
Molecular and antigenic characterization of the H3 hemagglutinin of H3N2 influenza A virus strains collected in the Czech Republic during the 2014/2015 epidemic season
Molekulární a antigenní analýza hemaglutininu virů chřipky A H3N2 detekovaných v České republice v epidemické sezoně 2014/2015
Epidemická sezona 2014/2015 byla charakterizována dominancí subtypu H3N2. Cílem této studie je analýza genetické a antigenní heterogenity kmenů H3N2 detekovaných v České republice v období listopad 2014 až březen 2015 na úrovni molekuly hemaglutininu (HA) H3. Za tímto účelem byla provedena fylogenetická analýza reprezentativních sekvencí H3 HA a glykosylační status a klíčové antigenní mutace byly porovnány s vakcinálními kmeny pro roky 2014 a 2015 (A/Texas/50/2012 a A/Switzerland/9715293/2013) a následně vizualizovány v krystalografické struktuře proteinu H3. Molekulární analýza byla doplněna o výsledky hemaglutinačně inhibičního testu (HIT) s patnácti H3N2 kmeny s využitím antisér A/Texas/50/2012 a A/Switzerland/9715293/2013. Výsledky analýzy českých H3N2 kmenů cirkulujících v sezoně 2014/2015 slouží jako doplnění výsledů oficiálních institucí o data z konkrétní geografické oblasti.
KLÍČOVÁ SLOVA:
influenza – chřipka – H3N2 – hemaglutinin
Authors:
A. Nagy 1,2; H. Jiřincová 2; J. Kynčl 3,4
![]() ; M. Havlíčková 2
; M. Havlíčková 2
Authors‘ workplace:
Laboratory of Molecular Methods, State Veterinary Institute Prague, Czech Republic
1; National Reference Laboratory for Influenza, National Institute of Public Health, Prague, Czech Republic
2; Department of Infectious Diseases Epidemiology, National Institute of Public Health, Prague, Czech Republic
3; Department of Epidemiology, Third Faculty of Medicine, Charles University in Prague, Czech Republic
4
Published in:
Epidemiol. Mikrobiol. Imunol. 65, 2016, č. 2, s. 92-101
Category:
Original Papers
Overview
The 2014/2015 influenza epidemic season was characterized by the predominance of the H3N2 subtype. The presented study investigated the genetic and antigenic heterogeneity of the H3N2 strains collected in the Czech Republic from November 2014 to March 2015. Phylogenetic analysis of the representative H3 hemagglutinin sequences was performed and the glycosylation status and crucial antigenic mutations were compared relative to the 2014 and 2015 vaccine strains (A/Texas/50/2012 and A/Switzerland/9715293/2013) and visualized in the H3 crystal structure. The molecular data were further supplemented by hemagglutination-inhibition test (HIT) results on fifteen H3N2 2014/2015 strains by using the A/Texas/50/2012 (H3N2) and A/Switzerland/9715293/13 (H3N2) antisera. Our data on the Czech H3N2 viruses from the 2014/2015 epidemic season could supplement the reports of official authorities with data from a particular geographical area.
KEYWORDS:
Influenza – H3N2 – hemagglutinin
INTRODUCTION
Influenza A and B viruses are important pathogens that cause epidemics of variable extent every year. High genetic plasticity of the virus is also accompanied, among others, by progressive changes at antigenic sites. Such antigenic drift reduces the affinity of pre-existing neutralizing antibodies and lead to escape from the immune response. Therefore, the influenza vaccine needs to be updated annually to match newly evolved viral strains. To be effective, the influenza vaccine has to be designed based on continuous monitoring of influenza virus circulation in the human population at the global scale along with the antigenic and molecular characterization of the newly emerging strains.
The recommended vaccines usually contain an H1N1pdm09, H3N2, and influenza B virus strains in a trivalent or quadrivalent composition. The actual vaccine composition is updated by the World Health Organization expert team twice a year, in September for the forthcoming influenza season in the southern hemisphere and in February for the awaited season in the north [1]. The H3N2 vaccine formula for the 2014/2015 epidemic season in the northern hemisphere contained the A/Texas/50/2012-like virus which replaced the A/Victoria/361/2011-like one used for the 2012/2013 and 2013/2014 seasons.
Rapid risk assessment [2] conducted by the European Centre for Disease Prevention and Control (ECDC) suggested that the H3N2 viruses detected from the beginning of the 2014/2015 epidemic season in the north also included strains that remarkably antigenically drifted from the vaccine strain used. Phylogenetically, these novel H3N2 viruses belonged to two H3 hemagglutinin (HA) sub-clades designated as 3C.2a and 3C.3a [2]. Consequently, the A/Switzerland/9715293/2013 (H3N2) virus representing the 3C.3a sub-clade was selected as the H3N2 component of the vaccine for the 2015 influenza season in the southern hemisphere [3] and then also for the 2015/2016 epidemic season in the north [4]. However, the ongoing monitoring revealed the presence of an additional 3C.3 sub-clade denoted as 3C.3b [5].
The Czech population is among the least vaccinated against influenza in Europe (Ministry of Health of the Czech Republic, Regional Public Health Authorities, personal communication). Therefore, based on the results of the rapid risk assessment, we investigated the genetic and antigenic heterogeneity of the H3N2 strains collected in the Czech Republic (further abbreviated as CZE) during the 2014/2015 epidemic season at the H3 HA level. To this end, phylogenetic and amino acid sequence analysis of the selected H3 sequences were performed and the pivotal antigenic mutations of the most prevalent sub-clades relative to the 2014 and 2015 vaccine strains were inferred from the solved crystal structure of the A/Victoria/361/2011 (H3N2) H3 molecule [6]. The data from the phylogenetic and molecular analyses were further correlated with the results of hemagglutination-inhibition tests (HIT).
MATERIALS AND METHODS
Virological surveillance
In the Czech Republic, virological surveillance of acute respiratory infections including influenza is conducted from calendar week (CW) 40 of the year to CW 20 of the following year, i.e. from early October to early May. The sentinel system requires weekly collection of not less than two samples from each of 14 districts of the republic. A nasopharyngeal swab (NPS) in PBS (7.4% with 2% BSA and antibiotics in a total volume of 3.5 ml) is usually collected from an adult and a pediatric patient with acute upper respiratory tract infection (codes J00, J02, J04, J05, J06) or influenza (codes J10.1, J10.8, J11.1, J11.8) as defined by the International Classification of Diseases [7] within the first three to four days after the onset of the disease. The non-sentinel system involves samples from inpatients diagnosed mainly with lower respiratory tract infections. Nasopharyngeal swabs are collected from these patients on admission or when further indicated by the deteriorating condition or a change in the clinical picture. In addition, inpatient sputum or bronchoalveolar lavage (BAL) fluid samples are often examined. All samples are incubated at +4°C and transported to the laboratory within 48 hours after the collection.
Virus isolation
Virus cultivation was performed from NPS on MDCK-SIAT 1 cells (Madin-Darby canine kidney; UltraMDCK Serum-free medium for MDCK Cells, Lonza, supplemented with gentamicin, L-Glutamine, and TPCK trypsin at a final concentration of 2 µg/ml) in 96-well plates. Fifty microliters of each swab suspension were transferred into five consecutive wells and the inoculated plate is centrifuged at 3500 revolutions per minute for 15 minutes. After that, each well was added with 150 microliters of the culture medium with trypsin and the plates were incubated at 35 °C in 5% CO2 for five days. The virus replication rate was measured using the HIT with 0.5% of guinea pig red blood cells [8]. Generally, two blind passages were included.
Sequence and phylogenetic analysis
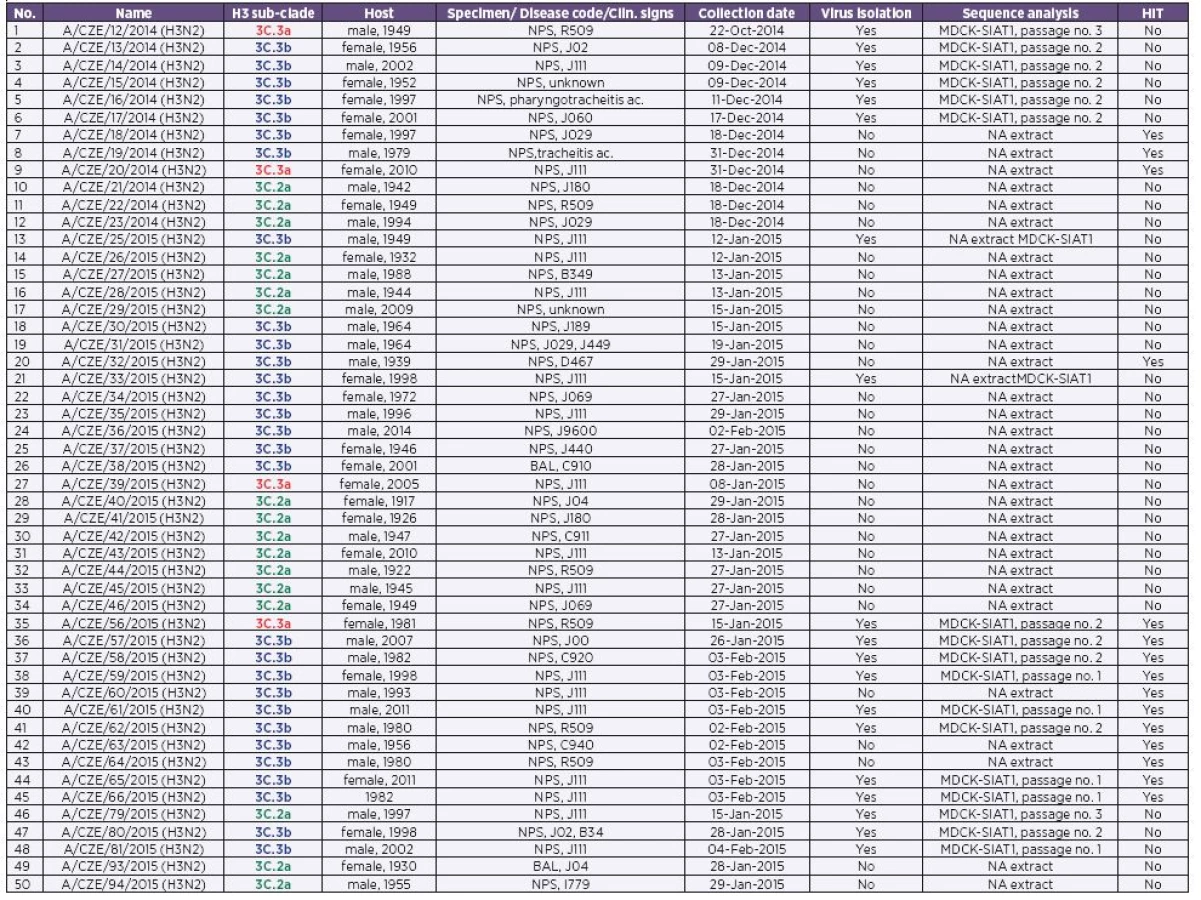
Fifty randomly selected H3N2 isolates and H3N2 positive clinical specimens (Table 1) collected through the influenza surveillance system in the CZE during the 2014/2015 epidemic season were subjected to molecular analysis. Nucleic acid (NA) extraction was performed using the MagNAPure Compact and MagNAPure LC (Total NA extraction Kit I, all from Roche) automatic extractors from input and elution volumes of 200-400 µl and 50-100 µl, respectively. The entire coding sequence of the H3 hemagglutinin (HA) was amplified by RT-PCR (OneStep RT-PCR Kit, Qiagen) and sequenced (BigDye terminator v3.1 Cycle Sequencing Kit, Genetic Analyser GA 3500; both form Life Technologies). For amplification and sequencing, various combinations of H3 consensus primers were employed. The list of the primers is available on request. The obtained data were assembled and edited with the SeqScape software (Life Technologies). Finally, the sequences were deposited in GenBank [9] under the following accession numbers: KR534269-KR534321.
In order to perform phylogenetic analysis, a H3 sequence data set was assembled containing sequences from our study as well as representative H3 sequences downloaded from the GISAID EpiFlu database [10]. The sequences were aligned via the MAFFT tool [11] and the alignment was trimmed by the BioEdit software [12]. For H3HA phylogeny inference the Mega 6 program [13] was applied. The maximum likelihood tree (100 replicates) was calculated on the basis of 1640 nucleotides long sequences (GenBank acc. no. KM069601, positions 43-1683) by using the Hasegawa-Kishino-Yano + discrete Gamma distribution with five rate categories substitution model. The glycosylation sequons were identified by using the NetGlyc 1.0 Server [14].
Structural analysis of the H3HA molecule
The solved crystal structure of the A/Victoria/361/2011 (H3N2) vaccine strain H3 trimer [6] selected for the use in the 2012-2013 northern hemisphere influenza season [15] was downloaded from the RCSB Protein Data Bank [16] (Accession codes: 4O5I, 4O5N). The structures were visualised by using the YASARA visualization tool [17].
Antigenic analysis
The HIT was conducted in accordance with the procedures recommended by the World Health Organization [8] without Oseltamivir addition by using 0,5% of guinea pig red blood cells. The test included the A/Texas/50/2012 and A/Switzerland/9715293/2013 (H3N2) strains and the respective post infection ferret antisera (kindly provided by the WHO Collaborating Centre in London). A four-fold or higher difference in comparison with the homologous titre was considered as significant.
RESULTS
The 2014/2015 influenza epidemic season in the Czech Republic
The influenza epidemic of the season 2014/2015 in the CZE spanned a period of six weeks from calendar week 4 to calendar week 9/2015. During this period a total of 1382 (sentinel and non-sentinel) samples were examined for influenza virus, with positivity rates of 37% for Influenza A virus (14% A/H1, 78% A/H3) and 16% for influenza B virus. The epidemic was moderately severe. Within the six weeks-long epidemic occurrence of acute respiratory infections including influenza in the Czech Republic altogether 1 083 000 persons presented to their general practitioners with cold symptoms. By 18 June 2015, 246 cases of severe influenza often requiring admission to an intensive care unit were reported. From of them 69 were influenza-associated fatal cases.
Phylogenetic analysis of the Czech 2014/2015 epidemic H3N2 viruses
To decipher the evolutionary relationships between the H3 strains, a maximum likelihood tree was constructed on the basis of 50 randomly selected Czech 2014/2015 H3 sequences (see Table 1) as well as those selected from the GISAID EpiFlu database. The phylogenetic analysis revealed that the HA sequences of the H3N2 viruses that circulated in the CZE during the 2014/2015 epidemic season were separated into three related but clearly recognizable 3C sub-clades (Figure 1). The first two grouped the already defined sub-clades 3C.2a and 3C.3a, with A/Hong Kong/5738/2014 and A/Switzerland/9715293/2013 (H3N2) as the reference strains [2]. The third sub-clade formed a clearly distinguishable cluster within the 3C.3 group and was designated as 3C.3b [5] (bootstrap support 98%). The phylogenetic analysis further suggested that the majority of the Czech H3N2 strains belonged to the 3C.2a (17 sequences) and 3C.3b (29 sequences) H3 sub-clades while the 3C.3a sub-clade contained only four sequences.

Molecular analysis of the Czech 2014/2015 epidemic H3N2 viruses
To reveal the differences between the sub-clades, the deduced amino acid sequences of the Czech H3 HAs were aligned relative to A/Texas/50/2012 (H3N2). The alignment (Figure 2) showed that the H3 sub-clades differed both in the expression of potential N-glycosylation (PNG) sites and amino acid substitutions altering the antigenic properties.

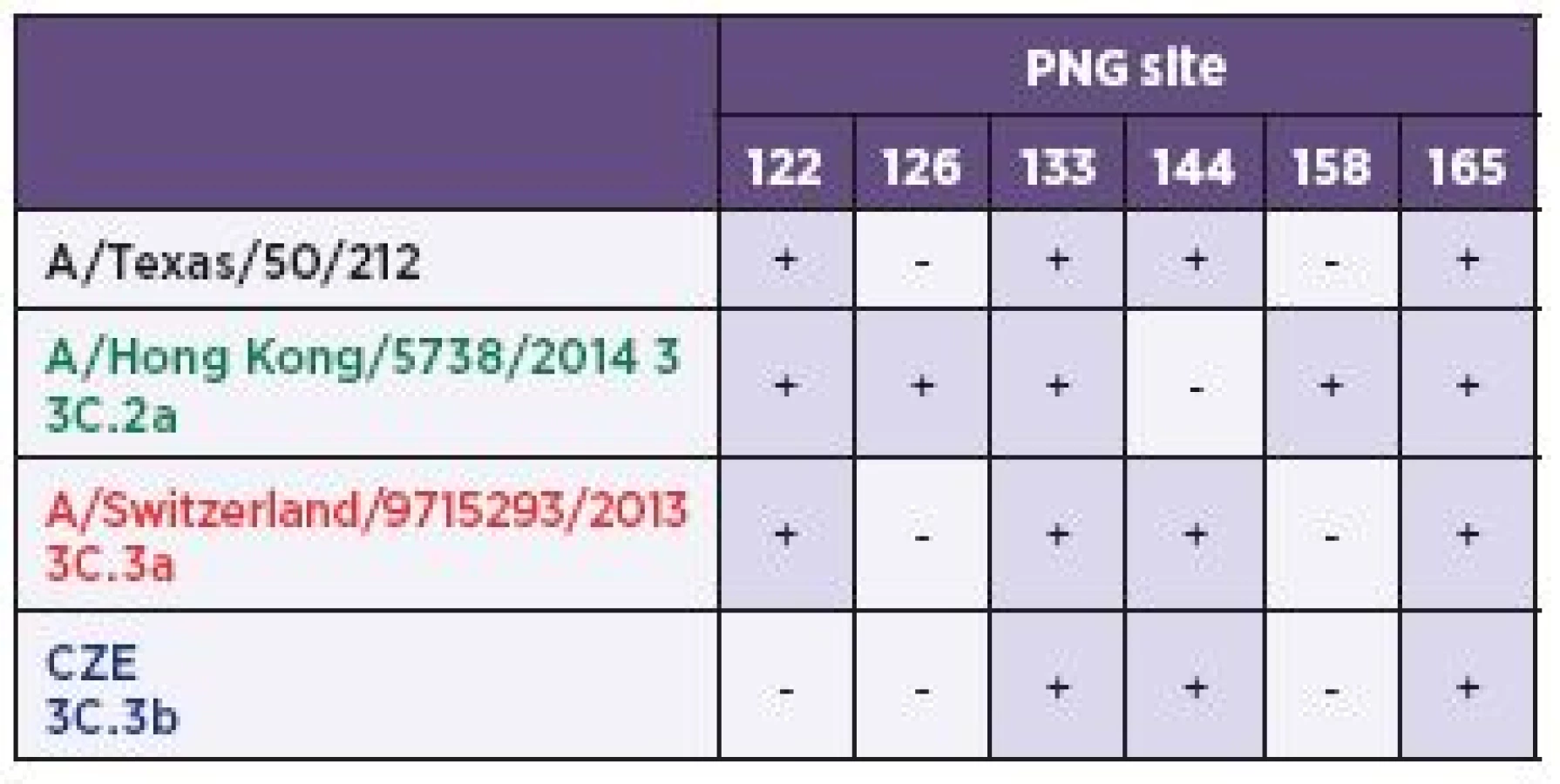
Generally, the H3 molecules express five conserved PNG sequons [6] (HA1 22, 38, 165, 285, and HA2 154). Moreover, all three H3 sub-clades shared five additional ones (8, 45, 63, 133, and 246) located in the HA1 subdomain. However, four PNG motifs (122, 126, 144, and 158) appeared to be expressed differently and forming, along with the common positions 133 and 165, a string of six consecutive PNG sequons. As can be further seen from the amino acid sequence alignment (see Figure 2), the six-PNG string is present in discernible patterns (Table 2) varying between the sub-clades. The A/Texas/50/2012 (H3N2) and the 3C.3a sequences, including A/Switzerland/9715293/2013 (H3N2), were equally lacking two sequons, 126 and 158. On the other hand, the sub-clade 3C.2a lost the PNG 144 but had two sites 126 and 158, which was unique between the sub-clades. Finally, the 3C.3b sub-lineage lacked three PNGs (122, 126, and 158) and represented the H3 group with the least number of glycosylation sites. Positioning of the six-PNG string in the crystal structure of A/Victoria/361/2011 showed that the four sequons (PNGs 122-144) were located on the HA1 surface across the arch-like antigenic site A while the PNG site 158 belonged to site B1 and was located at the molecular apex (Figure 3). All of the asparagine carbamoyl nitrogen atoms were exposed on the surface and were accessible to the solvent.

From the view of additional amino acid differences the two most abundant 3C groups 2a and 3b were further studied at the molecular level in more detail. The amino acid sequence alignment revealed that the 3C.2a and 3C.3b H3 strains had six or nine antigenic mainstream substitutions relative to A/Texas/50/2012 (H3N2) and six or seven substitutions in comparison with A/Switzerland/9715293/2013 (H3N2), respectively. These mutations spanned the antigenic sites A1, A2, A3, B1, B2, E1, and E2 [18, 19] (see Figure 2). Positioning the amino acid changes in the crystal structure of A/Victoria/361/2011 (H3N2) showed that they were localised in the solvent exposed back surface of the HA1 apical globular domain (Figure 4).

Visualisation of the 3C.2a differences relative to A/Texas/50/2012 (H3N2) revealed that three of the six antigenic mutations were located at the top (128N/T) and left edge protrusion (144N/S and 145N/S) of arc-like antigenic site A. The two N/S mutations were of especial interest, abolishing the glycosylation site 144 by changing the NNS/NSS sequon to generate a tetraserine motif. The last three alterations (159F/Y, 160K/T, and 198P/S) were situated at site B in the apex of the HA1 domain (Figure 4 a, b). Similarly, comparing the 3C.2a sub-clade differences relative to A/Switzerland/9715293/2013 (H3N2), changes were revealed in four identical positions like in 3C.2a-Texas although some of the residues were different. These alterations (128A/T, 144N/S, 159S/Y, and 160K/T) also included the SSSS motive starting in position 143. Of the three remaining 3C.2a versus Switzerland changes, the 142G/R was situated at the left edge protrusion of site A while the 138S/A and 186V/G were localised at the bottom right and left rims of the receptor binding site (Figure 4c, d).
Except one strain (A/CZE/63/2015), the Czech 3C.3b sub-lineages bared seven or nine antigenic substitutions relative to A/Switzerland/9715293/2013 (H3N2) and A/Texas/50/2012 (H3N2) respectively. Five of them (62E/K, 83K/R, 122N/D, 157L/S and 197Q/H) clearly differentiate them from both of the vaccine strains (see Figure 2). Positioning of these mutations in the crystal structure of A/Victoria/361/2011 (H3N2) revealed that they were located along the bottom of antigenic site E (mutations 62E/K and 83K/R), further at the right edge of the site A arc (122N/D), and finally in the molecular apex formed by site B (157L/S and 197Q/H). Beside these common changes, the Czech 3C.3b H3 molecules held four additional mutations (128N/A, 142R/G, 145N/S, and 198P/S) in comparison with A/Texas/50/2012 (H3N2). One was situated in the top and two in the left edge protrusion of antigenic site A (Figure 4 e, f). The remaining two amino acid changes between the Czech 3C.3b H3 molecules and A/Switzerland/9715293/2013 (H3N2) were located in the antigenic site A (138S/A) beyond the left edge protrusion, where they were forming a bottom rim of the receptor binding site, and on the globular apex (site B 159S/F; Figure 4 g, h).
Antigenic analysis of the Czech 2014/2015 epidemic H3N2 viruses
Antigenic analysis was carried out on a panel of 15 influenza virus isolates (Table 3) by using the A/Texas/50/2012 (H3N2) and A/Switzerland/9715293/13 (H3N2) sera. The limited number of strains analysed by the HIT was due to the reduced potential of the recent influenza virus isolates for agglutinating guinea pig red blood cells at the titres required for the test, i.e. higher than 1 : 8. For this reason unfortunately, the panel did not include any 3C.2a sub-clade representative and the HIT was targeted at H3 of sub-lineages 3C.3a and 3C.3b.
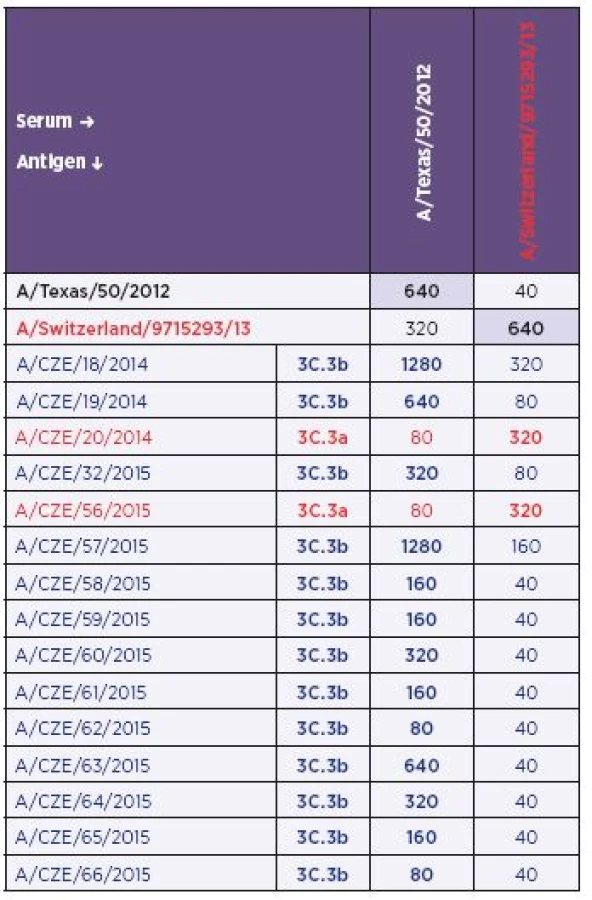
The antigenic analysis allowed unambiguous identification of H3N2 isolates belonging to the sub-clade 3C.3a (A/ CZE/20/2014 and A/CZE/56/2015). Reactivity of these strains with the anti-A/Switzerland/9715293/13 (H3N2) serum was similar to homologous reactivity (i.e., the prototype virus versus the prototype serum) while the reactivity with the anti-A/Texas/50/2012 (H3N2) serum was remarkably lower.
In comparison with group 3C.3a, the H3N2 strains phylogenetically identified as 3C.3b showed poor reactivity with the anti-A/Switzerland/9715293/13 (H3N2) serum for the majority of antigens with only exceptions for the A/CZE/18/2014 and A/CZE/57/2015 isolates. Interestingly, when the reactivity with the anti-A/Texas/50/2012 (H3N2) serum was investigated the data showed a wide range of titres from 1 : 80 to 1 : 1280. The highest titre was obtained with strains A/CZE/18/2014 and A/CZE/57/2015 and was twice as high as in the homologous reactivity. The lowest titre was observed with strains A/CZE/62/2015 and A/CZE/66/2015. Identical value was achieved only ones (A/CZE/19/2014). The remaining titres were two - to four-fold lower in comparison with the homologous reactivity. Nevertheless, despite the variability of the titre with the anti-A/Texas 50/2012 (H3N2) serum, the anti-A/Switzerland/9715293/13 (H3N2) serum allowed for clearly distinguishing the 3C.3b strains.
DISCUSSION
Phylogenetic analysis revealed that the influenza epidemic season 2014/2015 in the Czech Republic was characterised by co-circulation of H3N2 viruses with at least three clearly discernible H3 hemagglutinin variants. The phylogenetic data further suggests that the majority of the investigated H3 sequences belong to sub-clades 3C.2a and 3C.3b while the 3C.3a H3N2 viruses, which are the relatives of the 2015/2016 vaccine strain A/Switzerland/9715293/2013 (H3N2), were remarkably underrepresented. Although revealing the percentage proportion of H3 sub-lineages would require analysis of a broader panel of specimens, the random selection of the representative sequences indicates that the H3N2 viruses of the 3C.2a and 3C.3b sub-clades apparently co-dominated in the Czech Republic during the 2014/2015 season. In contrast, of the H3N2 viruses collected throughout the Europe till 20th March 2015, 63% belonged to sub-lineage 3C.2a represented by strain A/Hong Kong /738/2014 (H3N2) while the rest (36%) were classified as 3C.3 [20]. However, this data might be biased and countries might differ in the prevalence of H3 sub-clades during the same epidemic season.
The three H3 sub-clades differed at the molecular level both in the presence of potential glycosylation sites and amino acid substitutions at key antigenic positions. The number of glycosylation sequons across the H3 HA is cumulative over time. This tendency appears to being human H3N2 virus specific since gradual accumulation of PNG sequons was not observed in any other avian or human HA subtypes [21]. Attachment of new glycans on the H3 globular head evidently provides the ability to mask the antigenic sites and to evade the neutralizing antibodies [22, 23]. On the other hand, it may drastically reduce the receptor binding efficiency [23] and virulence [24].
The amino acid sequences suggested that the co-circulating H3 HAs differed in the presence of six-PNG-long sequon string located consecutively in the sequence. None of the three H3 sub-clades carried all six PNGs. Rather, sub-clade specific patterns could have been recognised. The highest number of PNGs, 13, was observed in the 3C.2a sub-clade while the 3C.3a harboured 12 motifs and the fewest PNGs, 11, was observed in 3C.3b sub-lineage. Such slight within-year variations in PNG sites have also been observed previously [21, 25]. Nevertheless, inferring of the glycosylation status directly from the number of PNGs has only predictive value since in reality not all positions may have been occupied by oligosaccharide moieties despite the fact that all of the aspragine cyrbamoyl nitrogens were exposed on the molecular surface as was implied from the crystal structure. The glycosylation efficiency is inherently dependent on the given PNG amino acid motif [26] and, to some extent it may also be influenced by the surrounding residues. Applying these assumptions to the H3 sub-lineages, it seems that the first two PNGs (122 and 126) of the six consecutive PNG string are weak N-glycosyl acceptors [26]. Moreover, their glycosylation would be sterically hindered by the glycosyl moieties situated in the neighbourhood. Similarly, the glycosylation status of the H3 HA cannot be fully inferred from the crystal structure of the A/Victoria/361/2011 (H3N2) [6] either. In this case the recombinant A/Victoria/361/2011 H3 sequence was expressed in a lepidopteral cell line, where processing of N-linked oligosaccharides occurring differently [27]. Therefore, the question whether the H3 sub-clades differ in the glycosylation status remains to be elucidated.
Further analysis of the amino acid sequences suggested that all three H3 sub-clades harboured a remarkably high number of changes relative to the A/Texas/50/2012 (H3N2) and A/Switzerland/9715293/2013 (H3N2) vaccine strains spanning the antigenic positions of the H3 molecule. From of them the 3C.2a and 3C.3b were studied in detail as the most abundant H3N2 strains of the 2014/2015 influenza season. Generally, the antigenic mutations of the 3C.2a sub-clade appeared to be concentrated on the apex of the HA1 subdomain while the changes of the 3C.3b sub-clade tend to be more scattered throughout the molecular surface. Study of escape mutations suggested that a single amino acid change allow the influenza virus to escape the neutralizing monoclonal antibody [28]. Similarly, a few or even a single amino acid change could apparently substantially alter the specific antibody response of the entire post infection sera [29]. The molecular mechanisms underlying the escape were well characterized and include steric hindrance or loss of an important hydrogen bond [30]. Interestingly both of the sub-clades contained mutations in such critical positions.
The HIT is the most widely used approach to detect influenza specific serum antibodies or to monitor antigenic changes among influenza viruses of the same subtype. We employed the HIT using A/Texas/50/2012 (H3N2) and A/Switzerland/9715293/13 (H3N2) specific antisera to discriminate between the 3C.3a and 3C.3b H3 sub-lineages collected in this study. Unfortunately, attempts to isolate the 3C.2a sub-clade viruses were not successful, which apparently is a more general problem [4, 5]. Therefore, the HIT assay could not have been supplemented by the data on the reactivity of specific antisera against the Czech 3C.2a isolates. This represents also the major limitation of the presented study. The HIT results showed that the representative 3C.3a strains reacted poorly with A/Texas/50/2012 (H3N2) antisera but reached titres comparable to the homologous A/Switzerland/9715293/13 (H3N2) ones. On the other hand, beside two exceptions, the 3C.3b sub-clade viruses reacted poorly with the A/Switzerland/9715293/13 (H3N2) specific antisera.
These data differed from the published HIT results [5] where generally lower titres have been achieved. The variability observed in the 3C.3b panel reactivity against the homologous antisera is difficult to explain. The amino acid sequence of the 3C.3b strains included in the HIT did not reveal significant changes with respect to other 3C.3a or 3C.3b viruses. In addition, sequence differences were not seen in the phylogenetic tree either. Hence, the HIT results for these H3N2 strains were apparently not on the antigen side and can be attributed mainly to technical aspects. The detection of antigenic differences by the HIT has been increasingly limited by the declining ability of the H3N2 virus to agglutinate red blood cells [31]. HIT data are not always easy to interpret: a homologous titre may be lower than a heterologous titre, the adequate quantity of the virus for the reaction (four agglutination units) is difficult to determine for poorly agglutinating isolates and a virus quantity lower than four agglutination units can bias the test results. Due to the difficulty of isolating A/H3N2 viruses in tissue culture and relatively unstable binding to erythrocytes, the HIT was performed with a borderline virus content and despite the adequate checks, a smaller volume of the virus in the actual reaction cannot be ruled out. Higher titres may have also been caused by the absence of oseltamivir; however, even the classical design of the test made it possible to clearly discriminate between sub-clades 3C.3a and 3C.3b. Hence the HIT data corresponded with the results of phylogenetic analysis.
Despite the objective limitations of the study presented we believe that our data could provide an insight into the diversity of the Czech H3N2 viruses from the 2014/2015 epidemic season and could also supplement the reports of official authorities with data from a particular geographical area.
Acknowledgements
We acknowledge epidemiologists from the Regional Public Health Authorities, virologists from regional laboratories as well as hospital physicians. This work was supported by the Ministry of Health, Czech Republic – conceptual development of research organization (NIPH, IN 75010330). Further we acknowledge the authors, originating and submitting laboratories of the sequences from GISAID's EpiFlu Database. All the sequence data are available through the GISAID and NCBI websites www.gisaid.org and http://www.ncbi.nlm.nih.gov/genomes/FLU/.
Do redakce došlo dne 10. 11. 2015.
Adresa pro korespondenci:
RNDr. Alexander Nagy, Ph.D.
Státní veterinární ústav Praha
Laboratoř molekulárních metod
Sídlištní 136/24
165 03 Praha6-Lysolaje
email: alexandernagy17@hotmail.com
Sources
1. Recommendations on the composition of influenza virus vaccines [online]. Geneva: World Health Organization, 2016 [cit. 2016-01-12]. Available at www: 2. European Centre for Disease Prevention and Control. Rapid risk assessment. Circulation of drifted influenza A(H3N2) viruses in the EU/EEA [online]. Stockholm: ECDC, 22 Dec 2014 [cit. 2016-01-12]. Available at www: 3. Recommended composition of influenza virus vaccines for use in the 2015 southern hemisphere influenza season [online]. Geneva: World Health Organization, 25 Sep 2014 [cit. 2016-01-12]. Available at www: 4. Recommended composition of influenza virus vaccines for use in the 2015 - 2016 northern hemisphere influenza season [online]. Geneva: World Health Organization, 26 Feb 2015 [cit. 2016-01-12]. Available at www: 5. European Centre for Disease Prevention and Control. Influenza virus characterization [online]. Stockholm: ECDC, Feb 2015 [cit. 2016-01-12]. Available at www: 6. Lee PS, Ohshima N, Stanfield LR, et al. Receptor mimicry by antibody F045–092 facilitates universal binding to the H3 subtype of influenza virus. Nat Commun, 2014;5 : 3614.
7. International Classification of Diseases (ICD). Version 2015 vaccines [online]. Geneva: World Health Organization, 2016 [cit. 2016-01-12]. Available at www: 8. WHO Global Influenza Surveillance Network. Manual for the laboratory diagnosis and virological surveillance of influenza. Geneva: World Health Organization; 2011. ISBN 978 92 4 154809 0.
9. National Center for Biotechnology Information (NCBI) [online]. Bethesda: NCBI [cit. 2016-01-12]. Available at www: 10. Global initiative on sharing avian influenza data, (GISAID) [online]. Munich: GISAID, 2015 [cit. 2016-01-12]. Available at www: 11. Katoh K, Misawa K, Kuma K, et al. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res, 2002;30(14):3059–3066.
12. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser, 1999;41 : 95–98.
13. Tamura K, Stecher G, Peterson D, et al. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol, 2013;30(12):2725–2729.
14. NetNGlyc 1.0 Server [online]. Lyngby: Center for Biological Sequence Analysis, 2015 [cit. 2016-01-12]. Available at www: 15. Recommended composition of influenza virus vaccines for use in the 2012-2013 northern hemisphere influenza season [online]. Geneva: World Health Organization, Feb 2012 [cit. 2016-01-12]. Available at www: 16. Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res, 2000;28(1):235–242.
17. Krieger E, Vriend G. YASARA View – molecular graphics for all devices – from smartphones to workstations. Bioinformatics, 2014;30(20):2981–2982.
18. Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature, 1981;289(5796):373–378.
19. Nakajima S, Nobusawa E, Nakajima K. Variation in response among individuals to antigenic sites on the HA protein of human influenza virus may be responsible for the emergence of drift strains in the human population. Virology, 2000;274(1):220–231.
20. Flu News Europe. Joint ECDC-WHO/Europe weekly influenza update [online]. Stockholm: ECDC, 2016 [cit. 2016-01-12]. Available at www: 21. Zhang M, Gaschen B, Blay W, et al. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology, 2004;14(12):1229–1246.
22. Skehel JJ, Stevens DJ, Daniels RS, et al. A carbohydrate side chain on hemagglutinins of Hong Kong influenza viruses inhibits recognition by a monoclonal antibody. Proc Natl Acad Sci USA, 1984;81(6):1779–1783.
23. Abe Y, Takashita E, Sugawara K, et al. Effect of the addition of oligosaccharides on the biological activities and antigenicity of influenza A/H3N2 virus hemagglutinin. J Virol, 2004;78(18):9605–9611.
24. Vigerust DJ, Ulett KB, Boyd KL, et al. N-linked glycosylation attenuates H3N2 influenza viruses. J Virol, 2007;81(16):8593–8600.
25. Seidel W, Künkel F, Geisler B, et al. Intraepidemic variants of influenza virus H3 hemagglutinin differing in the number of carbohydrate side chains. Arch Virol, 1991;120(3–4):289–296.
26. Shakin-Eshleman SH, Spitalnik SL, Kasturi L. The amino acid at the X position of an Asn-X-Sersequon is an important determinant of N-linked core-glycosylation efficiency. J Biol Chem, 1996;271(11):6363–6366.
27. Kulakosky PC, Hughes PR, Wood HA. N-Linked glycosylation of a baculovirus-expressed recombinant glycoprotein in insect larvae and tissue culture cells. Glycobiology, 1998;8(7):741–745.
28. Fleury D, Wharton SA, Skehel JJ, et al. Antigen distortion allows influenza virus to escape neutralization. Nat Struct Biol, 1998;5(2):119–123.
29. Koel BF, Burke DF, Bestebroer TM, et al. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science, 2013;342(6161):976–979.
30. Bizebard T, Barbey-Martin C, Fleury D, et al. Structural studies on viral escape from antibody neutralization. Curr Top Microbiol Immunol, 2001;260 : 55–64.
31. Medeiros R, Escriou N, Naffakh N, et al. Hemagglutinin residues of recent human A(H3N2) influenza viruses that contribute to the inability to agglutinate chicken erythrocytes. Virology, 2001;289(1):74–85.
Labels
Hygiene and epidemiology Medical virology Clinical microbiologyArticle was published in
Epidemiology, Microbiology, Immunology

2016 Issue 2
Most read in this issue
- A report of 10 cases of familial Creutzfeld-Jakob disease
- Novel approaches to control the rise in pertussis cases
-
The occurrence of Ixodes ricinus ticks and important tick-borne pathogens in areas with high tick-borne encephalitis prevalence in different altitudinal levels of the Czech Republic
Part I. Ixodes ricinus ticks and tick-borne encephalitis virus - The effect of oxygen on endotoxin production in bacteria of the Bacteroides fragilis group isolated from patients with colorectal carcinoma