mechanizmu, ale mají vlastní, tkáňově specifickou funkci. Genomové studie ukázaly, že až
10 % genové transkripce může být regulováno cirkadiánním mechanizmem [19].
VIP – vazoaktivní intestinální peptid<br>
Fig. 1. Transcriptional activators (BMAL1 and CLOCK) induce transcription of repressors (CRY1, CRY2, PER1, PER2) by binding to the E-box
element in their promoter sequences. The repressors inhibit the activity of CLOCK/BMAL1 after translocation to the nucleus and they
are simultaneously a substrate for post-translational modifications and controlled protein degradation. Upon significant decrease in the
repressors levels, the activators CLOCK/BMAL1 are released and the cycle commences again. The levels of transcripts and protein products
of clock genes oscillate with circadian period in the cells. The dynamics of these oscillations are changed by expression of Per1
and Per2 triggered by activation of CRE via VIP signalization and/or photic stimulation. The transcriptional activators induce transcription
of their own repressors as well as large groups of clock-controlled genes. The proteins of clock-controlled genes do not have direct
feedback to the circadian mechanism but exhibit their own tissue-specific functions. Genome studies have shown that up to 10% of the
whole gene transcription may be regulated by the circadian mechanism [19].
VIP – vasoactive intestinal peptide](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/0e471345af4c0f17e86728d0a8ffda85.jpeg)
Fig. 2. The suprachiasmatic nucleus receives the photic information from the retina, synchronizes rhythms in body temperature, melatonin
and cortisol secretion, sleep regulation and regulates the physiology of the peripheral organs both directly and indirectly.
RHT – retinohypothalamic tract; SCN – suprachiasmatic nucleus
Závěr
Narušení cirkadiánní rytmicity je podobně jako u většiny neurodegenerativních onemocnění zjevné u pacientů s HD ještě v premanisfestujících stadiích nemoci, před rozvojem motorického a kognitivního deficitu. Společným znakem pacientů i animálních modelů je úbytek VIP signalizace v cirkadiánním pacemakeru, který znamená desynchronizaci mezi jednotlivými buněčnými oscilátory SCN a snížení amplitudy cirkadiánních rytmů či jejich úplnou ztrátu. Protože buňky SCN produkující VIP jsou také receptivní pro signály z RHT, tento deficit mění i výchozí nastavení pacemakeru a jeho odpověď na světelné stimuly. Z dostupných studií jednoznačně vyplývá, že nedostatečný rozdíl v intenzitě světla mezi dnem a nocí přispívá k dalšímu rozvolnění interakcí mezi buněčnými oscilátory a desynchronizaci cirkadiánních rytmů. Je překvapivé, jak málo je známo o využití světelné terapie jak u pacientů s HD, tak u myších modelů této nemoci, ačkoliv u jiných typů neurodegenerativních onemocnění se již s úspěchem využívá pro zvýšení amplitudy cirkadiánních oscilací, a tím i zlepšení spánku, nálady, kognitivních a dokonce i motorických funkcí a pro zpomalení progrese onemocnění.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Dominika Pačesová
Národní ústav duševního zdraví
Topolová 748
250 67 Klecany
e-mail: dominika.pacesova@natur.cuni.cz
Přijato k recenzi: 27. 3. 2019
Přijato do tisku: 10. 5. 2019
Sources
1. Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993; 72(6): 971–983. doi: https://doi.org/10.1016/0092-8674(93)90585-E.
2. Saudou F, Humbert S. The biology of Huntingtin. Neuron 2016; 89(5): 910–926. doi: 10.1016/j.neuron.2016.02.003.
3. Fossale E, Seong IS, Coser KR et al. Differential effects of the Huntington’s disease CAG mutation in striatum and cerebellum are quantitative not qualitative. Hum Mol Genet 2011; 20(21): 4258–4267. doi: 10.1093/hmg/ddr355.
4. Langbehn DR, Brinkman RR, Falush D et al. International Huntington’s Disease Collaborative Group. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet 2004; 65(4): 267–277. doi: 10.1111/j.1399-0004.2004.00241.
5. Vonsattel JP, Myers RH, Stevens TJ et al. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 1985; 44(6): 559–577. doi: 10.1097/00005072-198511000-00003.
6. Eddy CM, Parkinson EG, Rickards HE. Changes in mental state and behaviour in Huntington’s disease. Lancet Psychiatry 2016; 3(11): 1079–1086. doi: 10.1016/S2215-0366(16)30144-4.
7. Aziz NA, Anguelova GV, Marinus J et al. Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington’s disease. Parkinsonism Relat Disord 2010; 16(5): 345–350. doi: 10.1016/j.parkreldis.2010.02.009.
8. Morton AJ. Circadian and sleep disorder in Huntington’s disease. Exp Neurol 2013; 243 : 34–44. doi: 10.1016/j.expneurol.2012.10.014.
9. Morton AJ, Wood NI, Hastings MH et al. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J Neurosci 2005; 25(1): 157–163. doi: 10.1523/JNEUROSCI.3842-04.2005.
10. Goodman AO, Rogers L, Pilsworth S et al. Asymptomatic sleep abnormalities are a common early feature in patients with Huntington’s disease. Curr Neurol Neurosci Rep 2011; 11(2): 211–217. doi: 10.1007/s11910-010-0163-x.
11. Maywood ES. Synchronization and maintenance of circadian timing in the mammalian clockwork. Eur J Neurosci 2018. doi: 10.1111/ejn.14279.
12. Shirakawa T, Honma S, Honma K. Multiple oscillators in the suprachiasmatic nucleus. Chronobiol Int 2001; 18(3): 371–387. doi: 10.1081/CBI-100103962.
13. Evans JA, Leise TL, Castanon-Cervantes O et al. Dynamic interactions mediated by nonredundant signaling mechanisms couple circadian clock neurons. Neuron 2013; 80(4): 973–983. doi: 10.1016/j.neuron.2013.08.022.
14. Aton SJ, Colwell CS, Harmar AJ et al. Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat Neurosci 2005; 8(4): 476–483. doi: 10.1038/nn1419.
15. Aton SJ, Huettner JE, Straume M et al. GABA and Gi/o differentially control circadian rhythms and synchrony in clock neurons. Proc Natl Acad Sci U S A 2006; 103(50): 19188–19193. doi: 10.1073/pnas.0607466103.
16. Harmar AJ, Marston HM, Shen S et al. The VPAC(2) receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell 2002; 109(4): 497–508. doi: 10.1016/S0092-8674(02)00736-5.
17. Brown TM, Colwell CS, Waschek JA et al. Disrupted neuronal activity rhythms in the suprachiasmatic nuclei of vasoactive intestinal polypeptide-deficient mice. J Neurophysiol 2007; 97(3): 2553–2558. doi: 10.1152/jn.01206.2006.
18. Colwell CS, Michel S, Itri J et al. Disrupted circadian rhythms in VIP - and PHI-deficient mice. Am J Physiol Regul Integr Comp Physiol 2003; 285(5): R939–R949. doi: 10.1152/ajpregu.00200.2003.
19. Panda S, Antoch MP, Miller BH et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002; 109(3): 307–320. doi: 10.1016/S0092-8674(02)00722-5.
20. Ramkisoensing A, Meijer JH. Synchronization of biological clock neurons by light and peripheral feedback systems promotes circadian rhythms and health. Front Neurol 2015; 6 : 128. doi: 10.3389/fneur.2015.00
128.
21. Golombek DA, Rosenstein RE. Physiology of circadian entrainment. Physiol Rev 2010; 90(3): 1063–1102. doi: 10.1152/physrev.00009.2009.
22. Travnickova-Bendova Z, Cermakian N, Reppert SM et al. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc Natl Acad Sci U S A 2002; 99(11): 7728–7733. doi: 10.1073/pnas.102075599.
23. Yan L, Silver R. Resetting the brain clock: time course and localization of mPER1 and mPER2 protein expression in suprachiasmatic nuclei during phase shifts. Eur J Neurosci 2004; 19(4): 1105–1109. doi: 10.1111/j.1460-9568.2004.03189.x.
24. Colwell CS. Linking neural activity and molecular oscillations in the SCN. Nat Rev Neurosci 2011; 12(10): 553–569. doi: 10.1038/nrn3086.
25. Kalliolia E, Silajdžić E, Nambron R et al. Plasma melatonin is reduced in Huntington’s disease. Mov Disord 2014; 29(12): 1511–1515. doi: 10.1002/mds.26003.
26. Baker CR, Domínguez D JF, Stout JC et al. Subjective sleep problems in Huntington’s disease: a pilot investigation of the relationship to brain structure, neurocognitive, and neuropsychiatric function. J Neurol Sci 2016; 364 : 148–153. doi: 10.1016/j.jns.2016.03.021.
27. Shan L, Dauvilliers Y, Siegel JM. Interactions of the histamine and hypocretin systems in CNS disorders. Nat Rev Neurol 2015;11(7): 401–413. doi: 10.1038/nrneurol.2015.99.
28. Williams RH, Morton AJ, Burdakov D. Paradoxical function of orexin/hypocretin circuits in a mouse model of Huntington’s disease. Neurobiol Dis 2011; 42(3): 438–445. doi: 10.1016/j.nbd.2011.02.006.
29. Bellosta Diago E, Pérez Pérez J, Santos Lasaosa S et al. Circadian rhythm and autonomic dysfunction in presymptomatic and early Huntington’s disease. Parkinsonism Relat Disord 2017; 44 : 95–100. doi: 10.1016/j.parkreldis.2017.09.013.
30. Aziz NA, Pijl H, Frölich M et al. Delayed onset of the diurnal melatonin rise in patients with Huntington’s disease. J Neurol 2009; 256(12): 1961–1965. doi: 10.1007/s00415-009-5196-1.
31. Aziz NA, Pijl H, Frölich M et al. Increased hypothalamic-pituitary-adrenal axis activity in Huntington’s disease. J Clin Endocrinol Metab 2009; 94(4): 1223–1228. doi: 10.1210/jc.2008-2543.
32. Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet 1999; 354(9188): 1435–1439. doi: 10.1016/S0140-6736(99)013-
76-8.
33. van Duijn E, Selis MA, Giltay EJ et al. Hypothalamic-pituitary-adrenal axis functioning in Huntington’s disease mutation carriers compared with mutation-negative first-degree controls. Brain Res Bull 2010; 83(5): 232–237. doi: 10.1016/j.brainresbull.2010.08.006.
34. van Wamelen DJ, Aziz NA, Anink JJ et al. Suprachiasmatic nucleus neuropeptide expression in patients with Huntington’s Disease. Sleep 2013; 36(1): 117–125. doi: 10.5665/sleep.2314.
35. van Wamelen DJ, Aziz NA, Anink JJ et al. Paraventricular nucleus neuropeptide expression in Huntington’s disease patients. Brain Pathol 2012; 22(5): 654–661. doi: 10.1111/j.1750-3639.2012.00565.x.
36. Emson PC, Fahrenkrug J, Spokes EG. Vasoactive intestinal polypeptide (VIP): distribution in normal human brain and in Huntington’s disease. Brain Res 1979; 173(1): 174–178. doi: 10.1016/0006-8993(79)91109-0.
37. Bates GP, Mangiarini L, Mahal A et al. Transgenic models of Huntington’s disease. Hum Mol Genet 1997; 6(10): 1633–1637. doi: 10.1093/hmg/6.10.1633.
38. Mangiarini L, Sathasivam K, Seller M et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996; 87(3): 493–506. doi: 10.1016/S0092-8674(00)81369-0.
39. Lione LA, Carter RJ, Hunt MJ et al. Selective discrimination learning impairments in mice expressing the human Huntington’s disease mutation. J Neurosci 1999; 19(23): 10428–10437. doi: 10.1523/JNEUROSCI.19-23-10428.1999.
40. Björkqvist M, Petersén A, Bacos K et al. Progressive alterations in the hypothalamic-pituitary-adrenal axis in the R6/2 transgenic mouse model of Huntington’s disease. Hum Mol Genet 2006; 15(10): 1713–1721. doi: 10.1093/hmg/ddl094.
41. Pallier PN, Maywood ES, Zheng Z et al. Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington’s disease. J Neurosci 2007; 27(29): 7869–7878. doi: 10.1523/JNEUROSCI.0649-07.2007.
42. van Wamelen DJ, Aziz NA, Roos RA et al. Hypothalamic alterations in Huntington’s disease patients: comparison with genetic rodent models. J Neuroendocrinol 2014; 26(11): 761–775. doi: 10.1111/jne.12190.
43. Lin M, Liao P, Chen HM et al. Degeneration of ipRGCs in mouse models of Huntington’s Disease disrupts non-image forming behaviors prior to motor impairment. J Neurosci 2019; 39(8): 1505–1524. doi: 10.1523/JNEUROSCI.0571-18.2018.
44. Fahrenkrug J, Popovic N, Georg B et al. Decreased VIP and VPAC2 receptor expression in the biological clock of the R6/2 Huntington’s disease mouse. J Mol Neurosci 2007; 31(2): 139–148. doi: 10.1385/JMN/31 : 02 : 139.
45. Fisher SP, Black SW, Schwartz MD et al. Longitudinal analysis of the electroencephalogram and sleep phenotype in the R6/2 mouse model of Huntington’s disease. Brain 2013; 136(Pt 7): 2159–2172. doi: 10.1093/brain/awt132.
46. Maywood ES, Fraenkel E, McAllister CJ et al. Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J Neurosci 2010; 30(30): 10199–10204. doi: 10.1523/JNEUROSCI.1694-10.2010.
47. Ouk K, Hughes S, Pothecary CA et al. Attenuated pupillary light responses and downregulation of opsin expression parallel decline in circadian disruption in two different mouse models of Huntington’s disease. Hum Mol Genet 2016; 25(24): 5418–5432. doi: 10.1093/hmg/ddw359.
48. Lucas RJ, Hattar S, Takao M et al. Diminished pupillary light reflex at high irradiances in melanopsin-knockout mice. Science 2003; 299(5604): 245–247. doi: 10.1126/science.1077293.
49. Paulus W, Schwarz G, Werner A et al. Impairment of retinal increment thresholds in Huntington’s disease. Ann Neurol 1993; 34(4): 574–578. doi: 10.1002/ana.410340411.
50. Kersten HM, Danesh-Meyer HV, Kilfoyle DH et al. Optical coherence tomography findings in Huntington’s disease: a potential biomarker of disease progression. J Neurol 2015; 262(11): 2457–2465. doi: 10.1007/s00415-015-7869-2.
51. Pakhotin P, Harmar AJ, Verkhratsky A et al. VIP receptors control excitability of suprachiasmatic nuclei neurones. Pflugers Arch 2006; 452(1): 7–15. doi: 10.1007/s00424-005-0003-z.
52. Vosko A, van Diepen HC, Kuljis D et al. Role of vasoactive intestinal peptide in the light input to the circadian system. Eur J Neurosci 2015; 42(2): 1839–1848. doi: 10.1111/ejn.12919.
53. Cuesta M, Aungier J, Morton AJ. Behavioral therapy reverses circadian deficits in a transgenic mouse model of Huntington’s disease. Neurobiol Dis 2014; 63 : 85–91. doi: 10.1016/j.nbd.2013.11.008.
54. Roenneberg T, Merrow M. The circadian clock and human health. Curr Biol 2016; 26(10): R432–R443. doi: 10.1016/j.cub.2016.04.011.
55. Madrid-Navarro CJ, Sanchez-Galvez R, Martinez-Nicolas A et al. Disruption of circadian rhythms and delirium, sleep impairment and sepsis in critically ill patients. Potential therapeutic implications for increased light-dark contrast and melatonin therapy in an ICU environment. Curr Pham Des 2015; 21(24): 3453–3468. doi: 10.2174/1381612821666150706105602.
56. Dowling GA, Graf CL, Hubbard EM et al. Light treatment for neuropsychiatric behaviors in Alzheimer’s disease. West J Nurs Res 2007; 29(8): 961–975. doi: 10.1177/0193945907303083.
57. Dowling GA, Hubbard EM, Mastick J et al. Effect of morning bright light treatment for rest-activity disruption in institutionalized patients with severe Alzheimer’s disease. Int Psychogeriatr 2005; 17(2): 221–236. doi: 10.1017/S1041610205001584.
58. Willis GL, Turner EJ. Primary and secondary features of Parkinson’s disease improve with strategic exposure to bright light: a case series study. Chronobiol Int
2007; 24(3): 521–537. doi: 10.1080/07420520701420
717.
59. La Morgia C, Ross-Cisneros FN, Sadun AA et al. Retinal ganglion cells and circadian rhythms in Alzheimer’s disease, Parkinson’s disease, and beyond. Front Neurol 2017; 8 : 162. doi: 10.3389/fneur.2017.00162.
60. Fifel K, Videnovic A. Chronotherapies for Parkinson’s disease. Prog Neurobiol 2019; 174 : 16–27. doi: 10.1016/j.pneurobio.2019.01.002.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2019 Issue 3
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Test mince v ruce k detekci předstírání oslabeného paměťového výkonu ve srovnání s mírnou kognitivní poruchou a s mírnou demencí u Alzheimerovy nemoci
- Neuromuskulární choroby a gravidita
- Měření terče zrakového nervu a sítnice pomocí optické koherentní tomografie u nově diagnostikované idiopatické intrakraniální hypertenze bez ztráty zraku
- Využití vakuově-kompresní terapie v léčbě syndromu karpálního tunelu jako součást fyzioterapie – pilotní studie