
Syndrom renálních cyst a diabetu (RCAD) – druhá nejčastější příčina ledvinných cyst u dětí
Renal cysts and diabetes syndrome
Renal cysts and diabetes syndrome (RCAD) is a relatively new nosological entity caused by anomalies of the gene for hepatocyte nuclear faktor 1 beta (HNF1B). It was also named as maturity-onset diabetes of the young type 5 (MODY5). In the recent years it is diagnosed in increasing number of children with different cystic kidney anomalies and seems to be the second most common etiology of cystic kidney diseases in childhood.
The clinical manifestations are multiorgan and include renal anomalies (renal cysts, hypo-dysplasia, hypomagnesemia), pancreatic anomalies (diabetes mellitus, pancreatic atrophy) and can include also liver and external genitalia. Therefore, RCAD syndrome should be considered in children with renal cysts, hypomagnesemia, diabetes mellitus without autoantibodies. The most important diagnostic test is DNA analysis of the gene for HNF1B.
The prognosis of the RCAD syndrome depends mainly on the severity of renal and pancreatic involvement (chronic renal insufficiency/failure, diabetes mellitus in the adolescent/young adult age). The treatment is symptomatic and focused on the complications of renal and pancreatic anomalies.
Key words:
renal cysts and diabetes syndrome, MODY5, hypomagnesemia, chronic renal insufficiency
:
T. Seeman; M. Malina; P. Dušátková; R. Kotalová; J. Lebl; Š. Průhová
:
Pediatrická klinika 2. LF UK a FN Motol, Praha
přednosta prof. MUDr. J. Lebl, CSc.
:
Čes-slov Pediat 2015; 70 (1): 40-45.
:
Review
Syndrom renálních cyst a diabetu (RCAD) je relativně novou nozologickou jednotkou, která je způsobená anomáliemi v genu pro hepatocytární nukleární faktor 1 beta (HNF1B). Dříve byl označovaný také jako 5. typ MODY (maturity-onset diabetes of the young, MODY5). S rozvojem metod molekulární genetiky je v posledních letech stále častěji diagnostikován zejména u dětí s nejrůznějšími cystickými anomáliemi ledvin a v současnosti se zdá být dokonce druhým nejčastějším cystickým onemocněním ledvin u dětí.
Postižení je multiorgánové a kromě ledvin (renální cysty, tvarové anomálie ledvin, hypo-dysplazie, renální hypomagnezémie) a pankreatu (diabetes mellitus, tvarové anomálie) může být postižení jater nebo genitálu. Na RCAD syndrom je tedy potřeba pomýšlet zejména u dětí s nejasným cystickým onemocněním ledvin, hypomagnezémií nebo diabetes mellitus bez autoprotilátek. Nejdůležitějším vyšetřením pro potvrzení diagnózy RCAD syndromu je DNA analýza genu pro HNF1B.
Prognóza RCAD syndromu závisí zejména na rozsahu postižení ledvin a pankreatu, děti mohou skončit až v chronickém selhání ledvin, diabetes mellitus se manifestuje většinou až v adolescentním věku. Léčba se zaměřuje na projevy a komplikace při postižení ledvin a pankreatu (chronická renální insuficience/selhání, diabetes mellitus).
Klíčová slova:
syndrom renálních cyst a diabetu, MODY5, hypomagnezémie, chronické selhání ledvin
Úvod
Cysty v ledvinách jsou velmi častou patologií ledvinného parenchymu, která se vyskytuje u 2–35 % dospělých jedinců [1]. U dětí jsou ledvinné cysty mnohem vzácnější, vyskytují se přibližně jen u 1 % [2]. Další rozdíl mezi dospělou a dětskou populací je v příčinách ledvinných cyst. Zatímco v dospělém věku jsou nejčastější tzv. jednoduché nebo solitární cysty, které se vyskytují až u 35 % dospělých a jsou v naprosté většině benigní, v dětském věku jsou solitární cysty mnohem vzácnější – vyskytují se jen u 0,2 %dětí, tj. asi 100krát méně často než u dospělých. U dětí jsou však častější geneticky podmíněné dědičné cystické choroby ledvin – zejména polycystická onemocnění (autozomálně dominantní a autozomálně recesivní), které mají na rozdíl od jednoduchých solitárních cyst progresivní charakter a způsobují závažné zdravotní komplikace včetně chronického selhání ledvin nebo i úmrtí pacienta [3, 4]. V posledních letech se objevuje stále více informací o onemocnění ledvin způsobeném anomáliemi genu pro hepatocytární nukleární faktor 1 beta (HNF1B) [5, 6].
Historie
Gen HNF1B byl objeven již v roce 1997 u jedné japonské rodiny s dědičnou formou diabetes mellitus (MODY – maturity-onset diabetes of the young) a onemocnění bylo nazváno 5. typem MODY (MODY5) [7]. Zpočátku se myslelo, že mutace HNF1B genu způsobuje jen diabetes a pacienti nemají změny jiných orgánů a postižení ledvin u japonské rodiny bylo považováno mylně za diabetickou nefropatii. Avšak již v roce 1998 publikovali autoři ze stejné japonské skupiny další rodinu s mutací v genu pro HNF1B, kde pacienti s MODY5 měli renální dysfunkci způsobenou ledvinnými cystami a nikoliv diabetickou nefropatií [8]. Navíc poukázali na to, že anomálie ledvin předcházejí vzniku diabetu, neboť u jedné z postižených pacientek byly renální cysty detekovatelné již při porodu a prenatálně měla pacientka zvětšené hyperechogenní ledviny.
Od té doby byla publikována řada prací, které jasně dokazují, že anomálie v genu pro HNF1B způsobují nejen dědičnou formu diabetu, ale také téměř vždy i poškození ledvin, zejména renální cysty. Proto se v posledních letech preferuje pro označení pacientů s mutacemi v HNF1B název syndrom renálních cyst a diabetu (renal cyst a diabetes syndrome, RCAD), který byl poprvé použit v roce 2001 [9, 10]. Také v diabetologii byl název MODY5 nahrazen za HNF1B-MODY.
V současné době představují anomálie HNF1B multisystémové onemocnění zahrnující změny na ledvinách, pankreatu, reprodukčních orgánech či na játrech.
Fenotypické projevy anomálií HNF1B genu
Renální fenotyp
Postižení ledvin je přítomno téměř u všech pacientů s anomáliemi HNF1B (90–100 %) [11]. Typ postižení ledvin je však velmi různorodý, renální cysty představují 70–80 %.Forma cystického postižení ledvin může být také velmi variabilní, a to od prostých renálních cyst přes cystickou dysplazii ledvin, multicystickou dysplazii ledvin až po glomerulocystickou chorobu ledvin.
Prvním důležitým projevem postižení ledvin může být již prenatálně zjištěná hyperechogenita ledvin (60–96 % případů) [11, 12]. Prenatálně většinou nebývají ledviny zvětšené a cysty ještě viditelné, avšak po porodu jsou často u pacientů s prenatálně zjištěnými hyperechogenními ledvinami již cysty sonograficky detekovatelné. Anomálie HNF1B jsou nejčastější příčinou prenatálně zjištěných oboustranně hyperechogenních ledvin (29 %), přičemž jsou překvapivě častější než mutace v PKHD1 genu (19 %) způsobující autozomálně recesivní polycystózu ledvin (ARPKD) a mutace v PKD1 a PKD2 genu (13 %) způsobující autozomálně dominantní polycystózu ledvin (ADPKD) [13]. V klinické praxi může být zrádné to, že anomálie HNF1B mohou sonograficky, klinicky i dědičností připomínat jak ADPKD (autozomálně dominantní dědičnost renálních cyst), tak i ARPKD (zdraví rodiče u pacientů s de novo mutacemi, mikrocysty v ledvinách, často již prenatálně detekované). Vzácně může anomálie HNF1B způsobit jednostrannou multicystickou dysplazii ledviny (multicystic dysplastic kidney, MCDK), přičemž ale téměř vždy je nějakým způsobem strukturálně postižena i druhá – funkčně solitární ledvina [5, 12]. Familiární hypoplastická forma glomerulocystické choroby ledvin (hypoplastic familial glomerulocystic kidney disease, FGCKD) je velmi vzácnou formou cystického poškození ledvin zjistitelná jen histopatologickým vyšetřením ze vzorku ledvinné tkáně získané renální biopsií.
Dalším možným renálním fenotypem je hypo-dysplazie ledvin bez viditelných cyst, která je přítomna asi u 3–10 % pacientů [11, 14, 15], jednostranná ageneze nebo hypoplazie ledviny, zdvojená či podkovovitá ledvina, případně i vrozená anomálie vývodných močových cest, např. vezikoureterální reflux (VUR) [11]. Téměř vždy je patologicky změněná i kontralaterální ledvina, takže postižení ledvin je oboustranné.
Mezi vzácné možné projevy defektu HNF1B patří také atypická familiární juvenilní hyperurikemická nefropatie (FJHN), která je, pokud není přítomen diabetes, fenotypicky neodlišitelná od 1. nebo 2. typu FJHN nebo medulární cystické choroby ledvin (medullary cystic kidney disease, MCKD) způsobené mutacemi v uromodulinovém, reninovém nebo mucinovém genu [16–18] a k její diagnóze je tedy nutné vyšetření HNF1B.
Funkce ledvin může být u dětí s RCAD syndromem normální, avšak u většiny pacientů (55–94 %) je funkce ledvin snížená a může vést již v dětském věku až k terminálnímu chronickému selhání vyžadujícímu léčbu dialýzou a transplantací ledviny (13–20 %) [11].
Jedním z patologických laboratorních nálezů, které jsou kromě snížených renálních funkcí často přítomné u pacientů s HNF1B anomáliemi, je hypomagnezémie (44–67 %) [19] způsobená sníženou tubulární resorpcí magnezia na podkladě poruchy regulace transkripce genu FXYD2, jehož produkt se podílí na tubulární resorpci magnezia. Podobný defekt nebyl dosud u jiných forem cystických chorob ledvin popsán, hypomagnezémie se tak jeví jako možný jednoduchý screeningový test pro diferenciální diagnostiku cystických chorob ledvin.
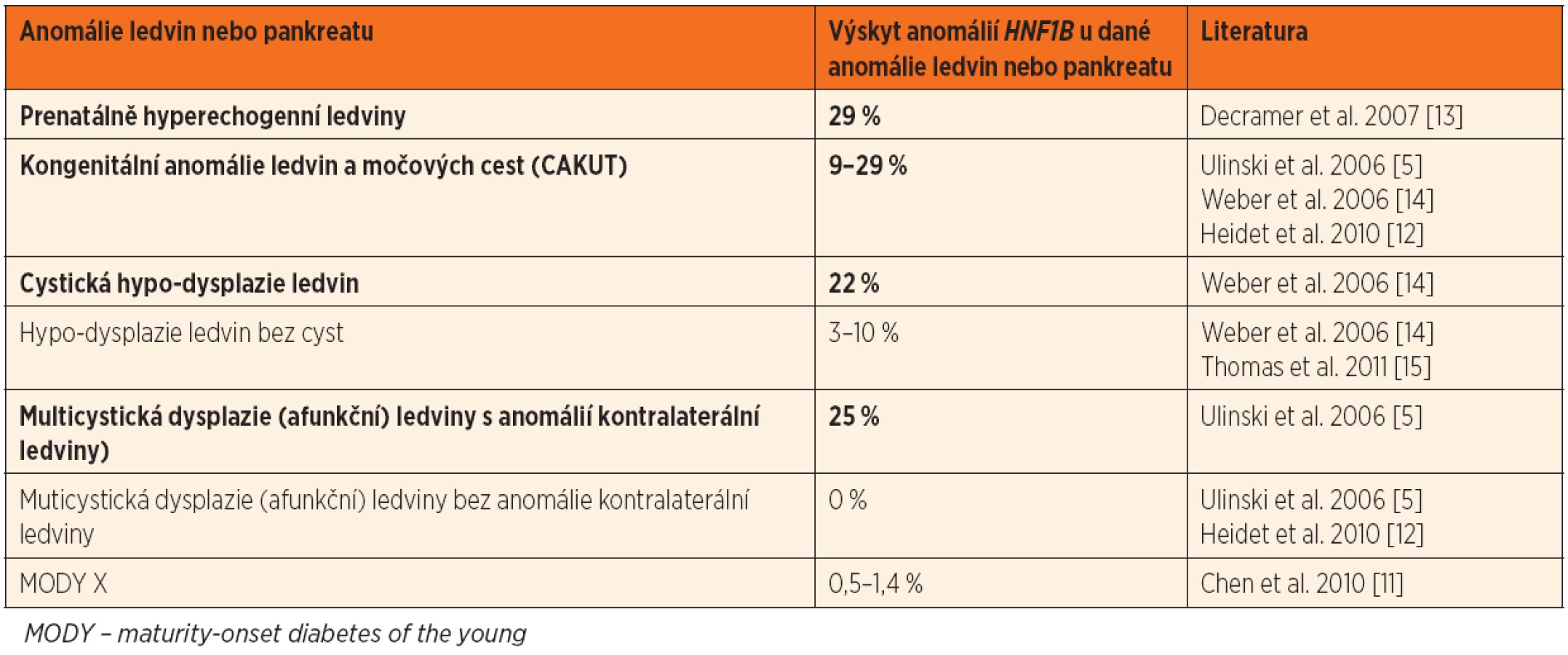
Přehled nejčastějších renálních fenotypů je zachycen v tabulce 1, výskyt HNF1B anomálií u pacientů s různými onemocněními ledvin je uveden v tabulce 2.
![Fenotyp pacientů s anomáliemi v HNF1B (upraveno dle Chen et al. 2010 [11]). Procenta ukazují, u jakého podílu nositelů anomálií v HNF1B byl daný projev přítomen. Tučně jsou zvýrazněny klinicky nejvýznamnější projevy.](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/d6bae51eab1cead598e91566c8f67f7e.png)
Diabetes mellitus jako součást RCAD
Mutace nebo delece genu HNF1B mohou způsobit zvláštní formu diabetes mellitus, který se dle klasifikace řadí do skupiny „diabetes způsobený genetickým defektem beta buněk“. V době svého objevu se označoval jako 5. typ tzv. MODY diabetu, pro který je typická pozitivní rodinná anamnéza diabetu, vznik diabetes mellitus v časném věku (do 40. roku života), negativní autoprotilátky (anti GAD, anti-IA2, antiinzulinové), poměrně pomalý nástup diabetu a často dobrá reakce na léčbu perorálními anti-diabetiky ze skupiny derivátů sulfonylurey. Diabetes, který je součástí RCAD, má navíc ještě další specifika: často se objevuje u pacienta v rodině poprvé (až v 50 % případů vzniká de novo delece celého genu), je většinou zjištěn náhodně jako cukr v moči, typický věk manifestace diabetu je 17 let a více. Diabetes však může vznikat přechodně již u mladších pacientů v případě zatížení léčbou např. růstovým hormonem, kortikoidy apod.
Celkově je pankreas strukturálně menší, je redukována endokrinní i exokrinní tkáň a většina pacientů má subklinickou nedostatečnost exokrinního pankreatu, která zpravidla nevyžaduje léčbu.
Jaterní fenotyp
Jaterní projevy delece nebo mutací genu HNF1B jsou studovány v experimentálních studiích na myších. Jsou prokazovány odchylky v diferenciaci tzv. duktální ploténky (DP), kdy vznikají tzv. malformace duktální ploténky (MDP). Duktální ploténka tvořená pruhy primitivních hepatocytů je embryonálním základem pro jejich remodelaci do žlučových kanálků, linie buněk uvnitř kanálků vyzrávají v cholangiocyty. Defektní gen vede k založení dysplastických žlučovodů nebo se žlučovody zakládají jen ve sníženém počtu (duktopenie). Ve vytvořených žlučovodech jsou defektní cholangiocyty s abnormálním počtem nebo strukturou cilií. To vede k ovlivnění funkce žlučovodů a je podkladem k reakci jaterního parenchymu na tyto změny. Jsou studovány i vztahy mezi geny HNF1B a PKHD1 (gen pro ARPKD) v diferenciaci DP.
V současnosti je popsáno jen málo klinických kazuistik s jaterní lézí na podkladě mutací v genu HNF1B. Důvodem je zřejmě to, že k zjištění etiologie léze vede až vyšetření při nálezu renálních nebo pankreatických morfologických či funkčních změn. Je popsán případ neonatální cholestázy při duktopenii [20] s mírnou hepatomegalií, dále to jsou u starších jedinců nálezy jaterní fibrózy provázené cholestázou nebo jen drobnými odchylkami v jaterních testech [21].
Ostatní fenotypické projevy
Postižení jiných orgánů než ledvin, pankreatu a jater je u pacientů s RCAD syndromem vzácné (1–10 %). Bývají popisovány vývojové vady pohlavního ústrojí (dělohy, vaginy, ovarií, kryptorchismus), testikulární hypoplazie, epididymální cysty, hypospádie. Postižení centrálního nervového systému a neuropsychického vývoje se může projevit jako mentální nebo motorická retardace, autismus – tyto projevy se objevují spíše u osob s rozsáhlejší delecí zahrnující mimo HNF1B také jiné geny. Intrauterinní růstová retardace se vyskytuje u 3 % pacientů.
Přehled postižení jednotlivých orgánů je znázorněn na obrázku 1.

Gen HNF1B a funkce proteinu
Gen HNF1B tvořený 9 exony (NM_000458.2) se nachází v oblasti centromery chromozomu 17 (17cen-q21.3). Transkripční faktor kódovaný HNF1B se skládá ze tří domén: na N-konci se nachází dimerizační doména, následuje ji DNA-vazebná doména složená ze specifické POU domény a POU homeodomény a transaktivační doména. Kauzální jednonukleotidové mutace se nejčastěji vyskytují v DNA-vazebné doméně [11], celkově však převažuje heterozygotní delece celého genu [22]. Vysvětlením může být kompozice regionu obklopujícího gen HNF1B, která je bohatá na AT, což vede ke vzniku sekundárních struktur a vyššímu riziku vzniku delecí [23]. Pravděpodobným mechanismem působení mutací v HNF1B je haploinsuficience, kdy porušená DNA-vazebná či transaktivační doména není schopná vázat koaktivátor, tj. některou z histon-acetyltransferáz [24]. Spekuluje se i o vlivu dominantně negativního efektu, jako u mutací v jiných transkripčních faktorech [25].
Exprese HNF1B byla detekována v játrech, pankreatu, ledvinách a žlučovodu, kde funguje jako transkripční faktor, který je důležitý pro správný embryonální vývoj těchto orgánů. V pankreatu a beta buňkách je HNF1B součástí složité regulační sítě transkripčních faktorů, funguje zvláště ve vazbě na HNF1A [26] a reguluje řadu genů včetně genu pro inzulin [27]. Je také prokázáno, že se faktor HNF1B váže na promotorovou oblast PKHD1 genu (jeho mutace způsobují ARPKD) a interaguje také s PKD2 genem (jeho mutace způsobují ADPKD) a stimuluje jejich genovou transkripci. Anomálie HNF1B tak postihuje i genovou transkripci genů, jejichž mutace vedou k cystickým onemocněním ledvin. Tím lze částečně vysvětlit tvorbu renálních cyst u pacientů s anomá-liemi HNF1B.
Studie myší s knockoutovaným genem hnf1b nejsou jednoduché, protože zvířecí modely umíraly v důsledku chybění hnf1b krátce po implantaci [28], což dokazuje zásadní roli tohoto faktoru v embryogenezi [29]. Speciální strategie ale umožnila generovat myši s chybějícím hnf1b genem výhradně v β-buňkách pankreatu, které krátce po narození projevily poruchu glukózové tolerance s tendencí k progresi během života a došlo i ke změně exprese jiných transkripčních faktorů [30].
Diagnostika
Diagnostika RCAD syndromu je postavena na ultrazvukovém vyšetření ledvin a pankreatu (cysty, tvarové anomálie, hypo-dysplazie), laboratorních metodách (hypomagnezémie, hyperglykemie) a zejména na molekulárně-genetickém vyšetření (analýza genu pro HNF1B). Pro diagnózu RCAD syndromu není potřeba přítomnosti všech orgánových projevů, ale stačí postižení alespoň jednoho orgánu (především ledvin) v kombinaci s průkazem anomálie genu pro HNF1B.
U kterých pacientů pomýšlet na RCAD syndrom
Na RCAD syndrom je potřeba pomýšlet zejména u dětí s těmito anomáliemi:
- prenatálně zjištěné hyperechogenní ledviny (i bez detekovatelných cyst);
- jakékoliv cystické onemocnění ledvin nejasné etiologie (např. cystická dysplazie ledvin, polycystické onemocnění ledvin, prosté cysty ledvin);
- multicystická dysplazie (afunkční) ledviny s jakýmkoliv poškozením kontralaterální ledviny;
- ageneze jedné ledviny s jakýmkoliv poškozením kontralaterální ledviny;
- hypomagnezémie ve spojení s jakýmkoliv strukturálním poškozením ledvin (cystickým i necystickým);
- diabetes mellitus s negativními autoprotilátkami (anti GAD, anti-IA2, antiinzulinovými) při manifestaci ve spojení s jakýmkoliv strukturálním nediabetickým poškozením ledvin (cystickým i necystickým), hypomagnezémií či hyperurikémií;
- autozomálně dominantně dědičné cystické onemocnění ledvin;
- cystické onemocnění ledvin s přítomností jakékoliv strukturální abnormality pankreatu, jater nebo pohlavních orgánů.
Samotné ultrazvukové vyšetření ledvin nebo jen klinicky diagnostikovaný MODY nemůže spolehlivě stanovit diagnózu RCAD syndromu, protože RCAD syndrom nelze klinicky, ultrasonograficky ani laboratorně spolehlivě odlišit od ADPKD, ARPKD nebo jiných cystických onemocnění ledvin, ani ho nelze bez DNA analýzy odlišit od jiných forem MODY. Hyperglykemie nebo přímo rozvoj diabetu je však velmi důležitým vodítkem pro klinické podezření na RCAD syndrom, zejména u pacientů s jakýmkoliv strukturálním postižením ledvin. Rozhodujícím vyšetřením pro konečnou diagnózu je však v dnešní době analýza genu pro HNF1B.
Přítomnost stejného nebo podobného postižení ledvin nebo pankreatu u rodičů pacienta (tj. autozomálně dominantní dědičnost afekce) zvyšuje pravděpodobnost RCAD syndromu u pacienta, avšak až 50 % případů vzniká de novo. Negativní rodinná anamnéza tedy nevylučuje možnost RCAD syndromu u dítěte.
Soubor dětských pacientů s RCAD syndromem v ČR
Mutační a celogenomovou analýzu HNF1B jsme zavedli na našem pracovišti v roce 2011 v rámci grantového projektu Interní grantové agentury MZ ČR. Během prvního roku vyšetřování jsme identifikovali celkem 12 dětí s anomáliemi HNF1B – RCAD syndromem z celé České republiky. Nejčastějším prvním příznakem u těchto pacientů bylo strukturální postižení ledvin (především cysty v ledvinách), u 3 dětí došlo k progresi do stadia chronického selhání ledvin a jsou po transplantaci ledviny. Diabetes mellitus se manifestoval zatím jen u 1 dívky ve věku 14 let při léčbě růstovým hormonem a následně v 17 letech po transplantaci ledviny.
Prognóza
Prognóza pacientů je závislá na závažnosti postižení jednotlivých orgánů, zejména ledvin a pankreatu. Postižení ledvin může vést již v dětském věku až k chronickému selhání s nutností léčby dialýzou a transplantací ledviny. Výskyt chronického selhání je u RCAD méně častý než u ARPKD, ale výrazně častější než u ADPKD, takže se dá renální prognózou zařadit mezi obě polycystická onemocnění ledvin.
Na druhou stranu pacienti s tvarovou anomálií ledvin a diabetem mají prognózu vcelku dobrou, protože diabetes mellitus má tendenci jen k pomalé progresi a při optimální léčbě může dlouho zůstávat bez projevů diabetických komplikací.
Léčba
Renální projevy
Léčba renálního postižení je symptomatická a řídí se běžnými zásadami léčby dětí s chronickým onemocněním ledvin (arteriální hypertenze, proteinurie, infekce močových cest, hypomagnezémie, projevy chronické renální insuficience, léčba chronického selhání ledvin dialýzou a transplantací ledviny). Po transplantaci ledviny je nutné pečlivé sledování a případná léčba nově vzniklého diabetes mellitus, vhodné jsou imunosupresivní protokoly s pouze krátkodobým podáváním kortikosteroidů nebo bez kalcineurinových inhibitorů.
Diabetes mellitus
Lékem volby diabetu bývá inzulin. Nicméně diabetes nastupuje většinou velmi pomalu a může být dlouho kompenzován jen dietou, u mladých osob následně volíme deriváty sulfonylurey, které mohou mít přechodně dobrý efekt. S věkem je pak spíše nutné přejít na léčbu inzulinem k udržení dobré kompenzace diabetu.
Péče o dětské pacienty s RCAD syndromem musí být vzhledem k multisystémovému postižení víceoborová a vyžaduje tedy týmovou spolupráci dětských nefrologů, diabetologů, genetiků, gastroenterologů, radiologů a v neposlední řadě i praktických lékařů pro děti a dorost. Díky léčbě dialýzou a transplantací přežívají i děti s chronickým selháním ledvin do dospělosti a brzy budou s tímto relativně novým geneticky podmíněným onemocněním konfrontováni v klinické praxi i nefrologové, diabetologové a praktičtí lékaři pečující o dospělé pacienty.
Závěr
Syndrom renálních cyst a diabetu (RCAD) způsobený anomáliemi v genu pro HNF1B je relativně novou nozologickou jednotkou, která je však v posledních letech stále častěji diagnostikována zejména u dětí s nejrůznějšími vrozenými cystickými anomáliemi ledvin. Zdá se být dokonce druhým nejčastějším cystickým onemocněním ledvin u dětí po ADPKD.
Pediatr by tedy měl na RCAD syndrom pomýšlet zejména u dítěte s nejasným cystickým onemocněním ledvin nebo diabetem (event. i v kombinaci s jinak neobjasněnou jaterní lézí), v případě kombinace těchto dvou (event. tří) anomálií je RCAD syndrom velmi pravděpodobný (viz výše a tab. 2). Nejdůležitějším vyšetřením pro potvrzení diagnózy RCAD syndromu je v dnešní době DNA analýza HNF1B, která je dostupná na pracovišti autorů.
Finanční podpora práce a poděkování
Podpořeno grantem IGA NT11457, IGA NT11402 a projektem (Ministerstva zdravotnictví) koncepčního rozvoje výzkumné organizace 00064203 (FN Motol).
Děkujeme všem kolegům za zasílání informací a biologického materiálu od pacientů s podezřením na RCAD syndrom.
Došlo: 7. 5. 2014
Přijato: 2. 9. 2014
Prof. MUDr. Tomáš Seeman, CSc.
Pediatrická klinika 2. LF UK
FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: tomas.seeman@lfmotol.cuni.cz
Sources
1. Chang CC, Kuo JY, Chan WL, et al. Prevalence and clinical characteristics of simple renal cyst. J Chin Med Assoc 2007; 70 : 486–491.
2. McHugh K, Stringer DA, Hebert D, et al. Simple renal cysts in children: diagnosis and follow-up with US. Radiology 1991; 178 : 383–385.
3. Janda J, Seeman T, Dušek J, et al. Autozomálně recesivní a autozomálně dominantní polycystická choroba ledvin u dětí. Čes-slov Pediat 1999; 54 : 399–405.
4. Janda J, Vondřichová H, Seeman T, et al. Sonografická charakteristika ledvin u dětí s autozomálně dominantní polycystickou chorobou ledvin (Korelace s klinickými a laboratorními nálezy). Čes-slov Pediat 1999; 54 : 255–259.
5. Ulinski T, Lescure S, Beaufils S, et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol 2006; 17 : 497–503.
6. Edghill EL, Oram RA, Owens M, et al. Hepatocyte nuclear factor-1beta gene deletions – a common cause of renal disease. Nephrol Dial Transplant 2008; 23 : 627–635.
7. Horikawa Y, Iwasaki N, Hara M, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet 1997; 17 : 384–385.
8. Nishigori H, Yamada S, Kohama T, et al. Frameshift mutation, A263fsinsGG, in the hepatocyte nuclear factor-1beta gene associated with diabetes and renal dysfunction. Diabetes 1998; 47 : 1354–1355.
9. Kolatsi-Joannou M, Bingham C, Ellard S, et al. Hepatocyte nuclear factor-1beta: a new kindred with renal cysts and diabetes and gene expression in normal human development. J Am Soc Nephrol 2001; 12 : 2175–2180.
10. Bingham C, Hattersley AT. Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol Dial Transplant 2004; 19 : 2703–2708.
11. Chen YZ, Gao Q, Zhao XZ, et al. Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chin Med J (Engl) 2010; 123 : 3326–3333.
12. Heidet L, Decramer S, Pawtowski A, et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol 2010; 5 : 1079–1090.
13. Decramer S, Parant O, Beaufils S, et al. Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J Am Soc Nephrol 2007; 18 : 923–933.
14. Weber S, Moriniere V, Knüppel T, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol 2006; 17 : 2864–2870.
15. Thomas R, Sanna-Cherchi S, Warady BA, et al. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol 2011; 26 : 897–903.
16. Bingham C, Ellard S, van’t Hoff WG, et al. Atypical familial juvenile hyperuricemic nephropathy associated with a hepatocyte nuclear factor-1beta gene mutation. Kidney Int 2003; 63 : 1645–1651.
17. Vylet’al P, Kublová M, Kalbácová M, et al. Alterations of uromodulin biology: a common denominator of the genetically heterogeneous FJHN/MCKD syndrome. Kidney Int 2006; 70 : 1155–1169.
18. Kirby A, Gnirke A, Jaffe DB, et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet 2013; 45 : 299–303.
19. Adalat S, Woolf AS, Johnstone KA, et al. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 2009; 20 : 1123–1131.
20. Beckers D, Bellanné-Chantelot C, Maes M. Neonatal cholestatic jaundice as the first symptom of a mutation in the hepatocyte nuclear factor-1beta gene (HNF-1beta). J Pediatr 2007; 150 : 313–314.
21. Roelandt P, Antoniou A, Libbrecht L, et al. HNF1B deficiency causes ciliary defects in human cholangiocytes. Hepatology 2012; 56 : 1178–1181.
22. Bellanné-Chantelot C, Chauveau D, Gautier JF, et al. Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med 2004; 140 : 510–517.
23. Chuzhanova N, Abeysinghe SS, Krawczak M, et al. Translocation and gross deletion breakpoints in human inherited disease and cancer II: Potential involvement of repetitive sequence elements in secondary structure formation between DNA ends. Hum Mutat 2003; 22 : 245–251.
24. Barbacci E, Chalkiadaki A, Masdeu C, et al. HNF1beta/TCF2 mutations impair transactivation potential through altered co-regulator recruitment. Hum Mol Genet 2004; 13 : 3139–3149.
25. Kitanaka S, Miki Y, Hayashi Y, et al. Promoter-specific repression of hepatocyte nuclear factor (HNF)-1 beta and HNF-1 alpha transcriptional activity by an HNF-1 beta missense mutant associated with type 5 maturity-onset diabetes of the young with hepatic and biliary manifestations. J Clin Endocrinol Metab 2004; 89 : 1369–1378.
26. Yamagata K, Oda N, Kaisaki PJ, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity - onset diabetes of the young (MODY3). Nature 1996; 384 : 455–458.
27. Okita K, Yang Q, Yamagata K, et al. Human insulin gene is a target gene of hepatocyte nuclear factor-1alpha (HNF-1alpha) and HNF-1beta. Biochem Biophys Res Commun 1999; 263 : 566–569.
28. Coffinier C, Barra J, Babinet C, et al. Expression of the vHNF1//HNF1beta homeoprotein gene during mouse organogenesis. Mech Dev 1999; 89 : 211–213.
29. Maestro MA, Boj SF, Luco RF, et al. Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Hum Mol Genet 2003; 12 : 3307–3314.
30. Wang L, Coffinier C, Thomas MK, et al. Selective deletion of the Hnf1beta (MODY5) gene in beta-cells leads to altered gene expression and defective insulin release. Endocrinology 2004; 145 : 3941–3949.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2015 Issue 1
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Rhabdomyolysis after heavy physical exercise at the fithess center
- Primary Care Paediatricians (PCP) and their options in providing services to newborns, infants, children and adolescent patients
- Renal cysts and diabetes syndrome
- Malignant tumours of thyroid gland in children