
Studium procesu homogenizace práškových směsí s využitím NIR spektroskopie
A study of the process of homogenization of powder mixtures using NIR spectroscopy
The paper focuses on the use of NIR spectroscopy in the study of mixing powder mixtures. The study made use of model mixtures composed of diclofenac sodium salt, cetyl alcohol, povidon-30, sucrose and magnesium stearate. The measurement of NIR spectra was carried out with the use of a probe making it possible to scan the spectra direct from the homogenization vessel. The measured spectra were mathematically adjusted and subsequently processed by means of chemometric methods in order to obtain the predicate data on the course of mixing. The obtained data were evaluated by means of statistical methods and subsequently they were plotted into graphs in dependence on the time of mixing in order that the course of mixing could be well observable. The resulting graphs describing the course of mixing clearly show the stages of convective and diffuse mixing. The spectra which were statistically evaluated as remote were further studied in order to identify the causes of their origin. The other measurements included the data of physical properties of powders, namely flow density, Hausner ratio, and average particle size. The obtained data well illustrate the possibilities of NIR spectroscopy in the examination of the variability of the mixing process in time and document the suitability of NIR spectroscopy in the control of the process of mixing of powder mixtures.
Key words:
NIR spectroscopy – homogenization – convective mixing – diffuse mixing – chemometric methods
Authors:
P. Doležel 1; J. Muselík 1; K. Dvořáčková 1; K. Šustová 2
Authors‘ workplace:
Veterinární a farmaceutická univerzita Brno, Farmaceutická fakulta, Ústav technologie léků
1; Mendelova univerzita v Brně, Agronomická fakulta, Ústav technologie potravin
2
Published in:
Čes. slov. Farm., 2010; 59, 263-271
Category:
Original Articles
Overview
Práce je zaměřena na využití NIR spektroskopie při studiu mísení práškových směsí. Pro studium se využily modelové směsi složené z diklofenaku sodné soli, cetylalkoholu, povidonu-30, sacharosy a stearanu hořečnatého. Pro měření NIR spekter se použila sonda umožňující snímání spekter přímo z homogenizační nádoby. Naměřená spektra se matematicky upravila a následně zpracovávala pomocí chemometrických metod za účelem získání vypovídajících dat o průběhu mísení. Získaná data se hodnotila pomocí statistických metod a následně se vynášela do grafů v závislosti na čase mísení tak, aby byl dobře pozorovatelný průběh mísení. Z výsledných grafů popisujících průběh mísení jsou zřetelné fáze konvektivního a difuzního mísení. Spektra statisticky ohodnocená jako odlehlá se dále studovala za účelem identifikace příčin jejich vzniku. Dále se měřila data fyzikálních vlastností prášků, a to setřesná hustota, Hausnerův poměr, průměrná velikost částic. Získaná data dobře ilustrují možnosti NIR spektroskopie při sledování variability mísícího procesu v čase a dokladují vhodnost NIR spektroskopie při kontrole procesu mísení práškových směsí.
Klíčová slova:
NIR spektroskopie – homogenizace – konvektivní mísení – difuzní mísení – chemometrické metody
Úvod
Blízká infračervená spektroskopie (NIR spektroskopie) je založená na principu absorpce infračerveného záření v oblasti 800–2500 nm. Absorpcí těchto vlnových délek dochází ke změnám rotačně-vibračních stavů molekul. Analyticky významné jsou především vibrační stavy molekul, mezi jejichž energetickými hladinami dochází k násobným přechodům, neboli overtonům a dále kombinaci jednoduchých přechodů 1). Většina pásů pozorovaných v NIR spektru pochází z vibrací lehkých atomů, jejichž vazby vibrují s vysokou frekvencí, např. O-H, C‑H, S-H, N-H. Výsledné spektrum tak vzniká z velkého množství překrývajících se pásů, které jeví nižší intenzitu v porovnání s infračervenou spektroskopií ve střední oblasti 2). Je tedy obtížnější vyvozovat ze spekter závěry o struktuře, a proto se spektroskopie v blízké infračervené oblasti používá zejména při kvantitativní analýze nebo při potvrzení identity látek. Mezi její hlavní výhody patří možnost analyzovat vzorky přímo, bez úprav jako je rozpouštění, nebo ředění. Je to rychlá, nedestruktivní metoda, vhodná pro analýzu kapalin a pevných látek, umožňující širokou aplikaci v potravinářství, zemědělství, analýze životního prostředí, nebo farmacii 3). Přirozeně tak NIR spektroskopie našla významné využití i při studiu homogenizace práškových směsí, kterému je ve farmaceutické technologii věnována velká pozornost. Jde totiž o jeden z klíčových stupňů při výrobě lékové formy, který je národními a nadnárodními autoritami v oblasti bezpečnosti léčiv ostře sledován 4).
POKUSNÁ ČÁST
Použité suroviny
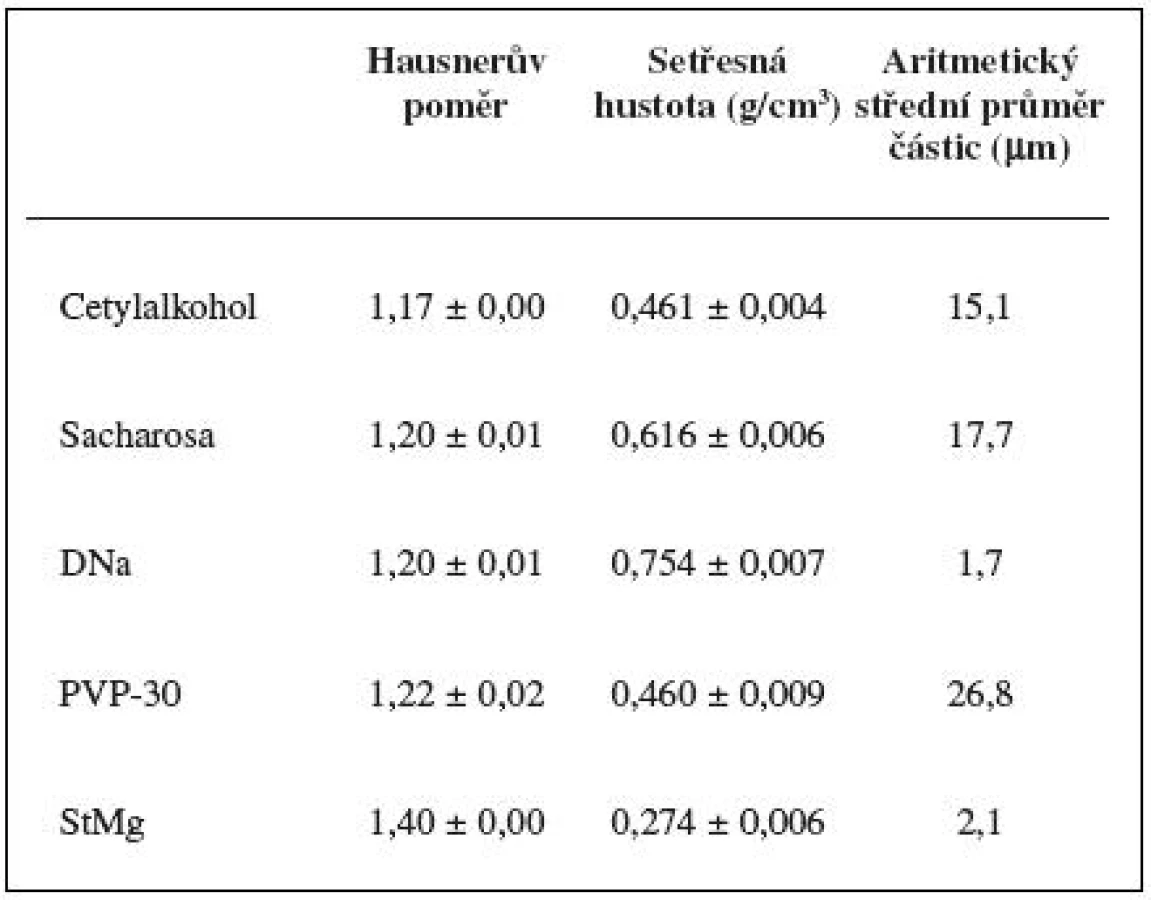
Jako modelové léčivo pro studium homogenizace se použil diklofenak sodná sůl – DNa (Amoli Organic, Indie), frakce < 250 μm. Jako pomocné látky se použily cetylalkohol (Cognis, Německo), frakce < 500 μm, povidon-30 – PVP-30 (BASF, Německo), frakce < 250 μm, sacharosa (Dobrovice, ČR), frakce < 80 μm, stearan hořečnatý – StMg (Peter Greven, Německo), frakce < 250 μm.
Metodika
Příprava vzorků
Pro studium procesu mísení byly zvoleny dvě směsi (tab. 1), které se lišily v časech, ve kterých se přidalo do směsi 4,5 % (m/m) stearanu hořečnatého 5). U vzorku SV2S100 se StMg přidal na začátku mísení a u vzorku V2S100 po 20 minutách mísení ostatních složek směsi. Jednotlivé složky směsí se opatrně vsypaly do homogenizační nádoby homogenizéru Turbula (WAB AG Machinefabrik, Švýcarsko) tak, aby se smísily co nejméně, a poté se započalo s homogenizací rychlostí 44 otáček za minutu.

Měření NIR spekter a fyzikálních vlastností prášků
Měření NIR spekter se provádělo sondou NIR spektroskopu Nicolet Antaris (Thermo Electron Corp., USA), která se pokládala k svrchní vrstvě vzorku v pěti místech homogenizační nádoby (ve středu a na okrajích v rozestupu 90 stupňů), tak aby se nezanořovala do hlouby vzorku a nedocházelo tím k rozvrstvení prášků ve směsi (obr. 1). Spektra se měřila metodou difuzní reflektance v rozsahu vlnočtů 10000–4000 cm-1 s rozlišením 4 cm-1 v různých časových rozestupech, které byly zvoleny tak, aby dobře popsaly jak fázi prvotního konvektivního mísení, při němž dochází k střetu toků a rychlému promísení jednotlivých prášků, tak pozdější fázi difuzního mísení, kdy jsou již prášky smíseny a dochází především k náhodnému přeskupování jednotlivých částic prášků 6). Ze začátku se tedy měřilo v kratších časových rozestupech.
Obrazová analýza (mikroskop DN45, Lambda, ČR; CCD kamera Alphaphot, Nikon, Japonsko; počítačový program IA32, Leco corp., USA) se použila k zjištění průměrné velikosti a tvaru částic. Dále se provádělo měření sypného a setřesného objemu po 1250 sklepnutích (SVM 102, Erweka, Německo). Vypočítaly se hodnoty Hausnerova poměru, sypné a setřesné hustoty.
Matematické úpravy a vyhodnocování spekter
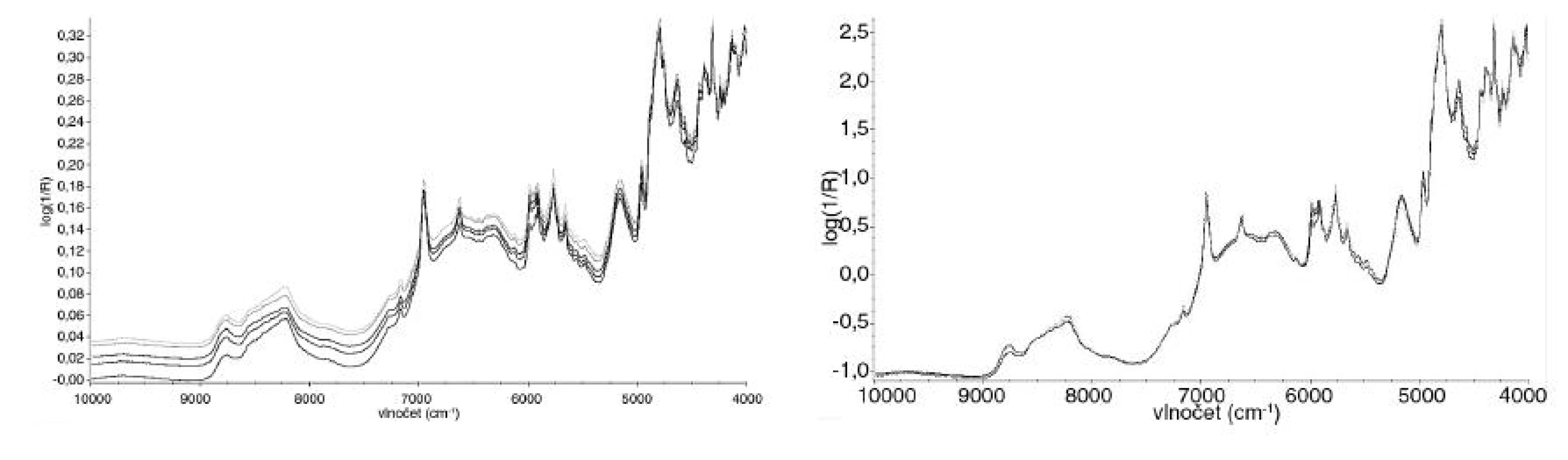
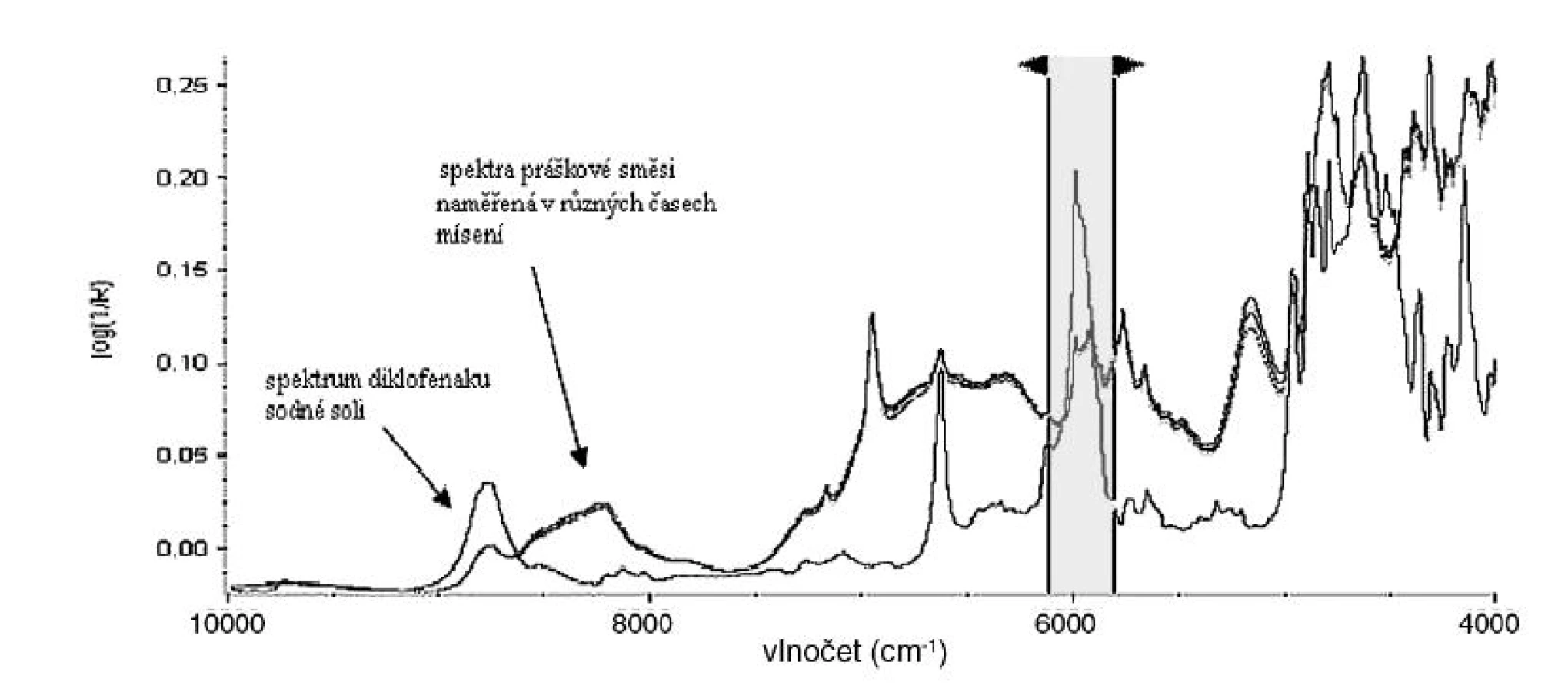
Naměřená spektra se nejprve matematicky upravovala v programu TQ Analyst 8. Prováděla se normalizace spekter pomocí SNV (standard normal variate) algoritmu (obr. 2), který umožňuje upravit spektra pocházející ze vzorků s různým posunem či náklonem základní linie způsobeným různou optickou dráhou ve vzorku, která vzniká zejména díky rozdílným velikostem částic, nebo rozdílnou tloušťkou vrstvy měřeného vzorku 7).
Ze spekter se v závislosti na použité metodě dále vypočítávala spektra varianční, která vyjadřují rozdílnost mezi jednotlivými spektry. Se zmenšujícími se rozdíly ve spektrech se zmenšuje i plocha pod variančním spektrem (obr. 3).
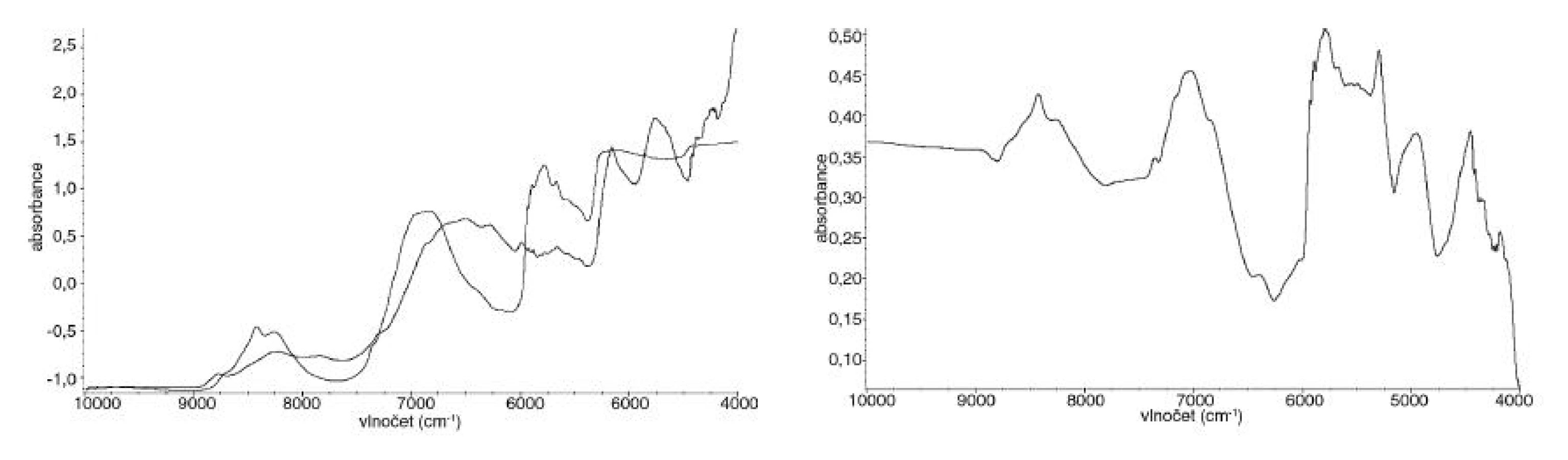
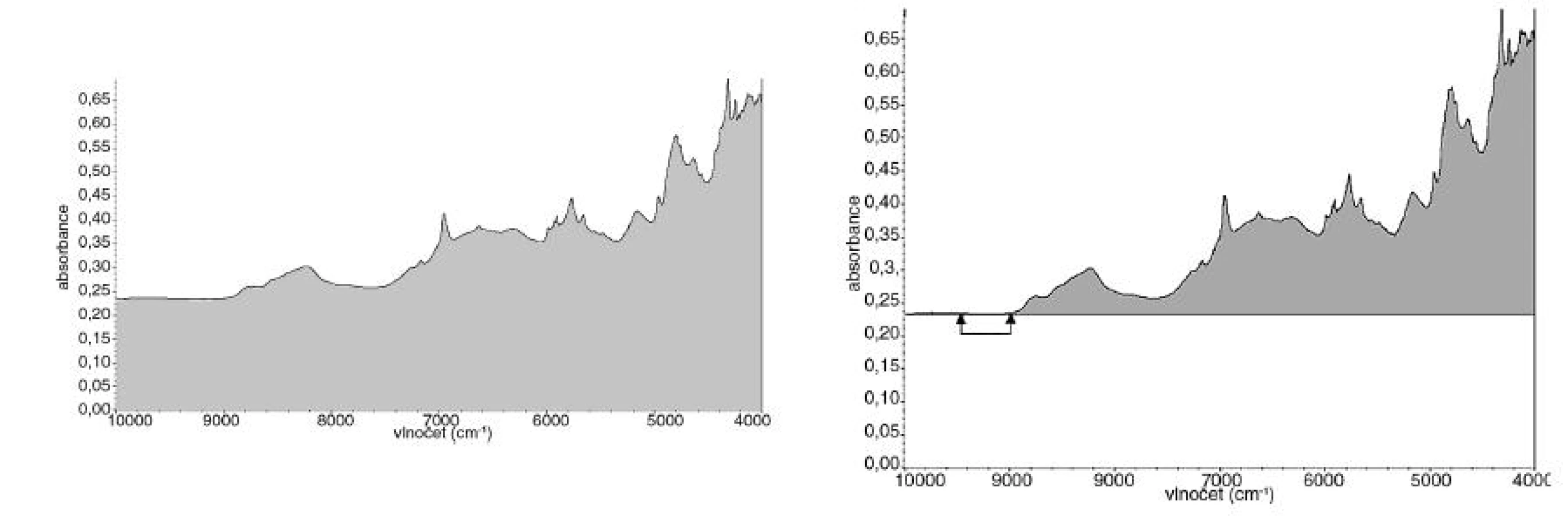
Další úprava spočívala v odečtu korigované a nekorigované plochy pod spektrem. Nekorigovaná plocha pod spektrem je plocha ohraničená NIR spektrem (případně variančním spektrem) a osou x. Korigovaná plocha pod spektrem je plocha ohraničená NIR spektrem (případně variančním spektrem) a rovnoběžkou s osou x protínající NIR spektrum v jeho minimu (obr. 4).
Metody hodnocení homogenity směsí
Program TQ Analyst 8 se použil k přípravě chemometrických metod. Metody SP1–4 byly založeny na předpokladu, že spektrum práškové směsi je kombinací spekter čistých prášků. Ze spektra práškové směsi lze tedy zjistit informace o zastoupení jednotlivých prášků, jak se v průběhu mísení měnilo. Metody porovnávají absorpční pásy ve zvolených rozsazích vlnočtů mezi normalizovaným spektrem čistého prášku a normalizovaným spektrem práškové směsi (obr. 5). Všechna naměřená spektra (pět v každém čase měření) se porovnala se spektrem čistého prášku. Takto se postupovalo u každé složky (prášku) zvlášť. Z každého spektra se získala hodnota podobnosti nabývající hodnot v rozmezí 0 až 1 (žádná podobnost až shoda). Tyto hodnoty se vynesly do grafů. Pokud by se stal prášek ideálně homogenním, srovnávací hodnoty by se ustálily na stejné hodnotě. Při reálných měřeních však tato hodnota kolísala v závislosti na aktuální koncentraci čisté složky (prášku) ve směsi. Metoda SP1 porovnávala spektra směsí se spektrem diklofenaku sodné soli v rozsahu vlnočtů 8565–9018 cm-1, 6532–6715 cm-1, 6108 až 5819 cm-1, 4589–4694 cm-1. Metoda SP2 porovnávala spektra směsí se spektrem cetylalkoholu v rozsahu vlnočtů 8948–7964 cm-1, 7216–7130 cm-1. Metoda SP3 porovnávala spektra směsí se spektrem povidonu-30 v rozsahu vlnočtů 5367–4991 cm-1. Metoda SP4 porovnávala spektra směsí se spektrem sacharosy v rozsahu vlnočtů 7034–6851 cm-1, 6465-6118 cm-1, 4654 až 4924 cm-1. Dále se použila metoda SPC, fungující na stejném principu jako metody SP1–4 s tím rozdílem, že normalizovaná spektra práškových směsí jsou porovnávána s normalizovaným průměrným spektrem směsi v celém rozsahu vlnočtů (10000–4000 cm-1). Hodnota podobnosti zde nabývá hodnot blízkých 1 (spektra práškových směsí jsou velmi podobná spektru průměrnému). Metoda je citlivá k odhalování všech odlehlých spekter.
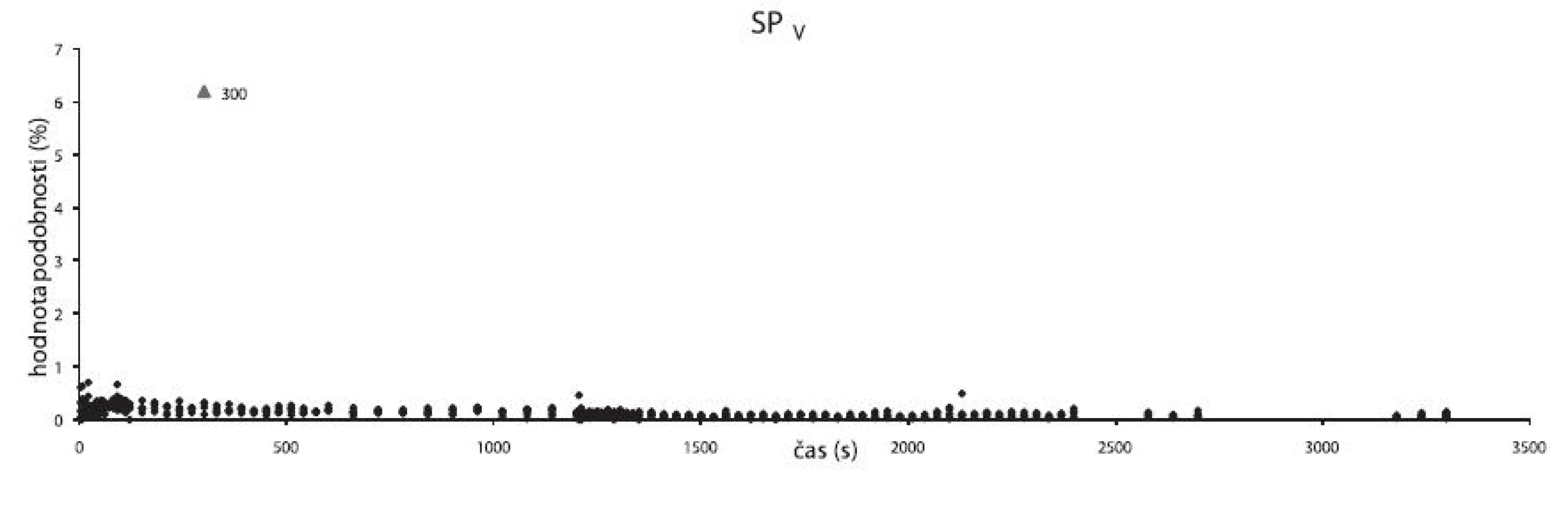
K odhalování odlehlých spekter zapříčiněných špatným přitlačením sondy ke vzorku sloužila metoda SPv fungující na stejném principu jako metody SP1–4 s tím rozdílem, že normalizovaná spektra práškových směsí jsou porovnávána s normalizovaným spektrem vzduchu v celém rozsahu vlnočtů (10000–4000 cm-1). Hodnota podobnosti nabývá hodnot blízkých 0 (spektra práškových směsí jsou nepodobná spektru vzduchu). Kladných hodnot budou nabývat pouze spektra vzniklá ze snímání, kdy mezi vzorkem a sondou byla vzduchová mezera.
Druhým použitým postupem pro hodnocení homogenity směsí jsou metody A1N a A1K, které k výpočtu využívaly pět spekter naměřených v každém měřeném čase homogenizace. Pro každý odběr vzorku v daném čase se tedy získalo pět normalizovaných spekter a z každé této pětice se vypočítalo spektrum varianční. Pro takto získaná varianční spektra se odečetly korigované (metoda A1K) a nekorigované plochy (metoda A1N). Hodnota plochy pod variančním spektrem je přímo úměrná podobnosti jednotlivých spekter. Pro pět stejných spekter je tedy hodnota plochy pod variančním spektrem 0. Odečet ploch se prováděl v celém měřeném rozsahu vlnočtů.
Data získaná ze všech metod se exportovala do programu Microsoft Excel 2007, v němž se provedlo grafické zobrazení a statistické hodnocení. Statistické hodnocení se zaměřovalo především na odhalování odlehlých hodnot a posuzování procesu mísení. Pro zjištění odlehlých hodnot se použil Grubbsův test. Byl aplikován na časy mísení přesahující 60 sekund, jelikož do 60 s probíhá intenzivní konvektivní mísení a dochází k prudké změně hodnot podobnosti. Hodnoty, které neprošly Grubbsovým testem, se vyřadily, poté byl test proveden znovu. Vyřazené hodnoty reprezentovaly odlehlá spektra, která se dále zkoumala v programu TQ Analyst 8 s cílem objasnit příčiny jejich odlehlosti. Pro orientační hodnocení variability v rámci všech časů měření a mezi jednotlivými časy měření se použila jednofaktorová analýza variance (ANOVA).
VÝSLEDKY A DISKUZE
Mísení DNa, cetylalkoholu, PVP-30, sacharosy metodami SP1–4 ve vzorku V2S100 zobrazuje obrázek 6. Je patrné, že v průběhu 1. minuty mísení se hodnoty podobnosti prudce mění, což odpovídá konvektivní fázi mísení. Ta přechází začátkem 2. minuty v difuzní fázi, kdy se již hodnoty podobnosti významně nemění a pohybují se kolem průměru. Ve 20. minutě dochází k prudké změně hodnot podobnosti, z důvodu přidání 4,5 % (m/m) stearanu hořečnatého.
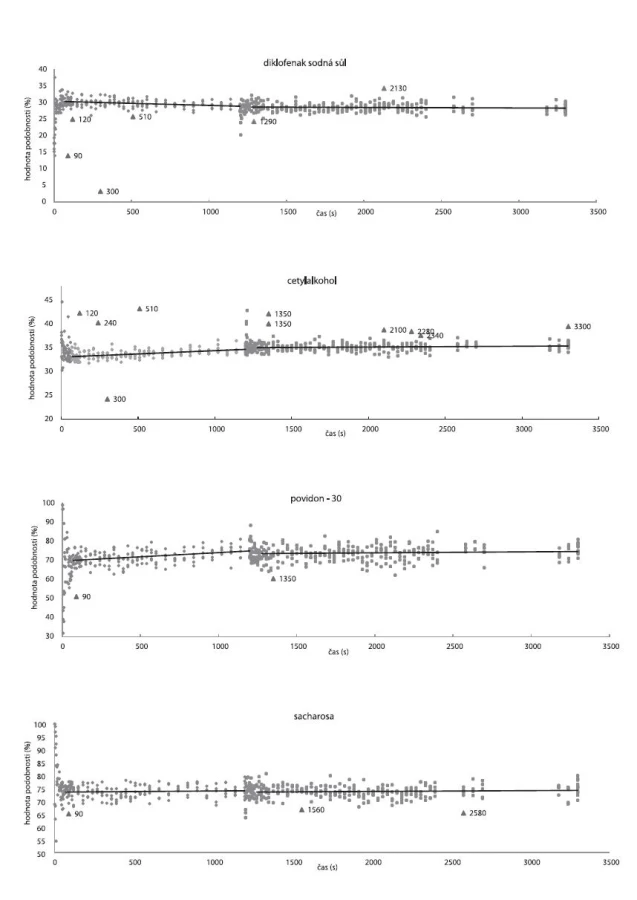
Hodnoty podobnosti (před a po přidání stearanu hořečnatého) se proložily přímkou lineární regrese, pro lepší znázornění rozptylu jednotlivých hodnot od průměru.
Z hodnot podobnosti pro cetylalkohol (obr. 6), jsou patrné kladně odchýlené hodnoty reprezentující spektra, jejichž hodnota podobnosti s cetylalkoholem je až o 17 % vyšší než průměr. Z toho lze usuzovat, že v průběhu mísení mohlo docházet ke shlukování částic cetylalkoholu.
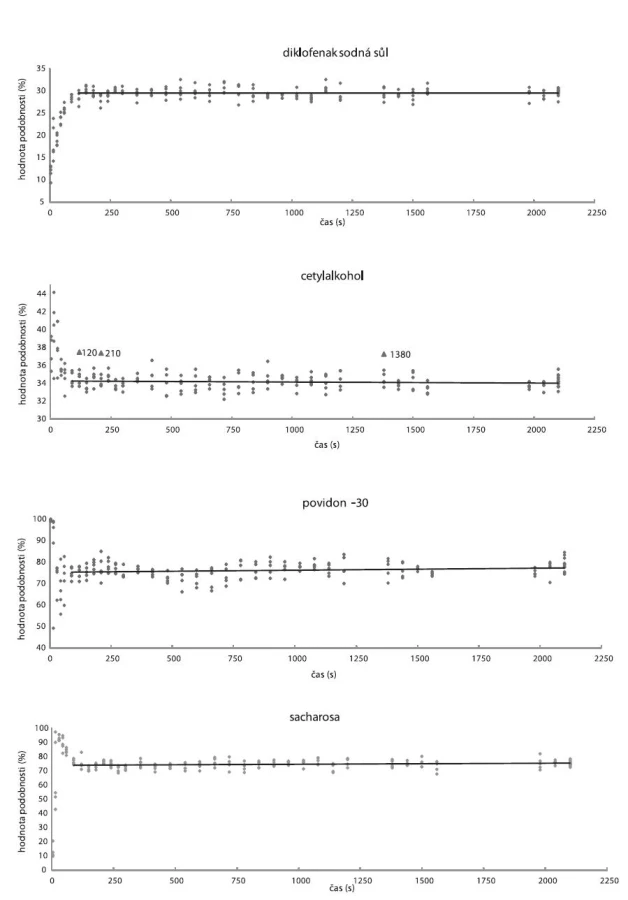
Průběh mísení vzorku SV2S100 (StMg přidán v čase 0 s) hodnocený metodami SP1–4 ukazuje obrázek 7. Stejně jako u vzorku V2S100 prudká změna hodnot podobnosti v průběhu 1. minuty popisuje mísení konvektivní, přecházející začátkem 2. minuty v mísení difuzní. Z hodnot podobnosti pro cetylalkohol (obr. 7) jsou patrné kladně odchýlené hodnoty reprezentující spektra, jejichž hodnota podobnosti s cetylalkoholem je až o 9 % vyšší než průměr. Z toho lze usuzovat, že docházelo ke shlukování částic cetylalkoholu, ale v menší míře než u vzorku V2S100, což mohlo být způsobeno přídavkem StMg již na začátku mísení.
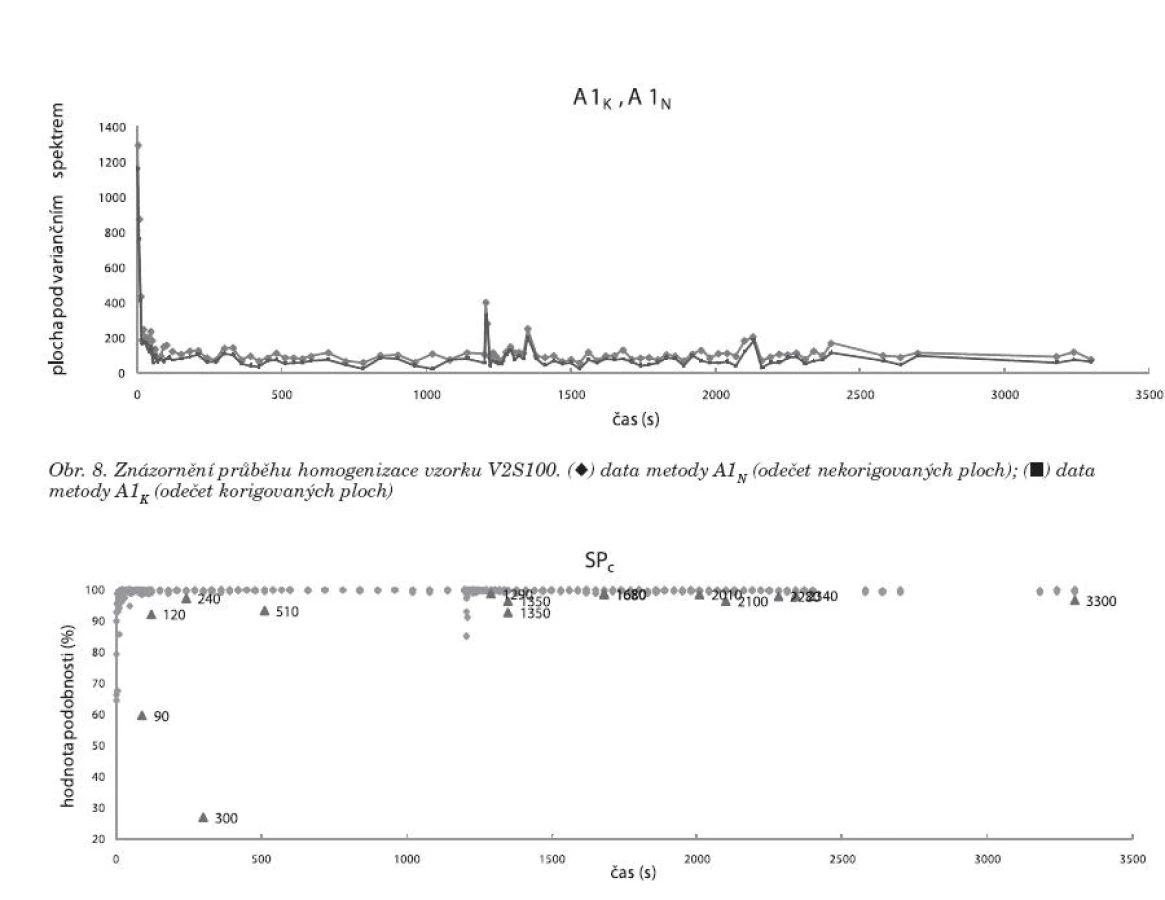
Metody A1N, A1K jsou založeny na principu, že pět spekter naměřených v jednom časovém bodě dává určité varianční spektrum. Z této hodnoty se odečetla plocha pod spektrem, a to buď korigovaná (obr. 8 – A1K), nebo nekorigovaná (obr. 8 – A1N), v každém čase měření. Tato metoda znázorňuje dostatečně variabilitu mezi spektry v jednotlivých časech měření. Mísení je ovšem proces, při kterém může docházet v průběhu času k segregaci částic, kterou způsobuje především odlišná velikost a hustota částic 8). Přestože je proces segregace velmi složitý, lze ho částečně objasnit efekty vyplývajícími z fyzikálních zákonů mechaniky. Jako příklad lze uvést efekt, na kterém se podílí trajektorie letu částice, valivý efekt, nebo efekt pronikání částic mezi sebe. V literatuře je popsáno, že vývojová tendence segregace je poměrně dlouhodobá (až desítky minut) 9). V našem případě mohl tento jev nastat u DNa, jehož průměrná velikost částic byla v porovnání s cetylalkoholem devětkrát menší a v porovnání s PVP-30 šestnáctkrát menší a navíc jevil DNa největší hodnotu setřesné hustoty (tab. 2). Pokud by tedy docházelo k segregaci DNa, tedy oddělování jeho částic od částic ostatních složek, byla by pravděpodobně rozeznatelná až porovnáním spekter v delším časovém rozestupu. Z toho vyplývá, že metody A1N, A1K nebudou příliš vhodné pro popis variability spekter mezi jednotlivými časy mísení.
Práce se dále zaměřovala na odhalování a interpretaci odlehlých spekter. Při homogenizaci se již v počátcích mísení (v prvních minutách) tvořily makroskopicky rozeznatelné shluky. Tyto shluky se modelově snímaly pomocí NIR sondy, protože se předpokládalo, že v průběhu času se budou rozbíjet na menší části neviditelné okem, které pak mohou být odhaleny už pouze pomocí NIR spektroskopie. Z dat získaných pomocí SP1–4, SPC (obr. 9) a SPV (obr. 10) metod se vyčlenila data odlehlá, reprezentující odlehlá spektra. Tato spektra se blíže zkoumala a porovnávala s modelovými spektry. Z těchto pozorování vyplývá, že složkou z větší části zodpovědnou za vznik shluků by mohl být cetylalkohol (viz obr. 6 – cetylalkohol; obr. 7 – cetylalkohol), protože jevil velké množství kladně odlehlých hodnot podobnosti, které byly až o 17 % vyšší než průměr. Z toho lze vyvodit, že takto odlehlá data prezentovala spektra, v nichž je vyšší obsah cetylalkoholu. Mohlo se tedy jednat o shluky s vyšším podílem cetylalkoholu oproti ostatním složkám. Dále se zjistilo, že některá odlehlá spektra mohla vzniknout z důvodu špatného přitlačení sondy ke vzorku, a proto vykazovala větší stupeň podobnosti se spektrem vzduchu (až 6 %). Lze tedy usuzovat, že při měření se nepodařilo sondu ideálně přitisknout ke vzorku a mezi vzorkem a sondou byla vzduchová mezera (obr. 10).
ZÁVĚR
V této práci se vyvinuly metody, které na základě dat z NIR spektroskopie umožnily vizualizovat proces mísení. Nejlépe proces mísení popisují metody SP1–4, které dovolují ze společného spektra směsi zjistit průběh mísení jednotlivých složek. Spojením těchto poznatků společně s daty o fyzikálních vlastnostech prášků se získalo cenné množství informací pro další úvahy o možných probíhajících dějích. Studiem odlehlých spekter se zjistilo, že prášky v průběhu mísení (především cetylalkohol) mohou tvořit shluky. Tomuto jevu lze částečně zabránit přídavkem stearanu hořečnatého. Některá odlehlá spektra mohou být způsobena i nedostatečným přitlačením sondy ke vzorku. Na výzkumu v dané oblasti bude třeba dále intenzivně pracovat a ověřit získaná data využitím metod kvantitativní NIR analýzy a kapalinové chromatografie. Práce ukazuje možnosti NIR spektroskopie při získávání informací o procesu mísení, které lze aplikovat při optimalizaci složení práškových směsí a technologických postupů.
Došlo 2. listopadu 2010 / Přijato 16. listopadu 2010
Adresa pro korespondenci:
Mgr. Petr Doležel
Ústav technologie léků FaF VFU
Palackého 1–3, 612 42 Brno
e-mail: petr.farmacie@seznam.cz
Sources
1. Brittain, H. G.: Spectroscopy of Pharmaceutical Solids. New York: Taylor & Francis Group 2006; 314–317.
2. Roggo, Y., Chalus, P., Maurer, L., Lema-Martinez, C., Edmond, A., Jent, N.: A review of near infrared spectroscopy and chemometrics in pharmaceutical technologies. J. Pharmaceut. Biomed. Anal., 2007; 44, 683–700.
3. Gauglitz, G., Vo-Dinh, T.: Handbook of Spectroscopy. Weinheim: Wiley-VCH, 2003; 105.
4. Berman, J., linski, D. E., onzales, C. R., ofer, J. D., imenez, P. J., lanchard, J. A., Tlachac, R. J.: Blend uniformity analysis: validation and in-process testing. Technical Report No. 25. PDA, PDA J. Pharm. Sci. Technol., 1997; 51, 1–99.
5. Dvořáčková, K., Rabišková, M., Masteiková, R., Okáčová, L.: Vliv koncentrace lipofilního nosiče na vlastnosti hydrofilně–lipofilních matricových systémů. Čes. slov. Farm., 2008; 57, 269–274.
6. Paul, E., Atiemo-Obeng, V., Kresta, S.: Handbook of Industrial Mixing. Hoboken: Wiley-interscience 2004; 887–982.
7. Siesler, H. W., Ozaki, Y., Kawata, S., Heise, H. M.: Near-Infrared Spectroscopy, Weinheim: Wiley-VCH 2002; 125–162.
8. Crowder, T. M., Hickey, A. J., Louey, M. D., Orr N.: A Guide to Pharmaceutical Particulate Science. New York: Interpharm/CRC, 2003; 75–79.
9. Masuda, H., Higashitani, K., Yoshida, H.: Powder Technology Handbook, 3. edition. New York: Taylor & Francis Group 2006; 371–382.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2010 Issue 6
Most read in this issue
- Orálně dispergovatelné lékové formy a technologie jejich výroby
- Studium procesu homogenizace práškových směsí s využitím NIR spektroskopie
- K dějinám lékárny Milosrdných bratří ve Skalici do roku 1919
- Používání humánních hromadně vyráběných léčivých přípravků ve veterinární praxi v České republice