
Fenotypizace enzymů podílejících se na metabolismu léčiv
Phenotyping of enzymes participating in drug metabolism
Pharmacogenetics is a rapidly developing field of science promising individualization of treatment through determination of genetic polymorphism in pharmacodynamics (receptors and other drug targets) and pharmacokinetics (carriers, metabolic enzymes). Enzyme activity may be predicted using genotyping or directly phenotyped – after administration of a probe substrate. This review article deals with some important metabolic enzyme polymorphisms and their phenotyping methods. Special consideration is given to the analytical methods described in the literature, which can be used to determine the metabolic rate.
Keywords:
pharmacogenetics, cytochrome P450, HPLC, drug biotransformation
Authors:
Svatopluk Světlík; Karolína Hronová; Ondřej Slanař
Authors‘ workplace:
Univerzita Karlova v Praze, 1. lékařská fakulta, Farmakologický ústav
Published in:
Čes. slov. Farm., 2012; 61, 115-126
Category:
Review Articles
Overview
Farmakogenetika je rychle se rozvíjející vědní obor slibující individualizaci léčby popsáním geneticky polymorfních aspektů farmakodynamiky (receptory a další cíle léčiv) i farmakokinetiky (přenašeče, biotransformační enzymy). Ke stanovení aktivity enzymů využívá jednak nepřímou metodu genotypizace, kdy je aktivita odhadována z genetické výbavy jedince, popř. přímo podáním substrátu daného enzymu a stanovením hladin parentní látky a příslušného metabolitu, jehož vznik enzym katalyzuje – fenotypizace. Tento přehledový článek se zabývá významnějšími enzymy podílejícími se na metabolismu léčiv, jejich polymorfismy a metodami jejich fenotypizace, přičemž zvláštní zřetel klade na analytické metody popsané v odborné literatuře, jichž je možno využít ke stanovení metabolického poměru parentní látka/metabolit.
Klíčová slova:
farmakogenetika, cytochrom P450, HPLC, biotransformace léčiv
Úvod
Farmakogenetika je poměrně novým výzkumným odvětvím především klinické farmakologie. Ačkoliv její kořeny sahají až do padesátých let 20. století k objevům deficitu butyrylcholinesterasy, senzitivity k primachinu a obecně známé pomalé acetylace isoniazidu, tj. nálezy podkládající vliv genetiky na lékovou odpověď, většího zájmu se farmakogenetice dostalo až s rozvojem molekulární biologie a projektu Human Genom Project1). Tento rozvoj umožnil nalezení vztahu polymorfismu genů s interindividuální variabilitou lékové odpovědí, resp. jeho hlubší pochopení.
Vliv genetické výbavy jedince na lékovou odpověď může probíhat na dvou úrovních: farmakokinetické a farmakodynamické. Samotná farmakokinetika pak může být ovlivněna jednak v oblasti biotransformace léčiv, tj. genový polymorfismus u metabolických enzymů typu oxidas se smíšenou funkcí (P450), thiopurinmetyltransferasy (TPMT), již zmíněné butyrylcholinesterasy a dalších, popř. v oblasti jejich transportu, kdy je ovlivněna exprese transportních proteinů typu p-glykoproteinu, přenašečů, iontových kanálů apod. Farmakodynamika je pak ovlivněna genetickým polymorfismem příslušných receptorů (opioidních, GABA atd.), nebo opět enzymů (topoizomerasa I, target cytostatika irinotekanu)2). Kombinací znalostí z obou těchto oblastí by bylo možné zvolit pro jednotlivce vhodné léčivo s potřebným dávkováním, tj. individualizovat léčbu.
Vymezení pojmů
V literatuře se můžeme setkat s definicí farmakogenetiky jakožto vědy sledující vliv jednoho genu na působení léčiv a farmakogenomiky jakožto aplikaci farmakogenetiky na celý genom, nicméně v naprosté většině odborných článků jsou tyto pojmy používány zaměnitelně, popř. je používána farmakogenetika pro oba případy. Této praxe se bude držet i tato rešerše.
Genetický polymorfismus je definován jako výskyt dvou či více variant (fenotypů, alel) v populaci v signifikantní frekvenci, které jej odlišuje od náhodné mutace. Aby se předešlo nejasnostem, je zvolena jako „signifikantní frekvence“ arbitrární mez 1 %. Pro představu, zrzavé vlasy mají frekvenci v populaci právě 1–2 %.
Jak vyplývá z předchozího textu, genetickému polymorfismu dávají vzniknout především bodové mutace – jedno nukleotidový polymorfismus (single nucleotide polymorphism – SNP). K tomuto může dojít v zásadě třemi mechanismy: substitucí, delecí či inzercí nukleotidu v řetězci DNA. Pokud se tak stane v rámci intronu, ačkoliv nemusí dojít k záměně AK ve vzniklém proteinu vzhledem k degeneraci genetického kódu (tichá mutace), ve většině případů dochází k ovlivnění konečného produktu daného genu. Například SNP309 v genu pro murine double minute 2 (MDM2) způsobuje posílení vazby transkripčního faktoru SP1 k MDM2 promotoru, a tedy větší expresi MDM2, což vede k silnější negativní regulaci tumor supresor proteinu p53, fenotypem pak je zřejmě větší náchylnost k nádorovým onemocněním3). Pokud k SNP dochází v exonu, nejčastěji je ovlivněna funkčnost výsledného proteinu, např. snížená katalytická aktivita enzymu v důsledku konformační změny okolí vazebné domény. Při deleci nebo inzerci nukleotidu dochází k posunu čtecího rámce a pokračující sekvence již nekóduje funkční produkt.
Polymorfismus biotransformačních enzymů vede ke čtyřem základním fenotypům, z nichž klinicky významně odlišné od běžné populace jsou především dva z nich: pomalý metabolizátor (PM) a ultrarychlý metabolizátor (UM). Pomalý metabolizátor je homozygot pro funkčně defektní alelu, zatímco ultrarychlý metabolizátor má funkční alely duplikované či multiplikované, nebo má expresi enzymu zvýšenou mutacemi/polymorfismy v regulačních oblastech genu. Třetím fenotypem je intermediární metabolizátor (IM) s aktivitou enzymu mezi PM a posledním fenotypem, homozygotem pro funkční nepozměněné (wild-type) alely, extensivním metabolizátorem (EM).
Metody farmakogenetiky
K zařazení daného jednotlivce do příslušné skupiny metabolizátorů a s tím související individualizaci dávkování se v klinické praxi využívají dvě metodiky: genotypizace a fenotypizace. Obecně lze říci, že fenotypizace odráží aktuální aktivitu enzymu, která se může měnit i vlivem jiných faktorů než pouze genetickým podkladem (věk, medikace, výživa a další), naproti tomu je nutno do těla zavádět exogenní látku, což dále může zatěžovat organismus (toto neplatí u krevních enzymů jako TPMT, butyrylcholinesterasa, jejichž fenotypizaci je možno provádět in vitro v odebrané krvi). Ne vždy je možné vůbec rozhodnout, zda je lepší v daném případě fenotypizace či genotypizace. Např. u azathioprinu pro fenotypizaci hovoří fakt, že i u homozygotů se aktivita TPMT může lišit až čtyřnásobně a v rasově různorodé populaci genotypizace nemusí odhalit nové popř. netestované mutace. Naproti tomu předešlá krevní transfuze může vést k špatnému určení fenotypu a další komplikací je fakt, že aktivita TPMT se zvyšuje, především u heterozygotů, s podáváním thiopurinových léčiv a tedy prvotně stanovená aktivita se může změnit po zahájení léčby1). Další nevýhodou fenotypizace je fakt, že není schopna odlišit UM od EM.
Genotypizace
Tato metodika je založena na stanovení genotypu a následném odhadu aktivity příslušného enzymu nepřímo predikcí fenotypu. Ke stanovení samotného genotypu se využívají metody molekulární biologie, po izolaci leukocytární DNA se provádí amplifikace příslušného úseku DNA pomocí PCR a vhodných primerů. Následuje analýza jednotlivých alel, ať už pomocí RFLP – restriction fragment lenght polymorphism založené na rozdílné délce restrikčních fragmentů, SSCP – single strand conformation polymorphism založené na rozdílné konformaci, sekundární struktuře řetězce DNA, jíž lze rozlišit i záměna jediné báze4), real-time PCR či jiných metod. Genotypizaci se věnuje řada článků, proto zde bude pojednáno především o fenotypizaci.
Fenotypizace
Princip této metodiky spočívá v podání vhodné exogenní (modelové) látky jedinci, u níž se následně stanoví farmakokinetické parametry. Vhodnou látkou se rozumí taková substance, která se metabolizuje pouze příslušným enzymem, jehož aktivitu si přejeme stanovit. Toho však lze v praxi dosáhnout jen stěží a je nutné se tedy spokojit alespoň s převažujícím metabolismem příslušným enzymem. Optimální modelová látka pak také není toxická a samozřejmě neinterferuje s další léčbou. Jak už bylo řečeno, takové stanovení odráží nejen genetickou výbavu jedince, ale i řadu dalších fyziologických, popř. patologických jevů, interakcí s jinými xenobiotiky (enzymové indukce, inhibice). Odraz těchto negenetických vlivů u fenotypizace je však většinou s výhodou, jelikož stejným vlivům pak bude podléhat i léčivá látka.
Cytochrom P450
Nadrodina enzymů cytochrom P450 (P450) zastřešuje oxidasy se smíšenou funkcí, které se dále dělí do rodin (40 % shoda v AK sekvenci) přiřazením arabského čísla ke zkratce CYP a do podrodin (55% shoda v AK sekvenci) přiřazením písmena. Jednotlivé enzymy se pak označují konečným přidáním dalšího arabského čísla, pro příklad: CYP2D6.
Je známo přes 11 500 jednotlivých enzymů, u lidí bylo popsáno 18 rodin a 43 podrodin P450 enzymů5). Pro metabolismus léčiv jsou významné první tři rodiny: CYP1, CYP2 a CYP3, ostatní se podílejí na metabolismu endogenních látek6). Až 80 % léčiv podstupuje oxidační reakce katalyzované P4507), z toho 50 % připadá na podrodinu CYP3A48), na CYP2D6 a CYP2C9 připadá 25, resp. 20 %9, 10).
CYP2D6
Tímto enzymem je metabolizována řada klinicky důležitých léčiv, ß-blokátory, antidepresiva, neuroleptika, antiarytmika a opiody, obecně vzato jsou to lipofilní báze1). Chemicky se jedná o polypeptid o 497 AK, tvořící 4 % z celkového množství enzymů P450 v játrech11).
Frekvence CYP2D6*3, CYP2D6*4, CYP2D6*5, CYP2D6*6, tedy hlavních variantních alel genu pro CYP2D6, v české populaci byla stanovena na 1,1 %, 22,9 %, 3,1 %, resp. 0,2 % a duplikace genu pro CYP2D6 byla nalezena u 3,1 % subjektů12).
K fenotypizaci je v klinické praxi používán spartein, debrisochin, tramadol13, 14), metoprolol15), dextrometorfan16), navíc desipramin, atemoxetin jsou doporučeny FDA jako substráty CYP2D6 k in vivo zkouškám17).
Dextrometorfan je opioidní agonista nejčastěji používaný proti kašli a bolesti. K fenotypizaci se ukázala jako nejlepší dávka 30 mg ve formě bromidové soli18), tedy dostatečně nízká dávka, která by u dospělého člověka neměla způsobit nežádoucí účinky. Ze všech modelových látek zmíněných výše je zřejmě nejpoužívanější díky svým mírným nežádoucím účinkům, má také nejvhodnější vlastnosti k fenotypizaci CYP2D6: je dostupný na trhu, má prokázanou in vitro specificitu používaného metabolického kroku, měřené koncentrace metabolitu korelují s aktivitou a obsahem CYP2D6 v mikrozomech lidských jater, projevují se rozdíly v metabolismu při onemocnění jater a při užívaní inhibitorů CYP2D619).
K fenotypizaci je možné použít stanovení metabolického poměru dextrometorfan/dextrorfan v moči, ta se sbírá nejčastěji během 8 hodin po podání dextrometorfanu20–22). Tato doba dobře koreluje se vzorkovacím intervalem 0–24 h (rs = 0,967; p < 0,0001) a má výhodu kratšího času, a tedy i menší pravděpodobnost chyby ve vzorkování23).
Dextrorfan se konjuguje s glukuronovou kyselinou, proto je nutné před samotné stanovení vložit hydrolytický krok (glukuronyltransferasa sama podléhá genetickému polymorfismu). Ten spočívá nejčastěji v enzymatické hydrolýze reakcí s ß-glukuronidasou24), nověji pak také v chemické hydrolýze reakcí s kyselinou chlorovodíkovou, jejíž výhodou je menší nákladnost, větší spolehlivost a rychlejší provedení (1 h oproti 18 až 24 h)25).
Další možností stanovení fenotypu je výpočet metabolického poměru dextrometorfan/dextrorfan z koncentrací v plazmě z jednoho vzorkovacího času: např. 1 h vzorek rozlišil mezi PM a EM, a to s menší variabilitou než metabolický poměr vypočtený z moči26). Malá korelace mezi hladinami v moči a v plazmě však také může poukazovat na možný bias vzniklý brzkým odběrem vzorku19). Dobrou korelaci vykazují vzorky odebrané v časech 2, 3, 4, 5 a 8 h po ingesci dextrometorfanu21, 27), přičemž například 3 h vzorek vykazuje korelaci k metabolickému poměru založenému na AUC 0–12 h 0,999 (p = 0,000; n = 6)28).
Zvlášť pro pacienty s poruchou ledvin byla zkoumána možnost stanovení fenotypu výpočtem metabolického poměru z koncentrací ve slinách. Nejmenší intraindividuální variabilitu vykázaly vzorkovací časy 2 h a 3 h po ingesci dextrometorfanu. Vzorkovací čas 3 h navíc dobře koreloval s močí odebíranou v intervalu 0–8 h (r = 0,704; p < 0,001; n = 62)29), z hlediska opakovatelnosti však vykazoval špatné výsledky, v tomto směru si vedl lépe vzorkovací čas 6 h21).
Metody stanovení shrnuje tabulka 1.
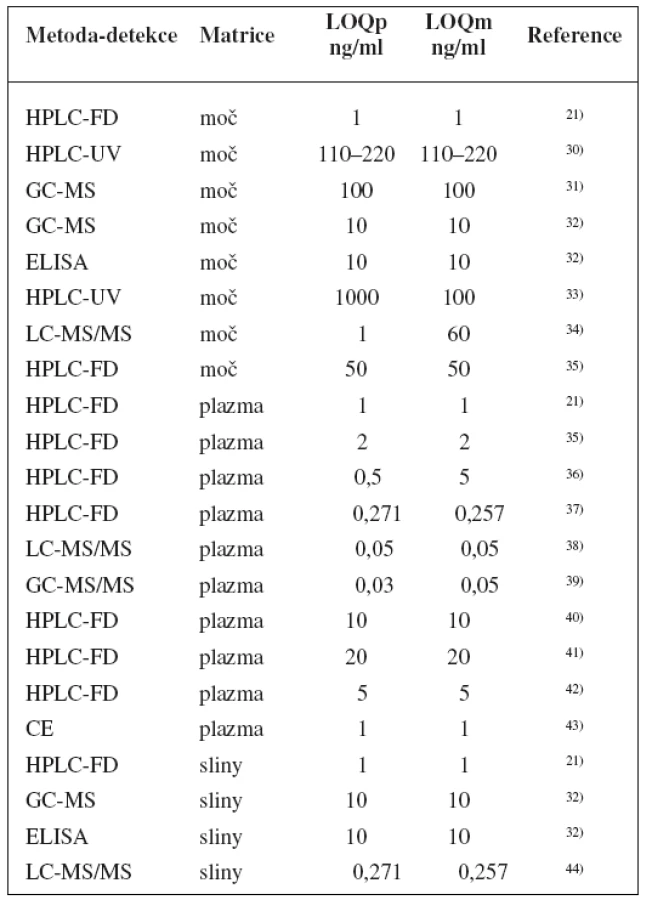
Nejnovějším zkoumaným způsobem k určení fenotypu je pak dechový test po podání [13C]-dextrometorfanu. V klinickém pokusu provedeném Leederem et al. bylo 30 dobrovolníkům podáno 0,5 mg/kg dextrometorfanu a měřeno obohacení 13CO2 infračervenou spektroskopií ve vydechovaném vzduchu během 4 h po podání, vztažené k obsahu 13CO2 ve vydechovaném vzduchu před podáním (DOB, delta over baseline). Výsledky byly srovnány s metabolickými poměry vypočtenými z koncentrací v moči. Při použití jednoho časového bodu 40 min po podání a definicí PM jako subjekty s DOB < 0,5 měla metoda 100% senzitivitu, 95% specificitu a 95% přesnost, přičemž jednoho IM nesprávně určila jako PM. Vzhledem k těmto výsledkům metoda slibuje rychlé, neinvazivní stanovení fenotypu CYP2D6, nicméně je nutný další vývoj45).
Ostatní látky k fenotypizaci CYP2D6 nevykazují jednu nebo více vlastností k tomuto účelu potřebných. Debrisochin již není k dostání na trhu a nemá prokázanou in vitro specificitu metabolické přeměny použité k fenotypizaci, spartein již také není na trhu a nejsou k dispozici data, zda se mění metabolismus u subjektů s onemocněním jater. Tramadol nemá prokázanou in vitro specificitu metabolické přeměny a tato přeměna dostatečně nepřevažuje nad jinými metabolickými kroky, jimž tramadol podléhá, tímto neduhem pak trpí i metoprolol, navíc k němu nejsou data prokazující odlišný metabolismus u lidí s onemocněním jater19).
CYP2C9
Tímto enzymem jsou metabolizována kumarinová antikoagulancia (warfarin, acenokumarol), deriváty sulfonylmočoviny (tolbutamid, glyburid, glimeprid), antagonisté AT II (losartan, irbesartan), nesteroidní protizánětlivá léčiva (diklofenak, ibuprofen, celekoxib), fenytoin a jiné.
Jedná se o polypeptid o 490 AK, tvoří asi 20 % všech P450 enzymů v játrech46) a zodpovídá za stejné poměrné množství reakcí katalyzovaných P45047).
Frekvence hlavních variantních alel, tj. CYP2C9*2 a CYP2C9*3, v české populaci byla stanovena na 12,2 %, resp. 5,9 %48).
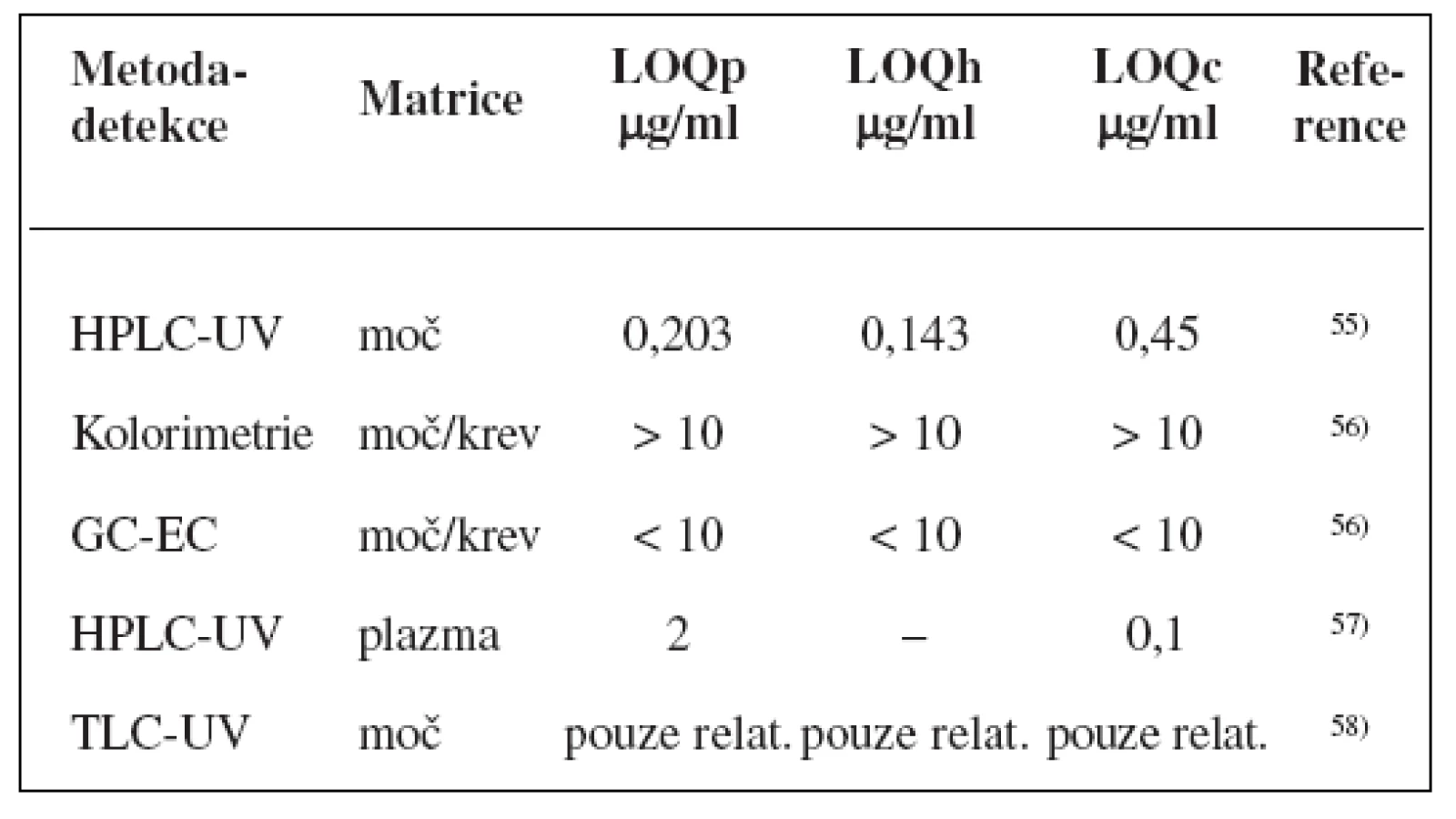
K fenotypizaci se nejčastěji používá tolbutamid49, 50), pro snížení rizika hypoglykémie je možné podat p. o. glukosu51, 52). Metabolický poměr vypočtený na základě koncentrací tolbutamidu a jeho dvou hlavních metabolitů hydroxytolbutamidu a karboxytolbutamidu v moči odebírané v intervalu 6–12 h po ingesci signifikantně predikoval clearence u zdravých subjektů53). Přestože parentní látka se vyskytuje v moči v poměru k metabolitům v nízké koncentraci, což snižuje její predikční schopnosti, zůstává tolbutamid doporučovanou látkou pro stanovení fenotypu CYP2C9, další navrhované látky (losartan, flurbiprofen) neprokázaly tak dobrý vztah mezi genotypem a jejich metabolismem (r2 = 0,42, resp. 0,53 vs. 0,64 pro tolbutamid)54).
Metody stanovení shrnuje tabulka 2.
CYP2C19
Tento enzym je důležitý v metabolismu inhibitorů protonové pumpy (omeprazol, lansoprazol), benzodiazepinů (diazepam, flunitrazepam), tricyklických antidepresiv (amitryptalin, imipramin), SSRI (fluoxetin, citalopram), barbiturátů (fenobarbital, hexobarbital), proguanilu, a dalších (chloramfenikol, indometacin).
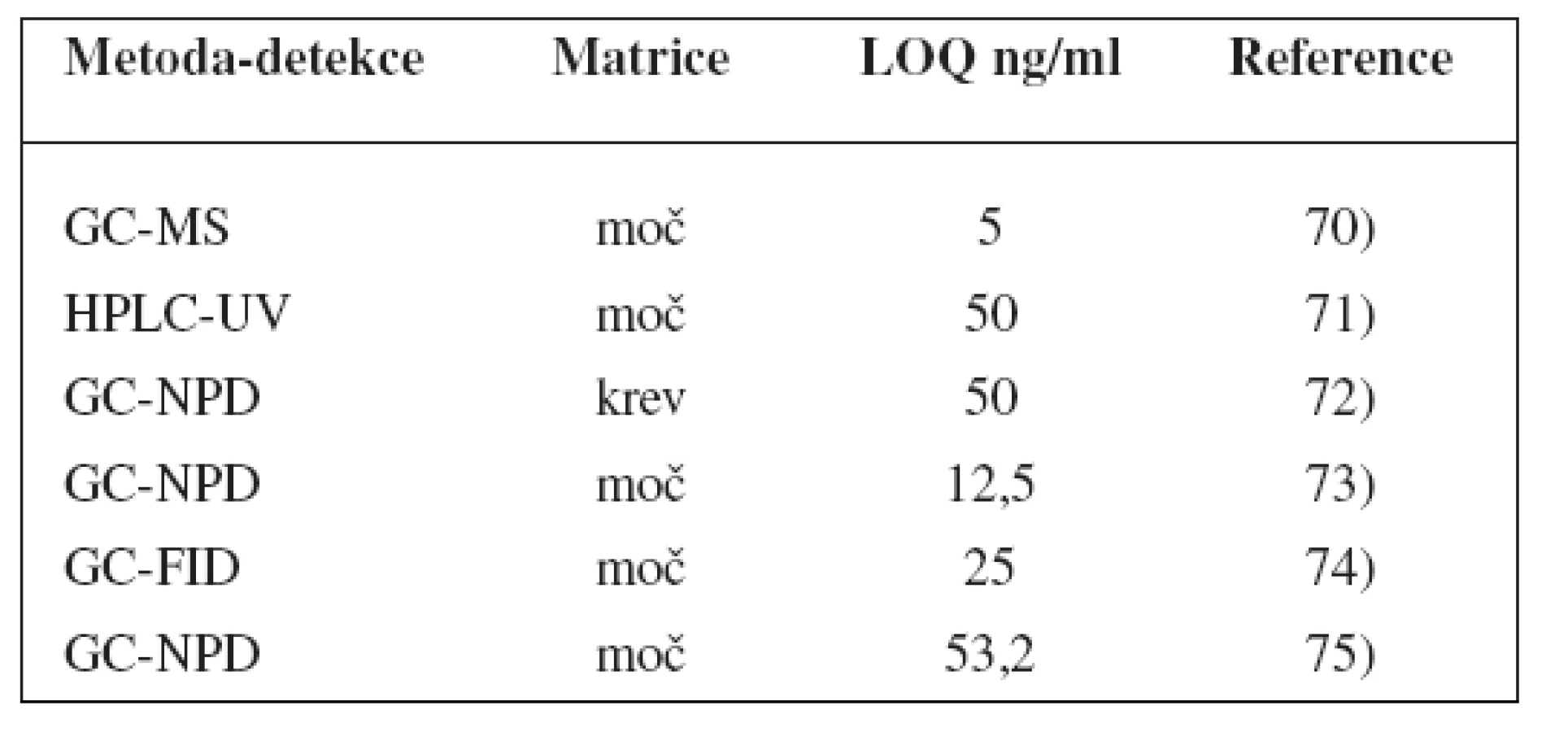
K fenotypizaci se velmi často používá stanovení poměru R-/S - mefenytoinu59–63). Kromě toho byly použity i další látky jako lansoprazol, omeprazol a proguanil28, 64–68). Lansoprazol však nedokázal zcela rozlišit mezi jednotlivými fenotypy64), proguanil vykázal shodu genotypu s fenotypem jen na základě probit analýzy poměrů proguanil/chlorofenylproguanid a proguanil//(cykloguanil + chlorofenylproguanid), ale ne proguanil/cykloguanil67), podobně omeprazol vykázal shodu genotypu s fenotypem jen u zdravých, mladých dobrovolníků, zatímco některé subjekty nad 65 let, popř. s poruchou jater označil jako PM, přestože se jednalo o genotyp EM69).
Metody stanovení shrnuje tabulka 3.
CYP2B6
Přestože je tímto enzymem metabolizováno 8–10 % používaných léčiv, mezi něž patří prasugrel, amitriptylin, halotan, ifosfamid, propofol a další, příspěvek CYP2B6 je většinou nízký (< 30 %) a dá se tedy předpokládat, že dopad genetického polymorfismu tohoto enzymu na metabolismus svých substrátů bude také nevýznamný76).
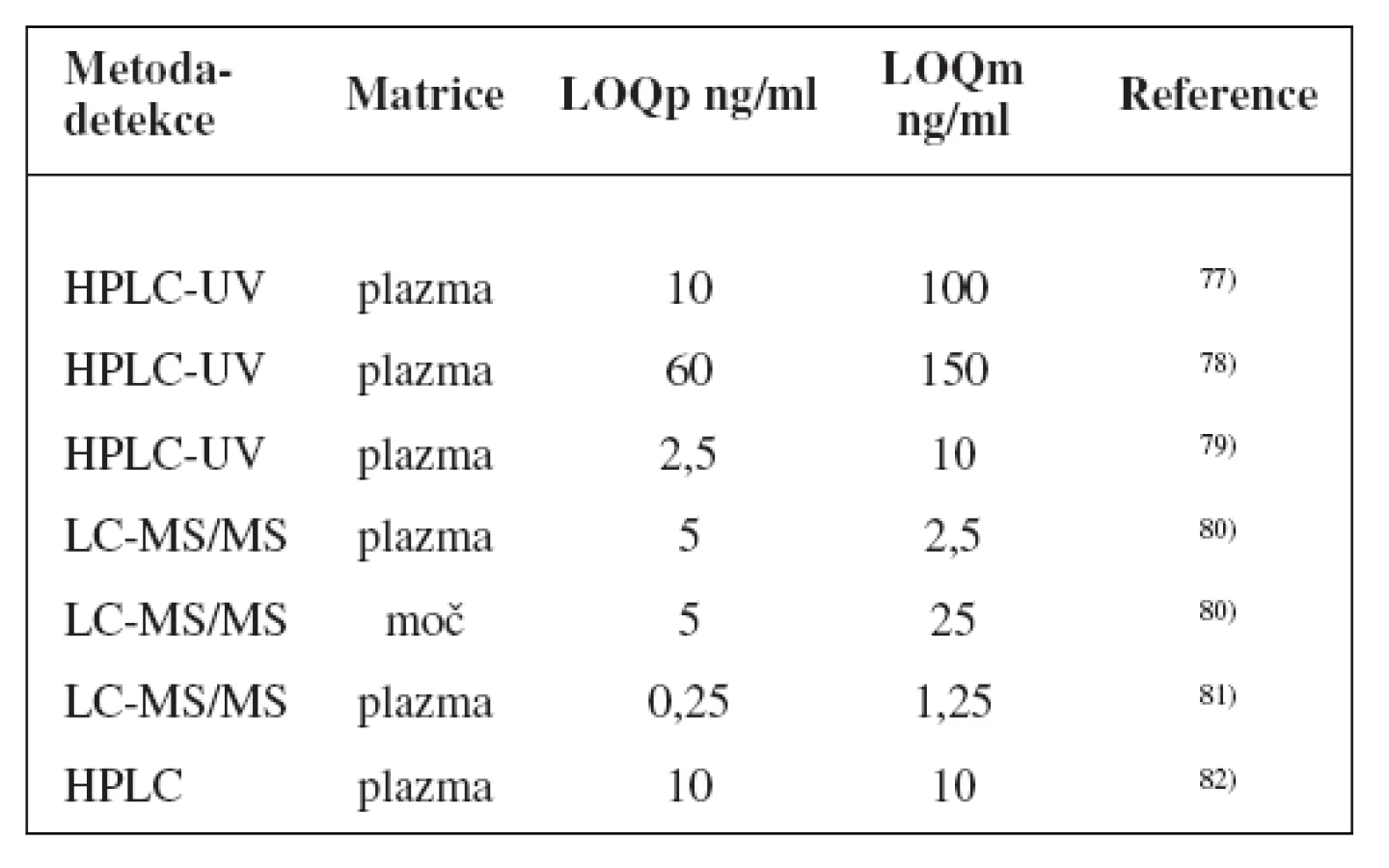
Přesto byla navržena řada látek k fenotypizaci CYP2B6 jako bupropion (hydroxylace), efavirenz (C8-hydroxylace), diazepam (N-demetylace), S-mefenytoin (N-demetylace za vzniku nirvanolu), nevirapin (C3-hydroxylace), testosteron (16ß-hydroxylace), efavirenz (doporučovaný také FDA k metabolickým studiím)17), nejčastěji se však používá první z nich76).
Metody stanovení shrnuje tabulka 4.
CYP2C8
Tímto enzymem je metabolizováno asi 5 % používaných léčiv, přičemž zaujímá asi 7 % celkového množství P450 v játrech83). K substrátům patří například R-ibuprofen (druhý stereoizomer je substrátem CYP2C9), repaglinid, ale především cytostatikum paklitaxel, který se zasloužil o obnovení zájmu o tento enzym1), u nějž se předpokládá možná predikce interindividuální variability na základě genotypu právě CYP2C8 spolu s genem pro P-gp84).
Výskyt variantních alel v české populaci byl stanoven na 0,3 % pro CYP2C8*2, 10,9 % pro CYP2C8*3 a 5,9 % pro CYP2C8*483).
K fenotypizaci se používá paklitaxel85), popř. jsou FDA doporučeny repaglinid a rosiglitazon jakožto substráty pro in vivo studie17). Nicméně žádné rozsáhlejší studie pro zhodnocení kterékoliv z nich provedeny nebyly.
Metody stanovení shrnuje tabulka 5.
CYP3A
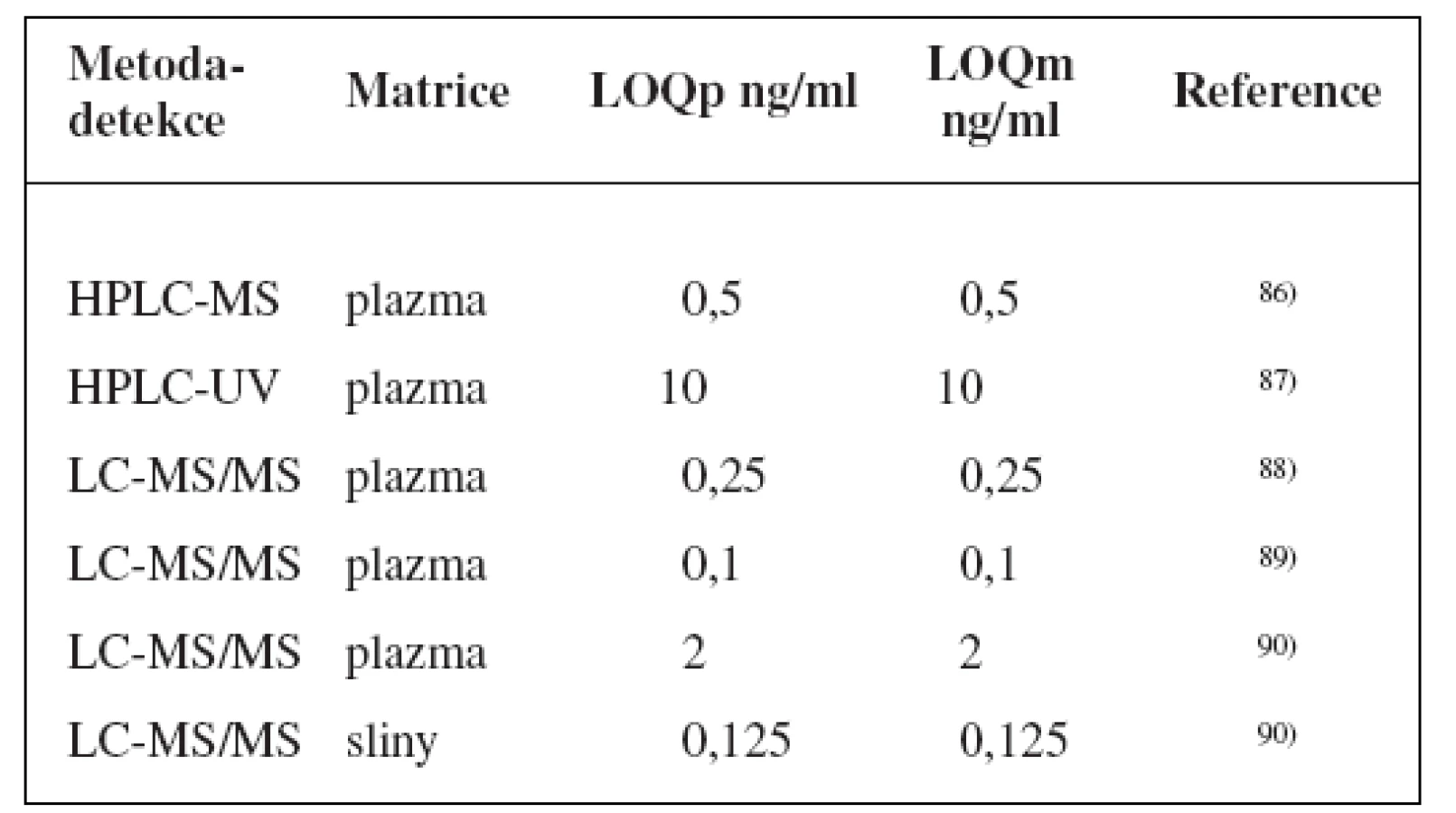
Tento lokus se skládá ze 4 genů kódujících 4 enzymy: CYP3A4, CYP3A5, CYP3A7 a CYP3A43, přičemž CYP3A7 se exprimuje jen ve fetálním období a CYP3A43 se vyskytuje jen ve velmi malém zastoupení a nízkou enzymatickou aktivitou. O zbylých dvou se pak často mluví prostě jako o „CYP3A“, protože mají širokou a navzájem jen velmi obtížně odlišitelnou substrátovou specificitu. Podílejí se na metabolismu 45–60 % používaných léčiv91), a vyskytují se v játrech v největším zastoupení ze všech enzymů P45092).
Díky široké substrátové specificitě je k dispozici také značné množství modelových látek: midazolam, felodipin, buspiron, lovastatin, eletriptan, sildenafil, simvastatin, triazolam, 14C-erytromycin, hydrokortizon, dexametason, antipyrin, chinin17, 50, 93–97). Nejvíce je však zřejmě používán midazolam1).
Metody stanovení shrnuje tabulka 6.
Další enzymy
Kromě P450 podléhají genetickému polymorfismu další enzymy, jejichž rozdílná aktivita může mít klinický dopad. Především se jedná o acetyltrasferasu, butyrylcholinesterasu, thiopurinmetyltransferasu, dihydropyrimidindehydrogenasu a uridindifosfátglukuronyltransferasu 1A11).
N-acetyltransferasa 2
Jak už bylo řečeno, tento enzym byl jedním z prvních, u kterého byl popsán genetický polymorfismus – již v padesátých letech 20. století brzy po zavedení isoniazidu, u nějž „pomalí acetylátoři“ jsou ve větším riziku pro vznik periferní neuropatie1).
Jak vyplývá z názvu enzymu, substrátem jsou obecně dusíkaté látky zahrnující hydraziny, aromatické aminy a amidy, hydrazidy, tj. kromě již zmíněného isoniazidu také prokainamid, aminoglutethimid, množství sulfonamidů, kyselina 5-aminosalicylová stejně jako její proléčivo sulfasalazin, dapson, hydralazin a další110).
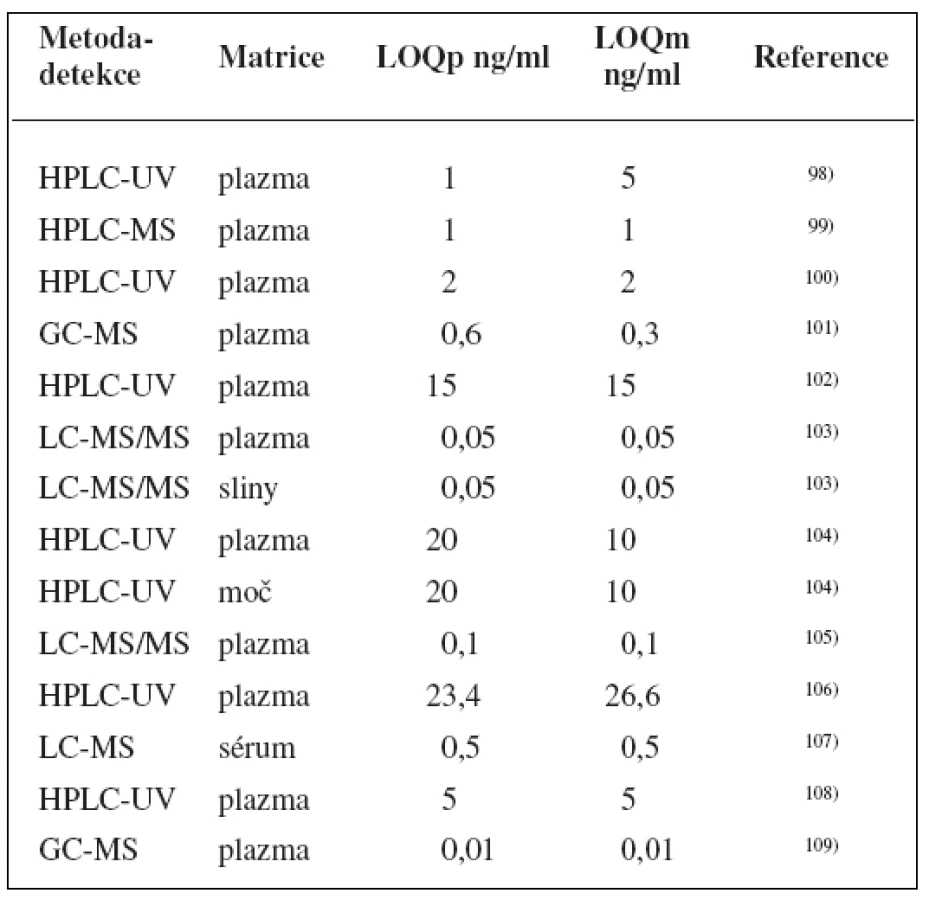
K fenotypizaci se zřejmě nejčastěji používá kofein111–115), ale je možné, že k acetylaci zde do určité míry přispívá NAT 1116). Kromě toho pak také isoniazid117), sulfamethazin118) či dapson119).
Metody stanovení shrnuje tabulka 7.
Butyrylcholinesterasa
Tento enzym se podílí na metabolismu jen několika léčiv, především myorelaxancií suxamethonia a mivokuria, kromě toho pak také kokainu130), prokainu (a příbuzných lokálních anestetik)131) a bambuterolu132). Endogenní funkce není jasná, ačkoliv byla zkoumána její role ve vývoji CNS133). Genetický polymorfismus, potažmo deficience butyrylcholinesterasy, má klinický význam hlavně u zmíněných myorelaxancií1).
Ke stanovení aktivity enzymu se využívají specifické inhibitory, s jejichž pomocí jsou získána pro příslušný fenotyp typická „inhibitorová čísla“. Ta vyjadřují poměrnou aktivitu náležící variantní formě enzymu, která je rezistentní k příslušnému inhibitoru, k aktivitě enzymu celkové. Jakožto inhibitor se již tradičně používá dibukain – „dibukainová čísla“134), fluorid sodný – „fluoridová čísla“135), ale ani jeden z těchto dvou inhibitorů není schopen určit všechny varianty. Tyto lze identifikovat pouze s pomocí třetího inhibitoru, karbamátu R0-02 0683, který je také používán nejčastěji a existuje pro něj nejvíce dat136). K dalším inhibitorům pak patří sukcinylcholin137), chlorid sodný138), propanolol139) a n-butanol140).
Samotná metoda je pak založena na stanovení úbytku absorpce záření o vlnové délce 240 nm při hydrolýze benzoylcholinu butyrylcholinesterasou zároveň v přítomnosti i nepřítomnosti inhibitoru136).
Další možností je využití metody založené na Ellmanově metodě původně určené ke stanovení aktivity acetylcholinesterasy adaptované ke měření aktivity butyrylcholinesterasy v práci Garryho a Routheho. Principem metody je štěpení acetylthiocholinu, sulfhydrylového analogu acetylcholinu, butyrylcholinesterasou, kdy uvolněná –SH skupina reaguje s 5,5-dithiobis-2-nitrobenzoovou kyselinou (DTNB, Ellmanova činidla), za vzniku žlutého 2-nitro-5-thiobenzoového aniontu (NTB-). Množství uvolněného NTB - je se stanovuje kolorimetricky a je úměrné množství –SH skupin z rozštěpeného acetylthiocholinu141). Tato metoda pak byla dále vylepšena použitím substrátů, které nejsou štěpeny acetylcholinesterasou jako butyrylthiocholin142), propanoylthiocholin143).
Thiopurinmethyltransferasa
Klinická důležitost genetického polymorfismu u tohoto enzymu je obecně uznávána a jeho aktivita rutinně stanovována před použitím léčiva, které je jeho substrátem. To se týká především azathioprinu a jeho prvního metabolitu 6-merkaptopurinu (6-MP), u nichž deficience toho enzymu předurčuje subjekt k léčivem indukované myelotoxicitě až s fatálním průběhem144).
Frekvence funkčně deficitních alel byla stanovena v české populaci na 0,1 % (TPMT*2), 4,3 % (TPMT*3A), 0,1 % (TPMT*3B) a 0,4 % (TPMT*3C)145).
Fenotypizaci tohoto enzymu, jak bylo zmíněno výše, je možno provést in vitro v odebrané krvi. Jako substrát pro TMPT je používán nejčastěji 6-MP metabolizovaný na 6-methylmerkaptopurin, popř. 6-thioguanin, který je pak metabolizován na 6-methylthioguanin. Jako donor methylové skupiny se používá S-adenosyl-L-metionin (SAM)146).
Dihydropyrimidindehydrogenasa (DPD)
Tento enzym je endogenně důležitý pro metabolismus pyrimidinů, tyminu a uracilu, z hlediska léčiv pak pro metabolismus 5-fluorouracilu a capecitabinu.
Fenotypizaci lze opět provádět ex vivo v odebrané krvi bez nutnosti vpravovat do těla xenobiotikum. K tomu se využívá inkubace s [14C]-fluorouracilem147, 148) popř. [14C]-tyminem149). Přesto existují i studie, kde byl fenotyp stanoven in vivo testem s [13C]-uracilem150). Další možností je opět stanovení vzestupu vydechovaného 13CO2 po dávce [13C]-uracilu151).
Uridindifosfátglukuronyltransferasa 1A1 (UGT1A1)
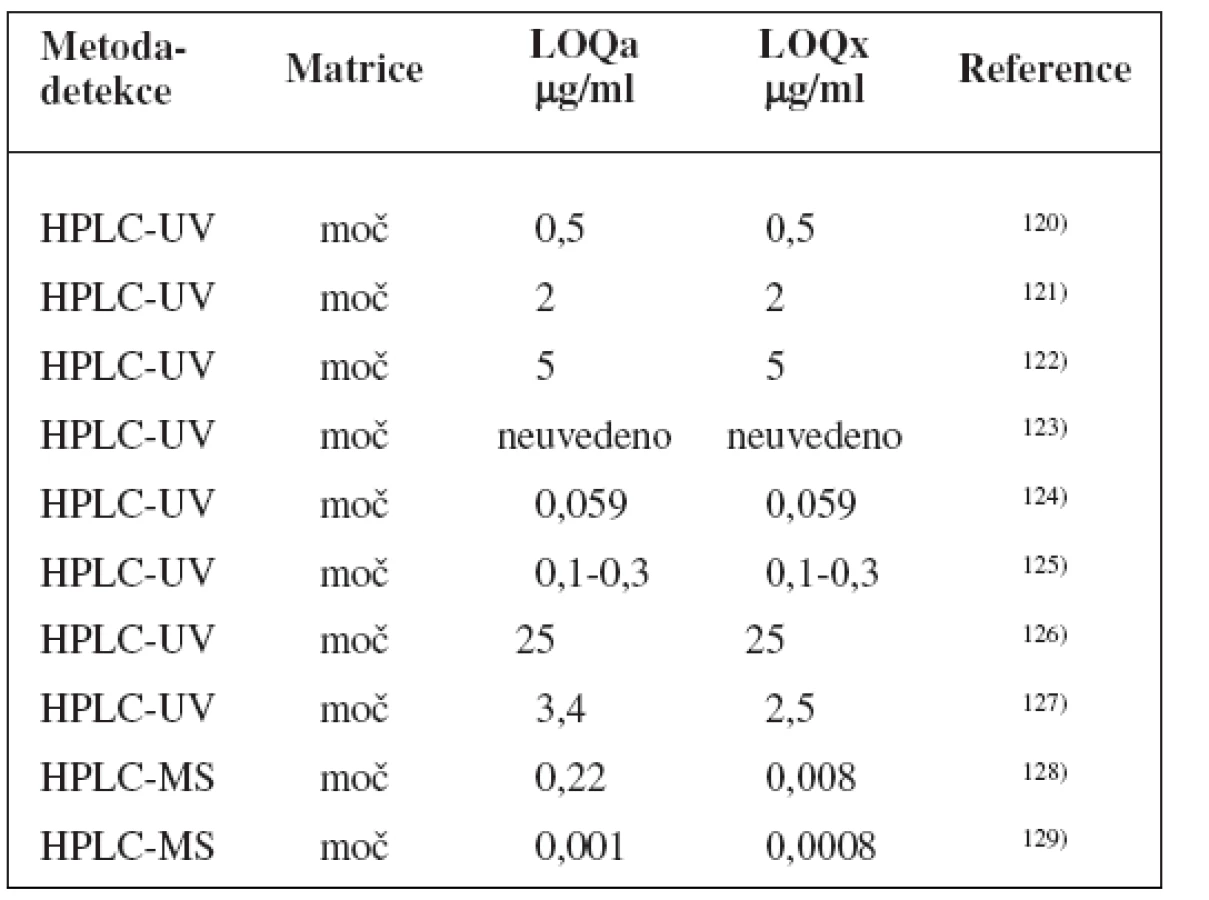
UGT1A1 vešla ve známost jakožto první enzym, pro nějž FDA vydala povolení farmakogenetického testování ve spojení s konkrétním lékem – irinotekanem, u kterého snížená aktivita UGT1A1 zvyšuje riziko diarei a myelosuprese1). Dalším substrátem je například etoposid92).
Ke stanovení fenotypu se používá paracetamol152), kromě toho byla popsána studie také s lamotriginem153).
Metody stanovení shrnuje tabulka 8.
„Koktejly“
V posledních letech se také začaly vyvíjet metody k determinaci aktivit více enzymů v jednom stanovení, kdy je subjektu podáno více léčiv najednou, tj. „koktejl“. Aby takový přístup byl možný, je samozřejmě nezbytné, aby jednotlivé modelové látky mezi sebou navzájem nijak neinteragovaly, tj. aby si navzájem neinhibovaly/neindukovaly metabolismus, nepotencovaly účinek apod.
„CIME koktejl“
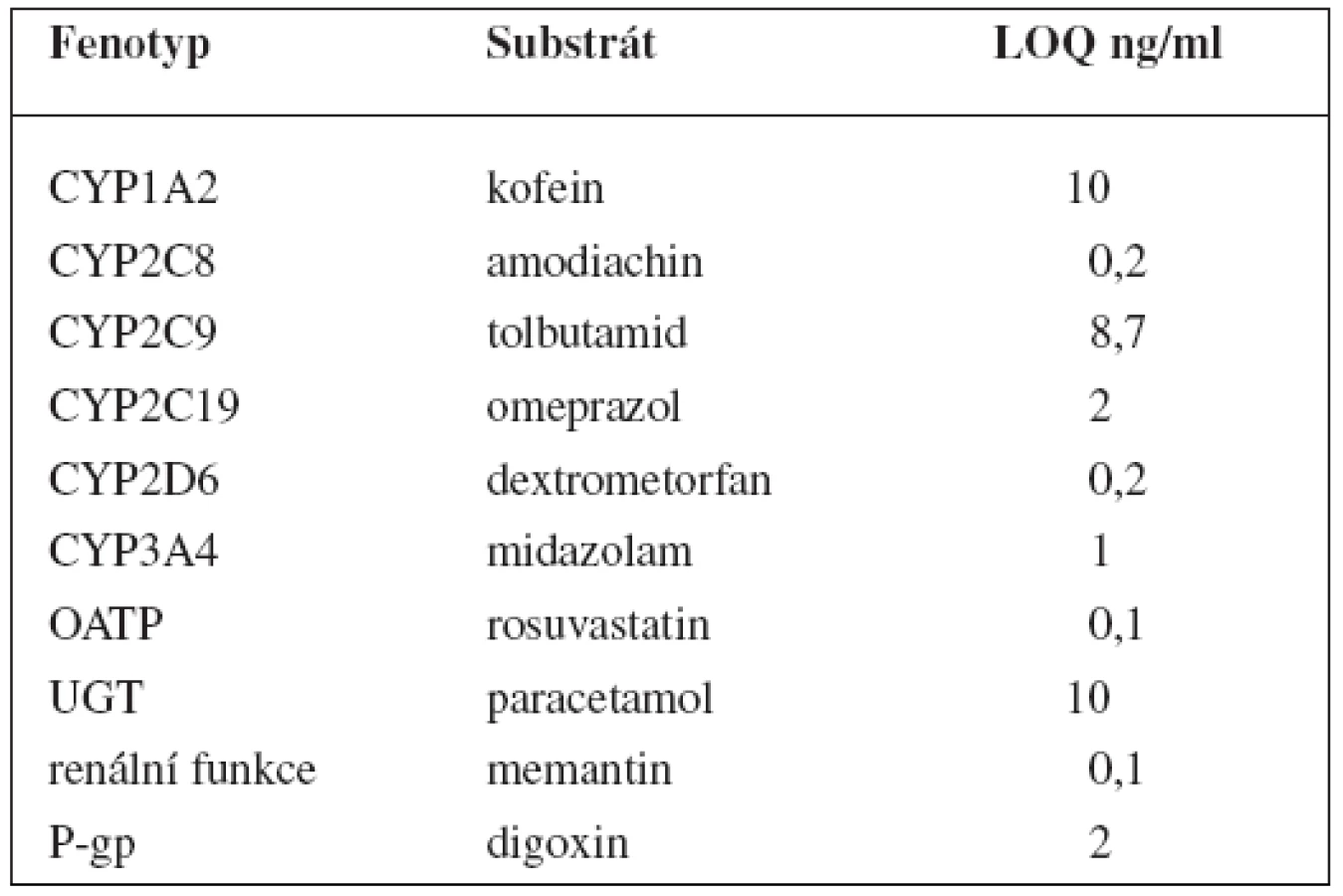
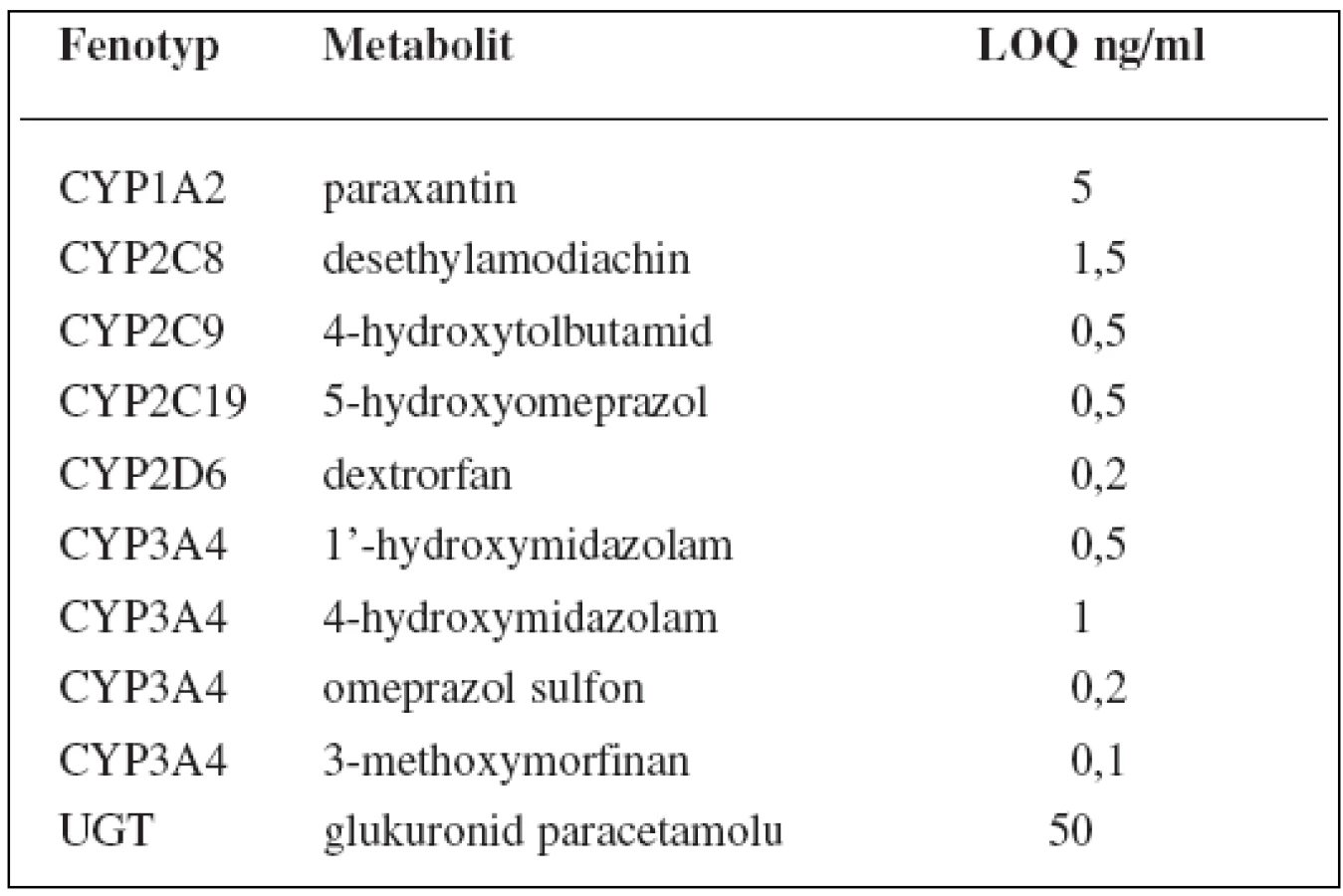
V tomto případě jsou v koktejlu obsaženy látky ke stanovení 6 enzymů P450: kofein pro CYP1A2, amodiachin pro CYP2C8, tolbutamid pro CYP2C9, omeprazol pro CYP2C19, dextrometorfan pro CYP2D6 a midazolam pro CYP3A, dále paracetamol pro UGT1A1. Kromě toho stanovuje tento koktejl fenotyp glykoproteinu P pomocí digoxinu, polypeptidu transportujícího organické anionty 1B1 (OATP1B1) rosuvastatinem a také renální funkci memantinem. Látky jsou pak stanovovány metodou UPLC-MS/MS, jejíž limity kvantifikace shrnují tabulky 9 a 10162).
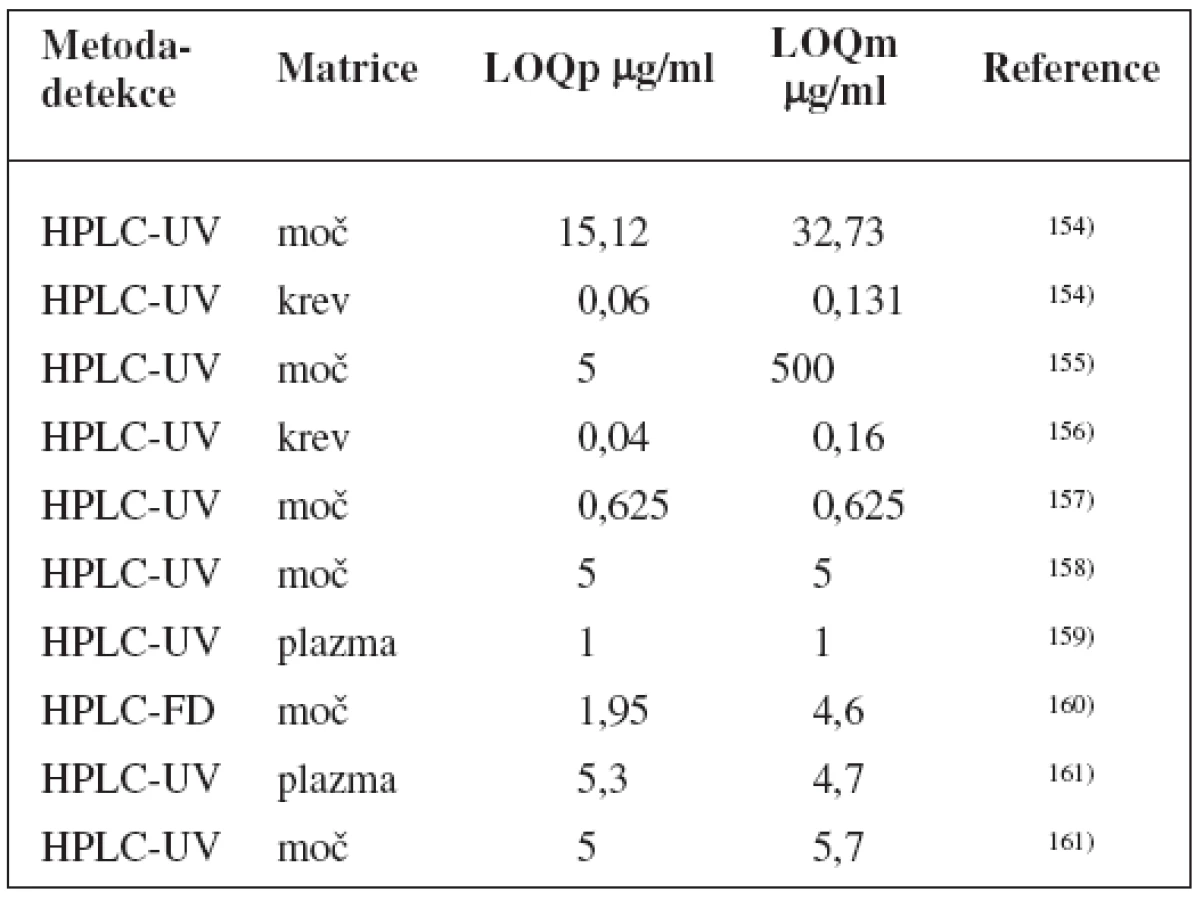
„Pittsburgh koktejl“
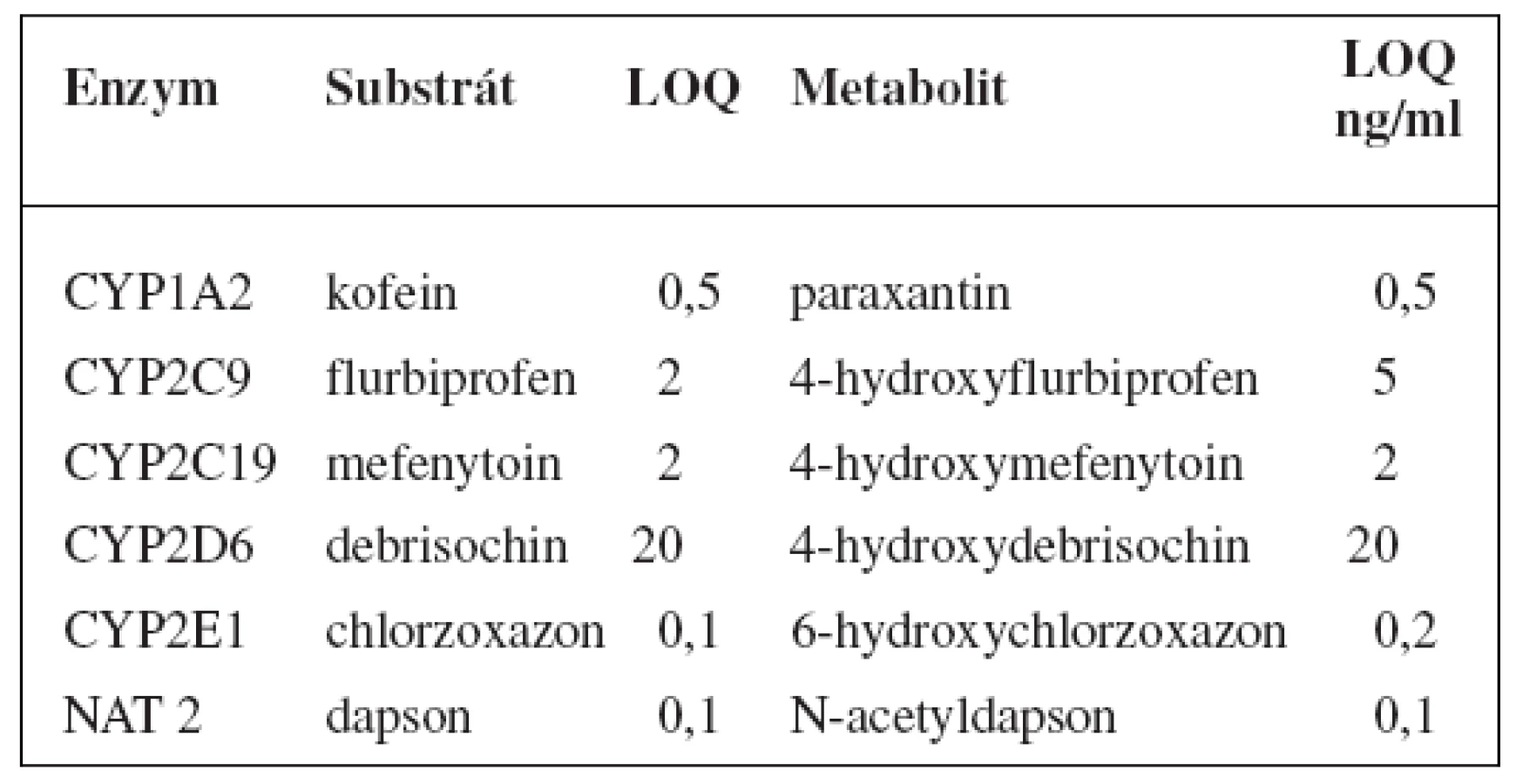
Tento koktejl stanovuje 5 enzymů P450: kofein pro CYP1A2, flurbiprofen pro CYP2C9, mefenytoin pro CYP2C19, debrisochin pro CYP2D6, chlorzoxazon pro CYP2E1 a navíc také NAT 2 stanovuje dapsonem. I zde je pak použito metody k UPLC-MS/MS ke stanovení substrátů i jejich metabolitů, LOQ shrnuje tabulka 11119).
„Koktejl Karolinska“
Ke stanovení fenotypu pouze enzymů P450, konkrétně CYP1A1 (kofein), CYP2C9 (losartan), CYP2C19 (omeprazol), CYP2D6 (debrisochin), CYP3A4 (chinin) byl navržen na Institutu Karolinska tento „koktejl“, tedy „Koktejl Karolinska“. Jak se ukázalo, metabolismus debrisochinu byl však inhibován některou z dalších látek163).
„Cooperstown koktejl“
Tento koktejl stanovuje fenotyp 6 enzymů pomocí 4 látek: kofein pro CYP1A2, xantinoxidasu a NAT 2, dextrometorfan pro CYP2D6, midazolam i.v. pro jaterní CYP3A a omeprazol pro CYP2C19164). Následně byl validován Cooperstown koktejl + 1, kdy byl do koktejlu přimíchán warfarin s vitaminem K pro stanovení CYP2C9165).
„Koktejl“ pro stanovení 5 enzymů P450
Tento koktejl stanovuje CYP1A2 pomocí kofeinu, CYP2E1 pomocí chlorzoxazonu, CYP2C19 pomocí mefenytoinu, CYP2D6 pomocí metoprololu a CYP3A pomocí midazolamu166).
Závěr
Fenotypizace je zavedená metoda v klinické praxi představující vhodnou alternativu ke genotypizaci. Obě metody mají své nevýhody a vhodnost té které z nich se liší případ od případu a mnohdy rozhoduje jen jejich dostupnost pro lékaře. Přesto zde rozdíly v použitelnosti existují: Genotypizace ze své podstaty může odhalit pouze deficience dědičného rázu a deficience způsobené například postižením jater, popř. inhibicí metabolických enzymů ko-medikací či enviromentálními vlivy jí uniká. Naproti tomu např. u CYP2D6 je schopna rozdělit populací do 4 skupin dle geneticky podmíněné aktivity enzymu, při fenotypizaci měla tato aktivita většinou jen bimodální (tj. EM a PM), výjimečně trimodální (tj. EM, IM, PM) rozdělení. Ultrarychlé metabolizátory rozlišit nedokáže, k tomu by patrně bylo potřeba použít jiných modelových látek, či jiných (kratších) vzorkovacích časů, které by pak pravděpodobně zase neodlišily PM.
Střet zájmů: žádný.
Došlo 16. dubna 2012 / Přijato 17. května 2012
Ing. Svatopluk Světlík, K. Hronová, O. Slanař
Univerzita Karlova v Praze, 1. lékařská fakulta, Farmakologický ústav
Albertov 4, 128 00 Praha 2
e-mail: svatopluk.svetlik@lf1.cuni.cz
Sources
1. Gardiner S. J., Begg E. J.: Pharmacogenetics, drug–metabolizing enzymes, and clinical practice. Pharmacol Rev. 2006; 58, 521–590.
2. Hoskins J. M., Marcuello E., Altes A., Marsh S., Maxwell T., van Booven D. J., Pare L., Culverhouse R., McLeod H. L., Baiget M.: Irinotecan pharmacogenetics: influence of pharmacodynamic genes. Clin Cancer Res. 2008; 14, 1788–1796.
3. Bond G. L., Hu W., Levine A.: A single nucleotide polymorphism in the MDM2 gene: from a molecular and cellular explanation to clinical effect. Cancer Res. 2005; 65, 5481–5484.
4. Hayashi K.: PCR–SSCP: a simple and sensitive method for detection of mutations in the genomic DNA. PCR Methods Appl. 1991; 1, 34–38.
5. Nelson D. R.: The cytochrome p450 homepage. Hum Genomics. 2009; 4, 59–65.
6. Hukkanen J.: Xenobiotic-metabolizing cytochrome P450 enzymes in human lung. Oulu: University of Oulu 2000; 69 s.
7. Eichelbaum M., Ingelman-Sundberg M., Evans W. E.: Pharmacogenomics and individualized drug therapy. Annu Rev Med. 2006; 57, 119–137.
8. Ingelman-Sundberg M., Sim S. C., Gomez A., Rodriguez-Antona C.: Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther. 2007; 116, 496–526.
9. Zanger U. M., Raimundo S., Eichelbaum M.: Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol. 2004; 369, 23–37.
10. Kirchheiner J., Brockmoller J.: Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin Pharmacol Ther. 2005; 77, 1–16.
11. Anzenbacher P., Anzenbacherova E.: Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci. 2001; 58, 737–747.
12. Buzkova H., Pechandova K., Slanar O., Perlik F.: Frequency of single nucleotide polymorphisms of CYP2D6 in the Czech population. Cell Biochem Funct. 2008; 26, 76–81.
13. Abdel-Rahman S. M., Leeder J. S., Wilson J. T., Gaedigk A., Gotschall R. R., Medve R., Liao S., Spielberg S. P., Kearns G. L.: Concordance between tramadol and dextromethorphan parent/metabolite ratios: the influence of CYP2D6 and non-CYP2D6 pathways on biotransformation. J Clin Pharmacol. 2002; 42, 24–29.
14. Subrahmanyam V., Renwick A. B., Walters D. G., Young P. J., Price R. J., Tonelli A. P., Lake B. G.: Identification of cytochrome P-450 isoforms responsible for cis–tramadol metabolism in human liver microsomes. Drug Metab Dispos. 2001; 29, 1146–1155.
15. Johnson J. A., Burlew B. S.: Metoprolol metabolism via cytochrome P4502D6 in ethnic populations. Drug Metab Dispos. 1996; 24, 350–355.
16. Schmid B., Bircher J., Preisig R., Kupfer A.: Polymorphic dextromethorphan metabolism: co–segregation of oxidative O-demethylation with debrisoquin hydroxylation. Clin Pharmacol Ther. 1985; 38, 618–624.
17. Draft FDA guidance for industry.
18. Streetman D. S., Ellis R. E., Nafziger A. N., Leeder J. S., Gaedigk A., Gotschall R., Kearns G. L., Bertino J. S. Jr.: Dose dependency of dextromethorphan for cytochrome P450 2D6 (CYP2D6. phenotyping. Clin Pharmacol Ther. 1999; 66, 535–541.
19. Frank D., Jaehde U., Fuhr U.: Evaluation of probe drugs and pharmacokinetic metrics for CYP2D6 phenotyping. Eur J Clin Pharmacol. 2007; 63, 321–333.
20. Duche J. C., Querol-Ferrer V., Barre J., Mesangeau M., Tillement J. P.: Dextromethorphan O-demethylation and dextrorphan glucuronidation in a French population. Int J Clin Pharmacol Ther Toxicol. 1993; 31, 392–398.
21. Hu O. Y., Tang H. S., Lane H. Y., Chang W. H., Hu T. M.: Novel single-point plasma or saliva dextromethorphan method for determining CYP2D6 activity. J Pharmacol Exp Ther. 1998; 285, 955–960.
22. Jacqz E., Dulac H., Mathieu H.: Phenotyping polymorphic drug metabolism in the French Caucasian population. Eur J Clin Pharmacol. 1988; 35, 167–171.
23. Chladek J., Zimova G., Martinkova J., Tuma I.: Intraindividual variability and influence of urine collection period on dextromethorphan metabolic ratios in healthy subjects. Fundam Clin Pharmacol Ther. 1999; 508–515.
24. Hoskins J. M., Shenfield G. M., Gross A. S.: Modified high-performance liquid chromatographic method to measure both dextromethorphan and proguanil for oxidative phenotyping. J Chromatogr B Biomed Sci Appl. 1997; 696, 81–87.
25. Daali Y., Cherkaoui S., Doffey-Lazeyras F., Dayer P., Desmeules J. A.: Development and validation of a chemical hydrolysis method for dextromethorphan and dextrophan determination in urine samples: application to the assessment of CYP2D6 activity in fibromyalgia patients. J Chromatogr B Analyt Technol Biomed Life Sci. 2008; 861, 56–63.
26. Kohler D., Hartter S., Fuchs K., Sieghart W., Hiemke C.: CYP2D6 genotype and phenotyping by determination of dextromethorphan and metabolites in serum of healthy controls and of patients under psychotropic medication. Pharmacogenetics. 1997; 7, 453–461.
27. Borges S., Li L., Hamman M. A., Jones D. R., Hall S. D., Gorski J. C.: Dextromethorphan to dextrorphan urinary metabolic ratio does not reflect dextromethorphan oral clearance. Drug Metab Dispos. 2005; 33, 1052–1055.
28. Tamminga W. J., Wemer J., Oosterhuis B., Brakenhoff J. P., Gerrits M. G., de Zeeuw R. A., de LeijL. F., Jonkman J. H.: An optimized methodology for combined phenotyping and genotyping on CYP2D6 and CYP2C19. Eur J Clin Pharmacol. 2001; 57, 143–146.
29. Hou Z. Y., Pickle L. W., Meyer P. S., Woosley R. L.: Salivary analysis for determination of dextromethorphan metabolic phenotype. Clin Pharmacol Ther. 1991; 49, 410–419.
30. Park Y. H., Kullberg M. P., Hinsvark O. N.: Quantitative determination of dextromethorphan and three metabolites in urine by reverse-phase high-performance liquid chromatography. J Pharm Sci. 1984; 73, 24–29.
31. Kim E. M., Lee J. S., Park M. J., Choi S. K., Lim M. A., Chung H. S.: Standardization of method for the analysis of dextromethorphan in urine. Forensic Sci Int. 2006; 161, 198–201.
32. Rodrigues W. C., Wang G., Moore C., Agrawal A., Vincent M. J., Soares J. R.: Development and validation of ELISA and GC-MS procedures for the quantification of dextromethorphan and its main metabolite dextrorphan in urine and oral fluid. J Anal Toxicol. 2008; 32, 220–226.
33. Ducharme J., Abdullah S., Wainer I. W.: Dextromethorphan as an in vivo probe for the simultaneous determination of CYP2D6 and CYP3A activity. J Chromatogr B Biomed Appl. 1996; 678, 113–128.
34. Vengurlekar S. S., Heitkamp J., McCush F., Velagaleti P. R., Brisson J. H., Bramer S. L.: A sensitive LC–MS/MS assay for the determination of dextromethorphan and metabolites in human urine––application for drug interaction studies assessing potential CYP3A and CYP2D6 inhibition. J Pharm Biomed Anal. 2002; 30, 113–124.
35. Chen Z. R., Somogyi A. A., Bochner F.: Simultaneous determination of dextromethorphan and three metabolites in plasma and urine using high-performance liquid chromatography with application to their disposition in man. Ther Drug Monit. 1990; 12, 97–104.
36. East T., Dye D.: Determination of dextromethorphan and metabolites in human plasma and urine by high-performance liquid chromatography with fluorescence detection. J Chromatogr. 1985; 338, 99–112.
37. Lin S. Y., Chen C. H., Ho H. O., Chen H. H., Sheu, M. T.: Simultaneous analysis of dextromethorphan and its three metabolites in human plasma using an improved HPLC method with fluorometric detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2007; 859, 141–146.
38. Kuhlenbeck D. L., Eichold T. H., Hoke S. H., 2nd, Baker T. R., Mensen R., Wehmeyer,K. R.: On-line solid phase extraction using the Prospekt-2 coupled with a liquid chromatography/tandem mass spectrometer for the determination of dextromethorphan, dextrorphan and guaifenesin in human plasma. Eur J Mass Spectrom (Chichester, Eng.. 2005; 11, 199–208.
39. Bagheri H., Es-haghi A., Rouini M. R.: Sol-gel-based solid-phase microextraction and gas chromatography-mass spectrometry determination of dextromethorphan and dextrorphan in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2005; 818, 147–157.
40. Afshar M., Rouini M. R., Amini M.: Simple chromatography method for simultaneous determination of dextromethorphan and its main metabolites in human plasma with fluorimetric detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2004; 802, 317–322.
41. Liu L. L., Wang Z., Feng X. T., Gao S.: [Column switching HPLC method for determination of dextrorphan, an active metabolite of dextromethorphan, in plasma]. Yao Xue Xue Bao. 1993; 28, 374–378.
42. Hartter S., Baier D., Dingemanse J., Ziegler G., Hiemke C.: Automated determination of dextromethorphan and its main metabolites in human plasma by high-performance liquid chromatography and column switching. Ther Drug Monit. 1996; 18, 297–303.
43. Kristensen H. T.: Simultaneous determination of dextromethorphan and its metabolites in human plasma by capillary electrophoresis. J Pharm Biomed Anal. 1998; 18, 827–838.
44. Lutz U., Volkel W., Lutz R. W., Lutz, W. K.: LC-MS/MS analysis of dextromethorphan metabolism in human saliva and urine to determine CYP2D6 phenotype and individual variability in N-demethylation and glucuronidation. J Chromatogr B Analyt Technol Biomed Life Sci. 2004; 813, 217–225.
45. Leeder, J. S., Pearce, R. E., Gaedigk, A., Modak, A., Rosen, D. I.: Evaluation of a [13C]-dextromethorphan breath test to assess CYP2D6 phenotype. J Clin Pharmacol. 2008; 48, 1041–1051.
46. Inoue K., Yamazaki H., Imiya K., Akasaka S., Guengerich F. P., Shimada T.: Relationship between CYP2C9 and 2C19 genotypes and tolbutamide methyl hydroxylation and S‑mephenytoin 4’-hydroxylation activities in livers of Japanese and Caucasian populations. Pharmacogenetics. 1997; 7, 103–113.
47. Miners J. O., Birkett D. J.: Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br J Clin Pharmacol. 1998; 45, 525–538.
48. Buzkova H., Pechandova K., Slanar O., Perlik F.: Genetic Polymorphism of Cytochrome CYP2C9 in the Czech population. Klin. Biochem. Metab. 2007; 15, 102–105.
49. Rendic S., Di Carlo F. J.: Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab Rev. 1997; 29, 413–580.
50. Streetman D. S., Bertino J. S., Jr., Nafziger A. N.: Phenotyping of drug-metabolizing enzymes in adults: a review of in-vivo cytochrome P450 phenotyping probes. Pharmacogenetics. 2000; 10, 187–216.
51. Kirchheiner J., Bauer S., Meineke I., Rohde W., Prang V., Meisel C., Roots I., Brockmoller J.: Impact of CYP2C9 and CYP2C19 polymorphisms on tolbutamide kinetics and the insulin and glucose response in healthy volunteers. Pharmacogenetics. 2002; 12, 101–109.
52. Kirchheiner J., Brockmoller J., Meineke I., Bauer S., Rohde W., Meisel C., Roots I.: Impact of CYP2C9 amino acid polymorphisms on glyburide kinetics and on the insulin and glucose response in healthy volunteers. Clin Pharmacol Ther. 2002; 71, 286–296.
53. Veronese M. E., Miners J. O., Randles D., Gregov D., Birkett D. J.: Validation of the tolbutamide metabolic ratio for population screening with use of sulfaphenazole to produce model phenotypic poor metabolizers. Clin Pharmacol Ther. 1990; 47, 403–411.
54. Lee C. R., Pieper J. A., Frye R. F., Hinderliter A. L., Blaisdell J. A., Goldstein J. A.: Tolbutamide, flurbiprofen, and losartan as probes of CYP2C9 activity in humans. J Clin Pharmacol. 2003; 43, 84–91.
55. Hansen L. L., Brosen K.: Quantitative determination of tolbutamide and its metabolites in human plasma and urine by high-performance liquid chromatography and UV detection. Ther Drug Monit. 1999; 21, 664–671.
56. Matin S. B., Rowland M.: Simultaneous determination of tolbutamide and its metabolites in biological fluids. Analytical Letters. 1973; 6, 865–876.
57. Raghow G., Meyer M. C.: High-performance liquid chromatographic assay of tolbutamide and carboxytolbutamide in human plasma. J Pharm Sci. 1981; 70, 1166–1168.
58. Thomas R. C., Ikeda G. J.: The metabolic fate of tolbutamide in man and in the rat. J Med Chem. 1966; 9, 507–510.
59. Klaassen T., Jetter A., Tomalik-Scharte D., Kasel D., Kirchheiner J., Jaehde U., Fuhr U.: Assessment of urinary mephenytoin metrics to phenotype for CYP2C19 and CYP2B6 activity. Eur J Clin Pharmacol. 2008; 64, 387–398.
60. Tamminga W. J., Wemer J., Oosterhuis B., Wieling J., Touw D. J., de Zeeuw R. A., de Leij L. F., Jonkman J. H.: Mephenytoin as a probe for CYP2C19 phenotyping:effect of sample storage, intra-individual reproducibility and occurrence of adverse events. Br J Clin Pharmacol. 2001; 51, 471–474.
61. Britzi M., Bialer M., Arcavi L., Shachbari A., Kapitulnik T., Soback S.: Genetic polymorphism of CYP2D6 and CYP2C19 metabolism determined by phenotyping Israeli ethnic groups. Ther Drug Monit. 2000; 22, 510–516.
62. Tamminga W. J., Wemer J., Oosterhuis B., Weiling J., Wilffert B., de Leij L. F., de Zeeuw, R. A., Jonkman J. H.: CYP2D6 and CYP2C19 activity in a large population of Dutch healthy volunteers: indications for oral contraceptive-related gender differences. Eur J Clin Pharmacol. 1999; 55, 177–184.
63. Jurima-Romet M., Goldstein J. A., LeBelle M., Aubin R. A., Foster B. C., Walop W., Rode, A.: CYP2C19 genotyping and associated mephenytoin hydroxylation polymorphism in a Canadian Inuit population. Pharmacogenetics. 1996; 6, 329–339.
64. Miura M., Motoyama S., Hinai Y., Niioka T., Hayakari M., Ogawa J., Suzuki T.: Correlation between R/S enantiomer ratio of lansoprazole and CYP2C19 activity after single oral and enteral administration. Chirality. 2010; 22, 635–640.
65. Panchabhai T. S., Noronha S. F., Davis S., Shinde V. M., Kshirsagar N. A., Gogtay, N. J.: Evaluation of the activity of CYP2C19 in Gujrati and Marwadi subjects living in Mumbai (Bombay). BMC Clin Pharmacol. 2006; 6, 8.
66. Kim M. J., Nafziger A. N., Zhang Y., Sellers E. M., Gaedigk A., Bertino J. S. Jr.: Lack of weight-based dose dependency and intraindividual variability of omeprazole for CYP2C19 phenotyping. J Clin Pharmacol. 2004; 44, 966–973.
67. Hoskins J. M., Shenfield G. M., Gross A. S.: Concordance between proguanil phenotype and CYP2C19 genotype in Chinese. Eur J Clin Pharmacol. 2003; 59, 611–614.
68. Kanazawa H., Okada A., Higaki M., Yokota H., Mashige F., Nakahara K.: Stereospecific analysis of omeprazole in human plasma as a probe for CYP2C19 phenotype. J Pharm Biomed Anal. 2003; 30, 1817–1824.
69. Kimura M., Ieiri I., Wada Y., Mamiya K., Urae A., Iimori E., Sakai T., Otsubo K., Higuchi S.: Reliability of the omeprazole hydroxylation index for CYP2C19 phenotyping: possible effect of age, liver disease and length of therapy. Br J Clin Pharmacol. 1999; 47, 115–119.
70. Nolin T. D., Frye R. F.: Stereoselective determination of the CYP2C19 probe drug mephenytoin in human urine by gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2003; 783, 265–271.
71. Huang S. L., Xie H. G., Wang W., Xu Z. H., Jiang C. H., Zhou H. H.: Determination of S/R ratio of mephenytoin in human urine by chiral HPLC and ultraviolet detection and its comparison with gas chromatography. Zhongguo Yao Li Xue Bao. 1998; 19, 548–550.
72. Wedlund P. J., Sweetman B. J., McAllister C. B., Branch R. A., Wilkinson G. R.: Direct enantiomeric resolution of mephenytoin and its N-demethylated metabolite in plasma and blood using chiral capillary gas chromatography. J Chromatogr. 1984; 307, 121–127.
73. de Morais S. M., Goldstein J. A., Xie H. G., Huang S. L., Lu Y. Q., Xia H., Xiao Z. S., Ile N., Zhou H. H.: Genetic analysis of the S-mephenytoin polymorphism in a Chinese population. Clin Pharmacol Ther. 1995; 58, 404–411.
74. Yao T. W., Zeng S., Wang T. W., Chen S. Q.: Phenotype analysis of cytochrome P450 2C19 in Chinese subjects with mephenytoin S/R enantiomeric ratio in urine measured by chiral GC. Biomed Chromatogr. 2001; 15, 9–13.
75. Kuang T. Y., Zhang J. M., Zou A. Q., Lou Y. Q.: A chiral capillary gas chromatographic method for direct determination of enantiomers of mephenytoin in human urine. Yao Xue Xue Bao. 1993; 28, 307–311.
76. Mo S. L., Liu Y. H., Duan W., Wei M. Q., Kanwar J. R., Zhou S. F.: Substrate specificity, regulation, and polymorphism of human cytochrome P450 2B6. Curr Drug Metab. 2009; 10, 730–753.
77. Cooper T. B., Suckow R. F., Glassman A.: Determination of bupropion and its major basic metabolites in plasma by liquid chromatography with dual-wavelength ultraviolet detection. J Pharm Sci. 1984; 73, 1104–1107.
78. Yeniceli D., Dogrukol-Ak D.: An LC method for the determination of bupropion and its main metabolite, hydroxybupropion in human plasma. Chromatographia. 2009; 70, 1703–1708.
79. Loboz K. K., Gross A. S., Ray J., McLachlan A. J.: HPLC assay for bupropion and its major metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2005; 823, 115–121.
80. Coles R., Kharasch E. D.: Stereoselective analysis of bupropion and hydroxybupropion in human plasma and urine by LC/MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2007; 857, 67–75.
81. Borges V., Yang E., Dunn J., Henion J.: High-throughput liquid chromatography–tandem mass spectrometry determination of bupropion and its metabolites in human, mouse and rat plasma using a monolithic column. J Chromatogr B Analyt Technol Biomed Life Sci. 2004; 804, 277–287.
82. Kirchheiner J., Klein C., Meineke I., Sasse J., Zanger U. M., Murdter T. E., Roots I., Brockmoller J.: Bupropion and 4-OH-bupropion pharmacokinetics in relation to genetic polymorphisms in CYP2B6. Pharmacogenetics. 2003; 13, 619–626.
83. Pechandova K., Buzkova H., Matouskova O., Perlik F., Slanar, O.: Genetic Polymorphisms of CYP2C8 in the Czech Republic. Genet Test Mol Biomarkers. 2012;
84. Green H., Soderkvist P., Rosenberg P., Mirghani R. A., Rymark P., Lundqvist E. A., Peterson C.: Pharmacogenetic studies of Paclitaxel in the treatment of ovarian cancer. Basic Clin Pharmacol Toxicol. 2009; 104, 130–137.
85. Dierks E. A., Stams K. R., Lim H. K., Cornelius G., Zhang H., Ball S. E.: A method for the simultaneous evaluation of the activities of seven major human drug-metabolizing cytochrome P450s using an in vitro cocktail of probe substrates and fast gradient liquid chromatography tandem mass spectrometry. Drug Metab Dispos. 2001; 29, 23–29.
86. Green H., Vretenbrant K., Norlander B., Peterson C.: Measurement of paclitaxel and its metabolites in human plasma using liquid chromatography/ion trap mass spectrometry with a sonic spray ionization interface. Rapid Communications in Mass Spectrometry. 2006; 20, 2183–2189.
87. Huizing M. T., Sparreboom A., Rosing H., van Tellingen O., Pinedo H. M., Beijnen J. H.: Quantification of paclitaxel metabolites in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1995; 674, 261–268.
88. Vainchtein L. D., Thijssen B., Stokvis E., Rosing H., Schellens J. H., Beijnen J. H.: A simple and sensitive assay for the quantitative analysis of paclitaxel and metabolites in human plasma using liquid chromatography/tandem mass spectrometry. Biomed Chromatogr. 2006; 20, 139–148.
89. Alexander M. S., Kiser M. M., Culley T., Kern J. R., Dolan J. W., McChesney J. D., Zygmunt J., Bannister S. J.: Measurement of paclitaxel in biological matrices: high-throughput liquid chromatographic-tandem mass spectrometric quantification of paclitaxel and metabolites in human and dog plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2003; 785, 253–261.
90. Mortier K. A., Renard V., Verstraete A. G., Van Gussem A., Van Belle S., Lambert W. E.: Development and validation of a liquid chromatography-tandem mass spectrometry assay for the quantification of docetaxel and paclitaxel in human plasma and oral fluid. Anal Chem. 2005; 77, 4677–4683.
91. Burk O., Wojnowski L.: Cytochrome P450 3A and their regulation. Naunyn Schmiedebergs Arch Pharmacol. 2004; 369, 105–124.
92. Opdam F. L., Gelderblom H., Guchelaar H. J.: Phenotyping drug disposition in oncology. Cancer Treat Rev. 2012;
93. Watkins P. B., Murray S. A., Winkelman L. G., Heuman D. M., Wrighton S. A., Guzelian P. S.: Erythromycin breath test as an assay of glucocorticoid-inducible liver cytochromes P-450. Studies in rats and patients. J Clin Invest. 1989; 83, 688–697.
94. Ged C., Rouillon J. M., Pichard L., Combalbert J., Bressot N., Bories P., Michel H., Beaune P., Maurel P.: The increase in urinary excretion of 6 beta-hydroxycortisol as a marker of human hepatic cytochrome P450IIIA induction. Br J Clin Pharmacol. 1989; 28, 373–387.
95. Gentile D. M., Tomlinson E. S., Maggs J. L., Park B. K., Back D. J.: Dexamethasone metabolism by human liver in vitro. Metabolite identification and inhibition of 6-hydroxylation. J Pharmacol Exp Ther. 1996; 277, 105–112.
96. Green H., Skoglund K., Rommel F., Mirghani R. A., Lotfi K.: CYP3A activity influences imatinib response in patients with chronic myeloid leukemia: a pilot study on in vivo CYP3A activity. Eur J Clin Pharmacol. 2010; 66, 383–386.
97. Michael M., Cullinane C., Hatzimihalis A., O’Kane C., Milner A., Booth R., Schlicht S., Clarke S. J., Francis P.: Docetaxel pharmacokinetics and its correlation with two in vivo probes for cytochrome P450 enzymes: the C(14)-erythromycin breath test and the antipyrine clearance test. Cancer Chemother Pharmacol. 2012; 69, 125–135.
98. Eeckhoudt S. L., Desager J. P., Horsmans Y., De Winne A. J., Verbeeck R. K.: Sensitive assay for midazolam and its metabolite 1’-hydroxymidazolam in human plasma by capillary high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 1998; 710, 165–171.
99. Rubio F., Miwa B. J., Garland W. A.: Determination of midazolam and two metabolites of midazolam in human plasma by gas chromatography-negative chemical-ionization mass spectrometry. J Chromatogr. 1982; 233, 157–165.
100. Lehmann B., Boulieu R.: Determination of midazolam and its unconjugated 1-hydroxy metabolite in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1995; 674, 138–142.
101. Martens J., Banditt P.: Simultaneous determination of midazolam and its metabolites 1-hydroxymidazolam and 4-hydroxymidazolam in human serum using gas chromatography-mass spectrometry. J Chromatogr B Biomed Sci Appl. 1997; 692, 95–100.
102. Mastey V., Panneton A. C., Donati F., Varin F.: Determination of midazolam and two of its metabolites in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1994; 655, 305–310.
103. Link B., Haschke M., Wenk M., Krahenbuhl S.: Determination of midazolam and its hydroxy metabolites in human plasma and oral fluid by liquid chromatography/electrospray ionization ion trap tandem mass spectrometry. Rapid Commun Mass Spectrom. 2007; 21, 1531–1540.
104. Chan K., Jones R. D.: Simultaneous determination of flumazenil, midazolam and metabolites in human biological fluids by liquid chromatography. J Chromatogr. 1993; 619, 154–160.
105. Jabor V. A., Coelho E. B., Dos Santos N. A., Bonato P. S., Lanchote V. L.: A highly sensitive LC-MS-MS assay for analysis of midazolam and its major metabolite in human plasma: applications to drug metabolism. J Chromatogr B Analyt Technol Biomed Life Sci. 2005; 822, 27–32.
106. ter Horst P. G. J., Foudraine N. A., Cuypers G., van Dijk E. A., Oldenhof N. J. J.: Simultaneous determination of levomepromazine, midazolam and their major metabolites in human plasma by reversed-phase liquid chromatography. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2003; 791, 389–398.
107. Marquet P., Baudin O., Gaulier J. M., Lacassie E., Dupuy J. L., Francois B., Lachatre G.: Sensitive and specific determination of midazolam and 1-hydroxymidazolam in human serum by liquid chromatography-electrospray mass spectrometry. J Chromatogr B Biomed Sci Appl. 1999; 734, 137–144.
108. Ha H. R., Rentsch K. M., Kneer J., Vonderschmitt D. J.: Determination of midazolam and its alpha-hydroxy metabolite in human plasma and urine by high-performance liquid chromatography. Ther Drug Monit. 1993; 15, 338–343.
109. Eap C. B., Buclin T., Cucchia G., Zullino D., Hustert E., Bleiber G., Golay K. P., Aubert A. C., Baumann P., Telenti A., Kerb R.: Oral administration of a low dose of midazolam (75 mu g. as an in vivo probe for CYP3A activity). European Journal of Clinical Pharmacology. 2004; 60, 237–246.
110. Walker K., Ginsberg G., Hattis D., Johns D. O., Guyton K. Z., Sonawane B.: Genetic polymorphism in N-Acetyltransferase (NAT): Population distribution of NAT1 and NAT2 activity. J Toxicol Environ Health B Crit Rev. 2009; 12, 440–472.
111. Luck H., Kinzig M., Jetter A., Fuhr U., Sorgel F.: Mesalazine pharmacokinetics and NAT2 phenotype. Eur J Clin Pharmacol. 2009; 65, 47–54.
112. Grant D. M., Tang B. K., Kalow W.: A simple test for acetylator phenotype using caffeine. Br J Clin Pharmacol. 1984; 17, 459–464.
113. Zusterzeel P. L., te Morsche R. H., Raijmakers M. T., Roes E. M., Peters W. H., Steegers-Theunissen R. P., Steegers E. A.: N-acetyl-transferase phenotype and risk for preeclampsia. Am J Obstet Gynecol. 2005; 193, 797–802.
114. Bolt H. M., Selinski S., Dannappel D., Blaszkewicz M., Golka K.: Re-investigation of the concordance of human NAT2 phenotypes and genotypes. Arch Toxicol. 2005; 79, 196–200.
115. Kennedy M. J., Abdel-Rahman S. M., Kashuba A. D., Leeder J. S.: Comparison of various urine collection intervals for caffeine and dextromethorphan phenotyping in children. J Clin Pharmacol. 2004; 44, 708–714.
116. Cribb A. E., Isbrucker R., Levatte T., Tsui B., Gillespie C. T., Renton K. W.: Acetylator phenotyping: the urinary caffeine metabolite ratio in slow acetylators correlates with a marker of systemic NAT1 activity. Pharmacogenetics. 1994; 4, 166–170.
117. Ellard G. A., Gammon P. T., Titinen H.: Determination of the acetylator phenotype using matrix isoniazid. Tubercle. 1975; 56, 203–209.
118. Pink J. C., Messing E. M., Reznikoff C. A., Bryan G. T., Swaminathan S.: Correlation between N-acetyltransferase activities in uroepithelia and in vivo acetylator phenotype. Drug Metab Dispos. 1992; 20, 559–565.
119. Stewart N. A., Buch S. C., Conrads T. P., Branch R. A.: A UPLC–MS/MS assay of the “Pittsburgh cocktail”: six CYP probe-drug/metabolites from human plasma and urine using stable isotope dilution. Analyst. 2011; 136, 605–612.
120. Bendriss E. K., Markoglou N., Wainer I. W.: Liquid chromatographic method for the simultaneous determination of caffeine and fourteen caffeine metabolites in urine. Journal of Chromatography B. 2000; 746, 331–338.
121. Rasmussen B. B., Brosen K.: Determination of urinary metabolites of caffeine for the assessment of cytochrome P4501A2, xanthine oxidase, and N-acetyltransferase activity in humans. Therapeutic Drug Monitoring. 1996; 18, 254–262.
122. Sinues B., Saenz M. A., Lanuza J., Bernal M. L., Fanlo A., Juste J. L., Mayayo E.: Five caffeine metabolite ratios to measure tobacco-induced CYP1A2 activity and their relationships with urinary mutagenicity and urine flow. Cancer Epidemiology Biomarkers & Prevention. 1999; 8, 159–166.
123. Kilbane A. J., Silbart L. K., Manis M., Beitins I. Z., Weber W. W.: Human N-acetylation genotype determination with urinary caffeine metabolites. Clinical Pharmacology & Therapeutics. 1990; 47, 470–477.
124. Krul C., Hageman G.: Analysis of urinary caffeine metabolites to assess biotransformation enzyme activities by reversed-phase high-performance liquid chromatography. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 1998; 709, 27–34.
125. Klassen R., Stavric B.: HPLC separation of theophylline, paraxanthine, theobromine, caffeine and other caffeine metabolites in biological-fluids. Journal of Liquid Chromatography. 1983; 6, 895–906.
126. Miceli J. N., Chapman W.: Measurement of Caffeine Metabolites in Urine Using Diode–Array Detection Hplc. Journal of Liquid Chromatography. 1990; 13, 2239–2251.
127. Nyeki A., Biollaz J., Kesselring U. W., Decosterd L. A.: Extractionless method for the simultaneous high-performance liquid chromatographic determination of urinary caffeine metabolites for N-acetyltransferase 2, cytochrome P450 1A2 and xanthine oxidase activity assessment. Journal of Chromatography B. 2001; 755, 73–84.
128. Schneider H., Ma L., Glatt H.: Extractionless method for the determination of urinary caffeine metabolites using high-performance liquid chromatography coupled with tandem mass spectrometry. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2003; 789, 227–237.
129. Weimann A., Sabroe M., Poulsen H. E.: Measurement of caffeine and five of the major metabolites in urine by high-performance liquid chromatography/tandem mass spectrometry. Journal of Mass Spectrometry. 2005; 40, 307–316.
130. Fang L., Pan Y., Muzyka J. L., Zhan C. G.: Active site gating and substrate specificity of butyrylcholinesterase and acetylcholinesterase: insights from molecular dynamics simulations. J Phys Chem B. 2011; 115, 8797–8805.
131. Dubbels R., Schloot W.: Studies on the metabolism of benoxinate by human pseudocholinesterase. Metab Pediatr Syst Ophthalmol. 1983; 7, 37–43.
132. Tunek A., Levin E., Svensson L. A.: Hydrolysis of 3H-bambuterol, a carbamate prodrug of terbutaline, in blood from humans and laboratory animals in vitro. Biochem Pharmacol. 1988; 37, 3867–3876.
133. Gorelick D. A.: Enhancing cocaine metabolism with butyrylcholinesterase as a treatment strategy. Drug Alcohol Depend. 1997; 48, 159–165.
134. Kalow W., Genest K.: A method for the detection of atypical forms of human serum cholinesterase; determination of dibucaine numbers. Can J Biochem Physiol. 1957; 35, 339–346.
135. Harris H., Whittaker M.: Differential inhibition of human serum cholinesterase with fluoride: recognition of two new phenotypes. Nature. 1961; 191, 496–498.
136. Goodall R.: Cholinesterase: phenotyping and genotyping. Ann Clin Biochem. 2004; 41, 98–110.
137. King J., Griffin D.: Differentiation of serum cholinesterase variants by succinyldicholine inhibition. Br J Anaesth. 1973; 45, 450–454.
138. Whittaker M.: The pseudocholinesterase variants. Differentiation by means of sodium chloride. Acta Genet Stat Med. 1968; 18, 556–562.
139. Picheth G., Fadel-Picheth C., Primo-Parmo S. L., Chautard-Freire-Maia E. A., Vieira M. M.: An improved method for butyrylcholinesterase phenotyping. Biochem Genet. 1994; 32, 83–89.
140. Whittaker M.: Differential Inhibition of Human Serum Cholinesterase with n-butyl alcohol: recognition of new phenotypes. Human Heredity. 1968; 18, 335–340.
141. Garry P. J., Routh J. I.: A Micro Method for Serum Cholinesterase. Clin Chem. 1965; 11, 91–96.
142. Silk E., King J., Whittaker M.: Scientific Review No. 5. Assay of cholinesterase in clinical chemistry. Ann Clin Biochem. 1979; 16, 57–75.
143. Dietz A. A., Rubinstein H. M., Lubrano T.: Colorimetric determination of serum cholinesterase and its genetic variants by the propionylthiocholine-dithiobis(nitrobenzoic acid.procedure. Clin Chem. 1973; 19, 1309–1313.
144. Slanar O., Chalupna P., Novotny A., Bortlik M., Krska Z., Lukas M.: Fatal myelotoxicity after azathioprine treatment. Nucleosides Nucleotides Nucleic Acids. 2008; 27, 661–665.
145. Slanar O., Bortlik M., Buzkova H., Donoval R., Pechandova K., Sebesta I., Lukas M., Perlik F.: Polymorphisms of the TPMT gene in the Czech healthy population and patients with inflammatory bowel disease. Nucleosides Nucleotides Nucleic Acids. 2008; 27, 835–838.
146. Anglicheau D., Sanquer S., Loriot M. A., Beaune P., Thervet E.: Thiopurine methyltransferase activity: new conditions for reversed-phase high-performance liquid chromatographic assay without extraction and genotypic-phenotypic correlation. J Chromatogr B Analyt Technol Biomed Life Sci. 2002; 773, 119–127.
147. Ridge S. A., Sludden J., Brown O., Robertson L., Wei X., Sapone A., Fernandez-Salguero P. M., Gonzalez F. J., Vreken P., van Kuilenburg A. B., van Gennip A. H., McLeod H. L.: Dihydropyrimidine dehydrogenase pharmacogenetics in Caucasian subjects. Br J Clin Pharmacol. 1998; 46, 151–156.
148. Ridge S. A., Sludden J., Wei X., Sapone A., Brown O., Hardy S., Canney P., Fernandez-Salguero P., Gonzalez F. J., Cassidy J., McLeod H. L.: Dihydropyrimidine dehydrogenase pharmacogenetics in patients with colorectal cancer. Br J Cancer. 1998; 77, 497–500.
149. van Kuilenburg A. B., Van Lenthe H., Tromp A., Veltman P. C., Van Gennip A. H.: Pitfalls in the diagnosis of patients with a partial dihydropyrimidine dehydrogenase deficiency. Clin Chem. 2000; 46, 9–17.
150. Ito S., Kawamura T., Inada M., Inoue Y., Hirao Y., Koga T., Kunizaki J., Shimizu T., Sato, H.: Physiologically based pharmacokinetic modelling of the three-step metabolism of pyrimidine using C-uracil as an in vivo probe. Br J Clin Pharmacol. 2005; 60, 584–593.
151. Mattison L. K., Fourie J., Hirao Y., Koga T., Desmond R. A., King J. R., Shimizu T., Diasio R. B.: The uracil breath test in the assessment of dihydropyrimidine dehydrogenase activity: pharmacokinetic relationship between expired 13CO2 and plasma [2–13C]dihydrouracil. Clin Cancer Res. 2006; 12, 549–555.
152. Bock K. W., Wiltfang J., Blume R., Ullrich D., Bircher J.: Paracetamol as a test drug to determine glucuronide formation in man. Effects of inducers and of smoking. Eur J Clin Pharmacol. 1987; 31, 677–683.
153. Burger D. M., Huisman A., Van Ewijk N., Neisingh H., Van Uden P., Rongen G. A., Koopmans P., Bertz R. J.: The effect of atazanavir and atazanavir/ritonavir on UDP-glucuronosyltransferase using lamotrigine as a phenotypic probe. Clin Pharmacol Ther. 2008; 84, 698–703.
154. Jensen L. S., Valentine J., Milne R. W., Evans A. M.: The quantification of paracetamol, paracetamol glucuronide and paracetamol sulphate in plasma and urine using a single high-performance liquid chromatography assay. Journal of Pharmaceutical and Biomedical Analysis. 2004; 34, 585–593.
155. Di Girolamo A., O’Neill W. M., Wainer I. W.: A validated method for the determination of paracetamol and its glucuronide and sulphate metabolites in the urine of HIV+/AIDS patients using wavelength-switching UV detection. Journal of Pharmaceutical and Biomedical Analysis. 1998; 17, 1191–1197.
156. Oliveira E. J., Watson D. G., Morton N. S.: A simple microanalytical technique for the determination of paracetamol and its main metabolites in blood spots. Journal of Pharmaceutical and Biomedical Analysis. 2002; 29, 803–809.
157. Alobaidy S. S., Po A. L. W., Mckiernan P. J., Glasgow J. F. T., Millership J.: Assay of paracetamol and its metabolites in urine, plasma and saliva of children with chronic liver-disease. Journal of Pharmaceutical and Biomedical Analysis. 1995; 13, 1033–1039.
158. Goicoechea A. G., Dealda M. J. L., Vilajato J. L.: A validated high-performance liquid-chromatographic method for the determination of paracetamol and its major metabolites in urine. Journal of Liquid Chromatography. 1995; 18, 3257–3268.
159. Adriaenssens P. I., Prescott L. F.: High-performance liquid-chromatographic estimation of paracetamol metabolites in plasma. British Journal of Clinical Pharmacology. 1978; 6, 87–88.
160. Baranowska I., Wilczek A.: Simultaneous RP-HPLC determination of sotalol, metoprolol, alpha-hydroxymetoprolol, paracetamol and its glucuronide and sulfate metabolites in human urine. Analytical Sciences. 2009; 25, 769–772.
161. Lau G. S., Critchley J. A.: The estimation of paracetamol and its major metabolites in both plasma and urine by a single high-performance liquid chromatography assay. J Pharm Biomed Anal. 1994; 12, 1563–1572.
162. Videau O., Delaforge M., Levi M., Thevenot E., Gal O., Becquemont L., Beaune P., Benech H.: Biochemical and analytical development of the CIME cocktail for drug fate assessment in humans. Rapid Commun Mass Spectrom. 2010; 24, 2407–2419.
163. Christensen M., Andersson K., Dalen P., Mirghani R. A., Muirhead G. J., Nordmark A., Tybring G., Wahlberg A., Yasar U., Bertilsson L.: The Karolinska cocktail for phenotyping of five human cytochrome P450 enzymes. Clin Pharmacol Ther. 2003; 73, 517–528.
164. Streetman D. S., Bleakley J. F., Kim J. S., Nafziger A. N., Leeder J. S., Gaedigk A., Gotschall R., Kearns G. L., Bertino J. S. Jr.: Combined phenotypic assessment of CYP1A2, CYP2C19, CYP2D6, CYP3A, N-acetyltransferase-2, and xanthine oxidase with the “Cooperstown cocktail”. Clin Pharmacol Ther. 2000; 68, 375–383.
165. Chainuvati S., Nafziger A. N., Leeder J. S., Gaedigk A., Kearns G. L., Sellers E., Zhang Y., Kashuba A. D., Rowland E., Bertino J. S. Jr.: Combined phenotypic assessment of cytochrome p450 1A2, 2C9, 2C19, 2D6, and 3A, N-acetyltransferase-2, and xanthine oxidase activities with the “Cooperstown 5+1 cocktail”. Clin Pharmacol Ther. 2003; 74, 437–447.
166. Zhu B., Ou-Yang D. S., Chen X. P., Huang S. L., Tan Z. R., He N., Zhou, H. H.: Assessment of cytochrome P450 activity by a five-drug cocktail approach. Clin Pharmacol Ther. 2001; 70, 455–461.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2012 Issue 3
Most read in this issue
- Fenotypizace enzymů podílejících se na metabolismu léčiv
- Extruze tavenin
- Modulácia leukotriénovej cesty – potenciálne ciele
- Mikrobiálne sekundárne metabolity ako inhibítory farmaceuticky významných oxidoreduktáz a transferáz