
Popis familiární formy Creutzfeldt-Jakobovy nemoci
A report of 10 cases of familial Creutzfeld-Jakob disease
Aim:
The analysis of the available reported data and epidemiological investigation data on the cases of Creutzfeld-Jakob disease (CJD) that emerged in the Nový Jičín district in 2001–2011, with a focus on the familial form of the disease.
Material and Methods:
Data from the Regional Public Health Service of the Moravian-Silesian Region, local Public Health Centre Nový Jičín, were used for the analysis.
Results:
This is a retrospective report of 10 cases of familial Creutzfeld-Jakob disease (CJD) in the Nový Jičín district in 2001–2011, based on the data from the public health service. Overall eight cases were notified and seven suspected cases were identified retrospectively. Out of these 15 cases, five were concluded to be sporadic CJD and 10 to be familial CJD. The PRNP D178N mutation was found in two fatal cases from 2002 and 2003. Genetic investigation of their relatives was not performed for ethical reasons.
Conclusion:
The high incidence of familial CJD in the Nový Jičín district in 2001–2011 is surprising, but the retrospective investigation failed to provide further evidence to understand this outbreak.
KEYWORDS:
Creutzfeldt-Jakob disease – familial form – cases in the Nový Jičín district – D178N mutation
:
J. Janoutová 1; L. Siváková 1; L. Máslová 2; A. Hozák 1; K. Vařechová 1,3; V. Janout 1
:
Ostravská univerzita v Ostravě, Lékařská fakulta, Ústav epidemiologie a ochrany veřejného zdraví, Ostrava
1; Krajská hygienická stanice Moravskoslezského kraje se sídlem v Ostravě
2; Stipendista města Ostravy
3
:
Epidemiol. Mikrobiol. Imunol. 65, 2016, č. 2, s. 145-148
:
Short Communication
Cíl:
Cílem této práce je analýza dat výskytu Creutzfeldt-Jakobovy nemoci (CJD) získaných hlášením a epidemiologickým šetřením v okrese Nový Jičín v letech 2001–2011 se zaměřením na familiární výskyt CJD.
Materiál a metodika:
Pro analýzu dat z okresu Nový Jičín byly použity informace z Krajské hygienické stanice Moravskoslezského kraje, územní pracoviště Nový Jičín.
Výsledky:
Jedná se o retrospektivní popis familiární formy Creutzfeld-Jakobovy nemoci (CJD) na Novojičínsku v letech 2001–2011 z dokumentace hygienické služby, kde bylo hlášeno celkem 8 onemocnění a zpětně vyhledáno dalších 7 pravděpodobných onemocnění. Z celkového počtu 15 onemocnění bylo 5 uzavřeno jako sporadická forma CJD a 10 jako familiární forma CJD. U dvou zemřelých v roce 2002 a v roce 2003 byla zjištěna mutace PRNP D178N. Genetické vyšetření příbuzných zemřelých z etických důvodů provedeno nebylo.
Závěr:
Vysoký výskyt familiární formy CJD v regionu Novojičínska je výjimečný, ale při retrospektivním vyšetřování nebyly zjištěny důvody tohoto vysokého výskytu.
KLÍČOVÁ SLOVA:
Creutzfeldt-Jakobova nemoc – familiární forma – výskyt na Novojičínsku – mutace D178N
ÚVOD
CJD je nejčastějším lidským prionovým onemocněním. Prionové nemoci patří do skupiny smrtelných neurodegenerativních onemocnění s charakteristickými spongiformními změnami mozkové tkáně a v současné době jsou neléčitelné. Klasická varianta CJD je poměrně vzácná, subakutní spongiformní encefalopatie se objevuje převážně u starších osob. Tato forma CJD je známa již od 20. let 20. století a vyskytuje se s celosvětovou incidencí asi 1 případ na milion osob ročně. V roce 1996 byla poprvé popsána nová varianta Creutzfeldt-Jakobovy nemoci (vCJD), u které se předpokládal přenos BSE (bovinní spongiformní encefalopatie) z krav na člověka [1, 2, 3, 4, 5]. U klasické varianty CJD jsou rozeznávány tři formy. Sporadická forma klasické varianty CJD, u které není známá příčina přeměny buněčného na patologický prionový protein [6], představuje přibližně 85 % případů CJD a postihuje zhruba stejně obě pohlaví [7, 8]. Genetická forma klasické varianty CJD, která představuje 10–15 % případů CJD, je genetickou formou CJD s mutací genu pro prionový protein (PRNP), který je lokalizován na krátkém raménku 20. chromozomu, nejčastěji v oblasti kodonu 200, ale i v jiných oblastech. Samotná E200K patogenní mutace je považována jako nedostatečná pro vznik prionového onemocnění a další dosud neobjasněné faktory by mohly vysvětlit penetraci E200K-dependentní CJD [9]. Při výskytu u více než dvou členů rodiny je genetická forma označována jako familiární forma CJD [10]. Třetí formou výskytu je iatrogenní forma (někdy označovaná jako accidentally transmitted CJD), která je vzácná a byla zjištěna po transplantaci rohovky, po použití nedostatečně sterilizovaných intracerebrálních elektrod, po transplantaci dury mater nebo užitím růstového hormonu a gonadotropinu z kadaverózních hypofýz. V souvislosti s prevencí této formy CJD probíhá od roku 2007 v Národní referenční laboratoři lidských TSE/CJD v Praze povinné testování CNS dárců rohovek [11].
Cílem této práce byla analýza dat výskytu Creutzfeldt--Jakobovy nemoci (CJD) získaných hlášením a epidemiologickým šetřením v okrese Nový Jičín v letech 2001–2011 se zaměřením na familiární výskyt CJD.
MATERIÁL A METODIKA
Pro analýzu dat z okresu Nový Jičín byly použity informace z Krajské hygienické stanice Moravskoslezského kraje, územní pracoviště Nový Jičín. Na toto pracoviště jsou zasílány pitevní protokoly z Národní referenční laboratoře transmisivních spongiformních encefalopatií a Creutzfeldt-Jakobovy nemoci České republiky, kde je prováděna definitivní diagnóza CJD, včetně uvedení konkrétní formy nemoci. Tyto údaje je možno považovat za validní.
VÝSLEDKY
V okrese Nový Jičín bylo od roku 2001 do konce roku 2011 hlášeno osm onemocnění Creutzfeldt-Jakobovou nemocí. Jednalo se o tři muže a pět žen. Z těchto osmi hlášených případů byla tři onemocnění diagnostikována jako familiární forma Creutzfeldt-Jakobovy nemoci, z nichž dva případy byly potvrzeny i histologicky (obr. 1).
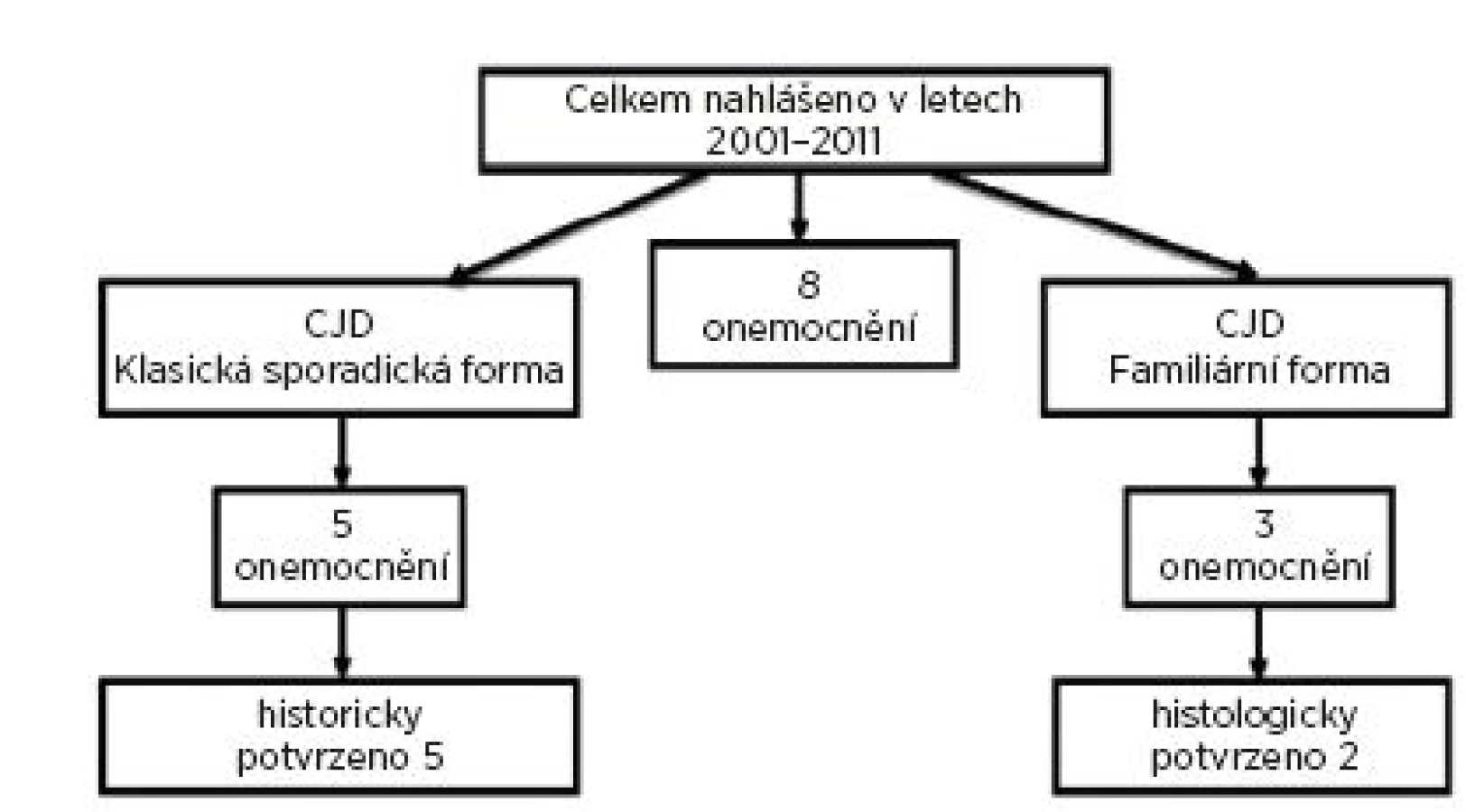
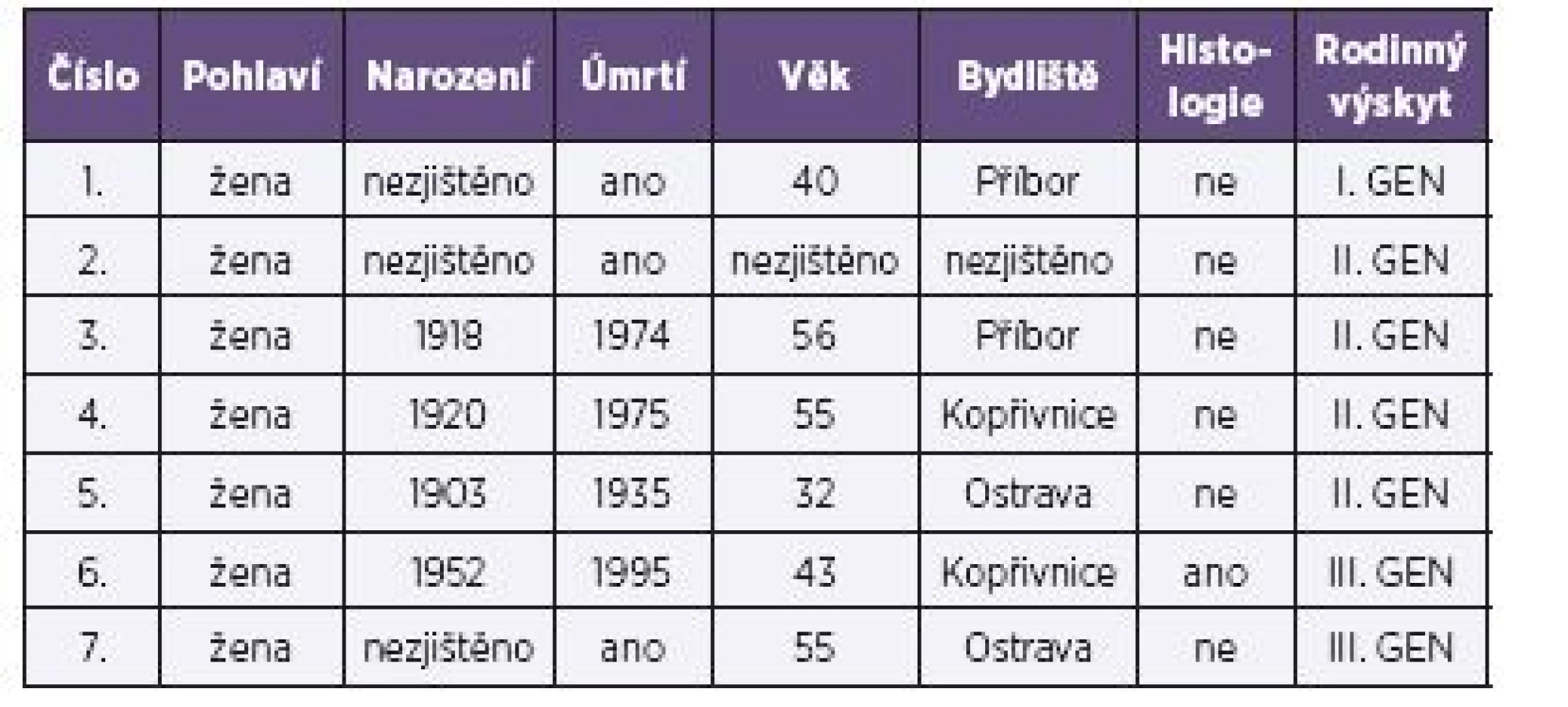
Na základě těchto hlášení bylo zahájeno retrospektivní epidemiologické šetření u příbuzných zemřelých pacientů. U žijících příbuzných se genetické vyšetření na přítomnost mutace prionového genu z etických důvodů neprovádělo. Pokud by výsledky testů vyšly pozitivně, měly by rodinní příslušníci jistotu, že v určité době mohou onemocnět. Vzhledem k tomu, že familiární formu CJD nelze nijak preventivně ovlivnit, znamenalo by to pro příbuzné velkou psychickou zátěž. Následným epidemiologickým šetřením bylo zjištěno dalších sedm nemocných. Familiární výskyt CJD na Novojičínsku zobrazují tabulky 1 a 2 a obrázek 1.

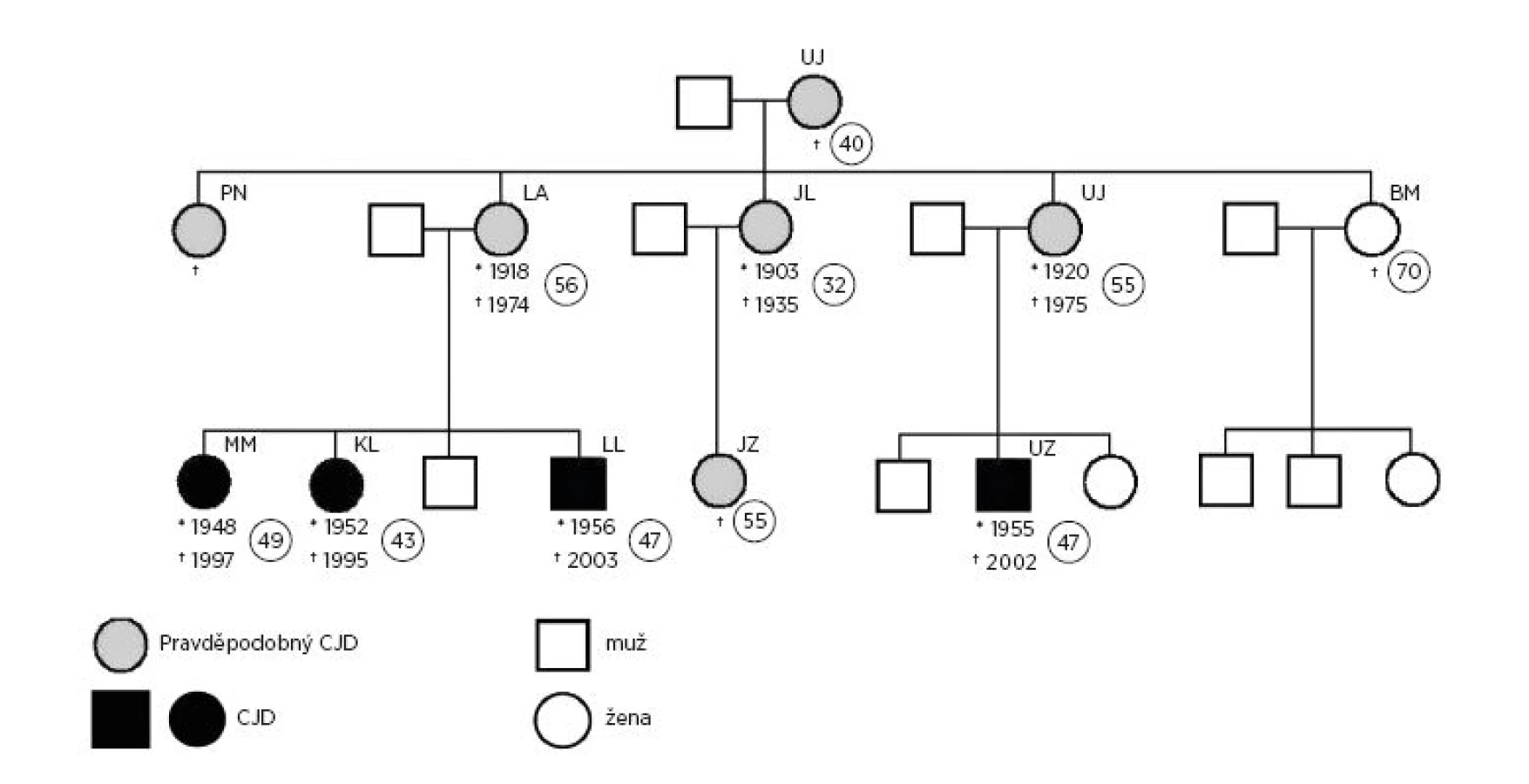
Familiární výskyt deseti případů CJD zahrnuje tři generace rodinných příslušníků. Pro přiblížení je na obrázku 2 znázorněn rodokmen.
I. generace
Pokud jde o nemocnou z první generace, jednalo se o ženu, která zemřela ve věku 40 let na tzv. „pomatení“. Měla 5 dcer, které po smrti rodičů vyrůstaly samy v péči nejstarší dcery.
II. generace
Z pěti sester druhé generace jedna zemřela ve 32 letech na podobné onemocnění jako její matka. Další tři sestry zemřely ve věku kolem 55 let (u jedné z nich se nepodařilo věk a rok úmrtí zjistit) na Alzheimerovu chorobu. Pouze poslední ze sester zemřela ve věku 70 let na srdeční selhání a v její rodině se doposud žádné neurodegenerativní onemocnění neobjevilo.
III. generace
Z osob třetí generace zemřela v roce 1995 43letá žena, jejíž mozek byl při pitvě fixován a diagnóza CJD byla potvrzena v Ústavu patologie Lékařské fakulty v Olomouci (prof. MUDr. R. Koďousek, DrSc.). V roce 1997 zemřela její 49letá sestra, u které byla diagnóza CJD uzavřena na základě klinických projevů, EEG nálezu a diagnózy úmrtí její sestry. Pitva nebyla provedena. V roce 2002 zemřel 47letý muž, který byl bratrancem výše uvedených žen a v roce 2003 umírá jejich bratr také ve věku 47 let. U obou těchto mužů byla zjištěna mutace PRNP D178N (Oddělení lékařské genetiky FN Ostrava a Oddělení patologie a molekulární medicíny FTNsP Praha). Epidemiologickým šetřením bylo dále zjištěno úmrtí 55leté ženy, která byla sestřenicí výše uvedených případů z třetí generace, dcerou zemřelé ženy z II. generace a vnučkou zemřelé ženy z I. generace.
První příznaky, průběh i klinický obraz onemocnění byly u všech případů familiárního výskytu Creutzfeldt--Jakobovy nemoci přibližně stejné. K úmrtí docházelo za 11–13 měsíců od prvních projevů onemocnění. Je pozoruhodné, že z 10 případů familiárního výskytu se v osmi případech jednalo o ženy.
Při pohledu na obrázek 2 je zřejmé, že v rodinách iniciálního případu (ženy, která zemřela ve věku 40 let) byl zjištěn jeden případ v jedné rodině, dva případy ve dvou rodinách a čtyři případy v jedné rodině. Jedna z pěti rodin byla nepostižena.
DISKUSE
Při následném epidemiologickém šetření bylo zjištěno 7 dalších pravděpodobných onemocnění CJD, u kterých ve 3 případech (sestry z druhé generace) byla jejich úmrtí označena jako úmrtí na Alzheimerovu chorobu. To mohlo být podpořeno i tím, že prionový protein PrPsc způsobuje tendenci vytvářet fibrilární nerozpustné agregáty, které se v mozkové tkáni chovají jako depozita amyloidu [6] a klinicky se onemocnění projevuje mimo jiné i rychle progredující demencí [12].
Familiární CJD je spojena s mutacemi genu prionového proteinu (PRNP). Nejčastější patogenní mutace je substituce aminokyselin na kodonu 200 (E200K), ale tato samotná mutace je považována jako nedostatečná pro vznik prionového onemocnění. Pravděpodobně další dosud neobjasněné faktory by mohly vysvětlit penetraci E200K-dependentní CJD [9]. Vedle kauzálních mutací jsou důležité i další změny v genu prionového proteinu – polymorfismy, které nemoc nezpůsobují, ale mohou ovlivnit rozvoj CJD a její průběh [8]. Je také pravděpodobná účast i non-PRNP genů v etiologii prionových onemocnění včetně CJD [13]. Mutace E200K je nejčastější na Slovensku, zatímco v západní Evropě převládá mutace D178N, tedy obdobná jako v popisovaném případu.
Diagnostika lidských prionových onemocnění probíhá od roku 2001 v Národní referenční laboratoři lidských TSE/CJD, kde byl zjištěn rostoucí podíl genetických forem [14]. Diagnostická kritéria pro epidemiologické potřeby rozlišují diagnózu CJD jako definitivní, možnou a pravděpodobnou [15, 16]. Také v této práci se kromě definitivně potvrzených 8 hlášených onemocní CJD objevuje i 7 pravděpodobných onemocnění CJD, které po uplynutí dlouhého období od jejich vzniku, případně úmrtí, již není možno přesněji specifikovat a diagnosticky uzavřít.
Celosvětová incidence Creutzfeldt-Jakobovy nemoci se udává v průměru 1 případ na milion obyvatel za rok. Familiární CJD představuje asi 10–15 % všech případů CJD. Zvýšený výskyt familiární CJD je uváděn u Židů libyjského původu (26 případů na milion), další ohniska jsou hlášena ze Slovenska (Orava, Liptov), z Maďarska a z Chile [17, 18, 19, 20].
V této sérii případů se u dvou zemřelých v roce 2002 a v roce 2003 podařilo zjistit mutaci PRNP D178N, takže potenciální vztah k případům ze Slovenska – mutace E200K – nebyl potvrzen. Rovněž genetické vyšetření žijících příbuzných nemocných a jejich potenciální další sledování nebylo z etických důvodů provedeno.
Popsané ohnisko je výjimečné nahromaděním pravděpodobných případů familiární CJD. V okrese Nový Jičín je nejvyšší výskyt CJD v Moravskoslezském kraji a tento kraj má druhý nejvyšší výskyt CJD (po Praze) v České republice. To ovšem může být také způsobeno tím, jakou pozornost věnují lékaři na všech stupních diagnostického procesu tomuto onemocnění. Teoreticky bylo možné uvažovat o příhraniční migraci osob se Slovenskem, ale zjištěná mutace genu prionového proteinu tuto možnost nepotvrdila.
Jiné důvody tohoto zvýšeného výskytu nejsou známy a v současné době jsou již obtížně zjistitelné.
ZÁVĚR
V letech 2001–2011 bylo v okrese Nový Jičín hlášeno 8 případů onemocnění CJD. Z těchto osmi onemocnění byly tři hlášené jako familiární forma CJD. Během epidemiologického šetření bylo zjištěno dalších 7 pravděpodobných případů familiární formy CJD, histologicky však bylo možno prokázat u těchto sedmi osob pouze jeden případ. Z etických důvodů genetické vyšetření žijících příbuzných nemocných nebylo provedeno. U dvou zemřelých případů z roku 2002 a z roku 2003 byla zjištěna mutace D178N, která případný vztah k onemocněním hlášeným ze Slovenska s mutací E200K nepotvrdila.
Do redakce došlo dne 30. 6. 2015.
Adresa pro korespondenci:
MUDr. Jana Janoutová, Ph.D.
Ústav epidemiologie a ochrany veřejného zdraví
Syllabova 19
703 00 Ostrava-Zábřeh
e-mail: jana.janoutova@osu.cz
Sources
1. Mitrová E. Diagnostika, výskyt, liečba a prevencia Creutzfeldtovej-Jakobovej choroby. Bratislava: Herba; 2008, 72 s.
2. Araújo AQ. Prionic diseases. Arq Neuropsiquiatr, 2013;71(9B):731–737.
3. Lee J, Kim SY, Hwang KJ, Ju YR, Woo HJ. Prion diseases as transmissible zoonotic diseases. Osong Public Health Res Perspect, 2013;4(1):57–66.
4. Sikorska B, Liberski PP. Human prion diseases: from Kuru to variant Creutzfeldt-Jakob disease. Subcell Biochem, 2012;65 : 457–496.
5. Smetana J, Vacková M, Chlíbek R. Prionové nemoci. Epidemiol Mikrobiol Imunol, 2007;56(3): 112–118.
6. Will RG, Alperovitch A, Poser S, et al. Descriptive epidemiology of Creutzfeldt-Jakob disease in six European countries, 1993-1995. EU Collaborative Study Group for CJD. Ann Neurol, 1998;43(6):763–767.
7. Mitrová E. Genetická forma humánnych prionových chorôb. Neurol. pro praxi, 2006;2 : 76–79.
8. Rusina R, Matěj R. Prionová onemocnění. Neurol. praxi, 2012;13(2):78–82.
9. Lee SM, Chung M, Hwang KJ, et al. Biological network inferences for a protection mechanism against familial Creutzfeldt-Jakob disease with E200K pathogenic mutation. BMC Med Genomics;7 : 52. doi: 10.1186/1755-8794-7-52.
10. Gdovinová Z. Creutzfeldtova-Jakobova choroba. Cesk Slov Neurol N, 2013;76/109(2):138–154.
11. Matěj R, Rusina R, Koukolík F. 5 let činnosti Národní referenční laboratoře lidských prionových onemocnění při Oddělení patologie a molekulární medicíny FTNsP: naše zkušenosti a přehled literatury. Cesk Slov Neurol N, 2007; 70/103(6):637–642.
12. Rohan Z, Rusina R, Marešová V, et al. Lidská prionová onemocnění v České republice. Epidemiol Mikrobiol Imunol, 2015;64(3):115–120.
13. Frontzek K, Moos R, Schaper E, et al. Iatrogenic and sporadic Creutzfeldt-Jakob disease in two sisters without mutation in the prion protein gene. Prion, 2015 Dec 3 : 0. [Epub ahead of print].
14. Rohan Z, Parobková E, Johanidesová S, et al. Lidské prionové nemoci v České republice – 10 let zkušeností s diagnostikou. Cesk Slov Neurol N, 2013;76/109(3):300–306.
15. Kovács GG. MRI mozgu v diagnostike Creutzfeldtovej-Jakobovej choroby. Neurol pro praxi, 2007;3 : 149–151.
16. World Health Organization. WHO manual for surveillance of human transmissible spongiform encephalopathies including variant Creutzfeldt-Jakob disease [on line]. Geneva, c2003 [cit. 2015-12-15]. Dostupný na www:
< http://www.who.int/bloodproducts/TSE-manual2003.pdf>. ISBN 92 4 154588 7.
17. Maďar R, Maslenová D, Ranostajová K, et al. Analysis of Creutzfeldt-Jacob disease cases in Orava and Liptov regions (nothern Slovak focus) 1983–2000. Cent Eur J Public Health, 2003;11(1):19–22.
18. Korczyn AD, Chapman J, Goldfarb LG, et al. A mutation in the prion protein gene in Creutzfeldt-Jakob disease in Jewish patients of Libyan, Greek, and Tunisian origin. Ann N Y Acad Sci, 1991;640 : 171–176.
19. Kovács GG, Bakos A, Mitrova E, et al. Human prion diseases: the Hungarian experience. Ideggyogy Sz, 2007;60(11–12):447–452.
20. Brown P, Gálvez S, Goldfarb LG, et al. Familial Creutzfeldt-Jakob disease in Chile is associated with the codon 200 mutation of the PRNP amyloid precursor gene on chromosome 20. J Neurol Sci, 1992;112(1–2):65–67.
Labels
Hygiene and epidemiology Medical virology Clinical microbiologyArticle was published in
Epidemiology, Microbiology, Immunology

2016 Issue 2
Most read in this issue
- A report of 10 cases of familial Creutzfeld-Jakob disease
- Novel approaches to control the rise in pertussis cases
-
The occurrence of Ixodes ricinus ticks and important tick-borne pathogens in areas with high tick-borne encephalitis prevalence in different altitudinal levels of the Czech Republic
Part I. Ixodes ricinus ticks and tick-borne encephalitis virus - The effect of oxygen on endotoxin production in bacteria of the Bacteroides fragilis group isolated from patients with colorectal carcinoma