
Implementace a využití metody sekvenace celého genomu (WGS) v surveillance invazivního pneumokokového onemocnění, Česká republika, 2017–2019
Implementation and use of whole genome sequencing (WGS) in the surveillance of invasive pneumococcal disease, Czech Republic, 2017–2019
Aim: In order to improve the surveillance of invasive pneumococcal disease (IPD), the National Reference Laboratory (NRL) for Streptococcal Infections implemented whole genome sequencing (WGS) of Streptococcus pneumoniae. This article reports the first WGS data on S. pneumoniae isolates in the Czech Republic.
Material and Methods: Thirty-five isolates of S. pneumoniae from IPD recovered in 2017–2019 were selected for WGS. These were serotypes 4, 8, 9V, 19A, and 22F, which were determined by the Quellung reaction in combination with endpoint multiplex PCR (mPCR). Multilocus sequence typing (MLST) is routinely used for more detailed analysis termed sequence typing. The selected isolates were analysed by WGS on the Illumina MiSeq platform. The sequences obtained were processed using the Velvet de novo Assembler software. The assembled genomes were uploaded into the PubMLST database, using the BIGSdb platform, and then scanned automatically and molecularly characterized. The isolates were compared at three resolution levels: seven MLST genes, 53 ribosomal genes (rMLST), and 1420 genes (all loci). The all loci scheme covers MLST genes, ribosomal genes, and core genome MLST genes (cgMLST). These are all currently defined genes of S. pneumoniae available in the PubMLST database. Distance matrices based on the number and variability of all loci analysed were generated automatically using the Genome Comparator tool. Phylogenetic networks were created and edited with the SplitsTree4 package, using the NeighborNet algorithm. The final graphics were edited with the Inkscape software.
Results: Based on an overall view of the phylogenetic networks, it can be concluded that the genetic lines within each of S. pneumoniae serotypes 4, 8, 9V, 19A, and 22F are highly unrelated, to the same extent as if the isolates were of different serotypes. S. pneumoniae isolates of the same serotype, whether or not of the same sequence type, can be described, based on the results, as a non-homogeneous group with a number of unrelated genetic clusters that share genes assigning them to a specific serotype. WGS has also shown its discriminatory power, allowing the assignment of isolates of the same serotype and sequence type to different genetic clusters.
Conclusion: Of the methods used so far in the Czech Republic, WGS allows the most detailed characterization of S. pneumoniae isolates. It is highly desirable to integrate it in the molecular surveillance of IPD in the Czech Republic, similarly to other countries in Europe and in the world.
Keywords:
Streptococcus pneumoniae – MLST – serotype – WGS – whole genome sequencing – genomic surveillance – sequence type
Authors:
J. Kozáková
![]() ; M. Honskus
; M. Honskus
![]() ; Z. Okonji
; Z. Okonji
![]()
Authors‘ workplace:
Centrum epidemiologie a mikrobiologie, Státní zdravotní ústav, Praha
Published in:
Epidemiol. Mikrobiol. Imunol. 69, 2020, č. 3, s. 134-141
Category:
Original Papers
Overview
Cíl práce: Za účelem zkvalitnění surveillance invazivního pneumokokového onemocnění (IPO) zavedla Národní referenční laboratoř (NRL) pro streptokokové nákazy metodu celogenomové sekvenace (WGS) Streptococcus pneumoniae. Tato publikace prezentuje první výsledky WGS izolátů S. pneumoniae v České republice.
Materiál a metodika: Pro WGS bylo vybráno 35 izolátů S. pneumoniae z IPO z let 2017–2019. Jednalo se o sérotypy 4, 8, 9V, 19A a 22F, které byly určeny pomocí Quellung reakce v kombinaci s metodou end-point multiplexPCR (mPCR). K detailnější charakterizaci, určení sekvenačních typů, se rutinně v NRL používá multilokusová sekvenační typizace (MLST). U vybraných izolátů následovala WGS na platformě Illumina MiSeq. Získané sekvence byly upraveny softwarem Velvet de novo Assembler. Sestavené genomy byly vloženy do PubMLST databáze, využívající platformu BIGSdb, a následně automaticky skenovány a molekulárně charakterizovány. Dále proběhlo vzájemné porovnání izolátů na třech úrovních rozlišení: podle sedmi MLST genů, na podkladě 53 ribozomálních genů (rMLST) a porovnáním 1420 genů (“all loci“). Schéma “all loci“ zahrnuje MLST geny, ribozomální geny a geny core genom MLST (cgMLST). Jedná se o aktuálně všechny definované geny S. pneumoniae v PubMLST databázi. “Distance matrix“, založené na počtu a variabilitě alel všech analyzovaných lokusů, byly vygenerovány automaticky programem Genome Comparator. Fylogenetické sítě byly vytvořeny a editovány v programu SplitsTree4, který využívá algoritmus NeighborNet. Konečná grafická úprava proběhla programem Inkscape.
Výsledky: Při celkovém pohledu na fylogenetické sítě lze říci, že genetické linie uvnitř jednotlivých sérotypů (4, 8, 9V, 19A a 22F) jsou u S. pneumoniae vysoce nepříbuzné, a to v míře stejné, jako by se jednalo o izoláty rozdílných sérotypů. Izoláty S. pneumonie stejného sérotypu, ač shodných nebo rozdílných sekvenačních typů, můžeme dle výsledků popsat jako nehomogenní skupinu o určitém počtu nepříbuzných genetických klastrů, které společně sdílí geny, jež jsou zodpovědné za jejich zařazení ke konkrétnímu sérotypu. WGS demonstruje svou detailnost i tím, že umožnila rozdělit izoláty totožného sérotypu i sekvenačního typu do odlišných genetických klastrů.
Závěr: Metoda WGS přinesla doposud nejdetailnější charakterizaci izolátů S. pneumoniae z využívaných metod v České republice. Je velice žádoucí její zařazení do molekulární surveillance IPO v České republice, stejně jak probíhá již v některých jiných státech v Evropě i ve světě.
Klíčová slova:
Streptococcus pneumoniae – WGS – celogenomová sekvenace – genomová surveillance – sérotyp – sekvenační typ – MLST
ÚVOD
Streptococcus pneumoniae (S. pneumoniae) je příkladem vysoce invazivního grampozitivního extracelulárního bakteriálního patogenu, který způsobuje vysokou nemocnost a úmrtnost v Evropě i ve světě. Běžně se nachází na sliznici horních cest dýchacích, kde je součástí normální mikroflóry. V případě, že se vyskytuje pneumokok v primárně sterilním prostředí, způsobuje tzv. invazivní pneumokokové onemocnění (IPO), přičemž nejvyšší výskyt je zaznamenán u malých dětí a starších osob.
Od roku 2008 je v České republice (ČR) zaveden program surveillance IPO. Všechny případy IPO odpovídají platné evropské i české definici případu IPO [1]: závažné onemocnění s laboratorním průkazem pneumokoka z klinického materiálu, který je za normálních podmínek sterilní. Jedinou účinnou prevencí tohoto závažného onemocnění je vakcinace. Od roku 2010 je v ČR zavedeno doporučené a hrazené očkování dětí pneumokokovými konjugovanými vakcínami (PCV). Od roku 2018 je očkování vakcínou PCV13 rozšířeno pro pacienty se zdravotní indikací i pro věkovou skupinu seniorů 65 a starších bez poplatku. Podkladem účinné vakcinační strategie je provádění kvalitní surveillance IPO. Kvalita surveillance IPO je výrazně zlepšována zaváděním molekulární charakterizace izolátů S. pneumoniae. Podle variant polysacharidového obalu lze nyní určit více než 90 sérotypů této bakterie. Určení sérotypu S. pneumoniae je nejdůležitější krok a probíhá v Národní referenční laboratoři pro streptokokové nákazy (NRL) kombinací sérologické Quellung reakce a end-point multiplexové polymerázové řetězové rekce (mPCR) [2]. Poté následuje proces klonální charakterizace, metodou multilokusové sekvenační typizace (Multilocus Sequence Typing, MLST), která je založená na amplifikaci a sekvenaci oblastí souboru genů základních metabolických struktur [3]. V současné době NRL testuje odklon od MLST metody a využívání sekvenace celého genomu (WGS). Zařazení WGS metody do molekulární surveillance infekčních onemocnění je doporučeno Evropským centrem pro kontrolu infekčních onemocnění (ECDC) a má v Evropě v posledních letech významný vzestupný trend [4].
Data surveillance IPO jsou publikována ve Zprávách Centra epidemiologie a mikrobiologie, Státního zdravotního ústavu [5] a jsou každoročně zasílána do ECDC TESSy – The European Surveillance System [6]. Sekvenační data jsou předkládána do mezinárodní databáze PubMLST (Public databases for molecular typing and microbial genome diversity) [7].
NRL v rámci řešení výzkumného projektu zavedla v roce 2019 metodu WGS s cílem její implementace do celorepublikové surveillance IPO. Tato práce prezentuje první použití metody WGS v molekulární surveillance IPO v České republice
MATERIÁL A METODY
Bakteriální izoláty Streptococcus pneumoniae
Byl vybrán soubor 35 izolátů S. pneumoniae z IPO z let 2017–2019. Jednalo se o sérotypy 4, 8, 9V, 19A a 22F. Izoláty byly uchovány v mrazicím boxu při teplotě -80 °C (KryobankaB, Itest s.r.o.). Kultivace probíhala v CO2 atmosféře ve 37 °C společně s optochinovým diskem a v kombinaci s testem rozpustnosti ve žluči. Určení sérotypů S. pneumoniae bylo provedeno pomocí Quellung reakce v kombinaci s reakcí end-point multiplexPCR (mPCR). K detailnější charakterizaci, určení sekvenačních typů, byla použita multilokusová sekvenační typizace (MLST).
Extrakce DNA
Z izolátů byla izolována deoxyribonukleová kyselina (DNA) pomocí izolačního kitu QIAamp DNA Mini Kit (QIAGEN). Postup izolace probíhal podle pokynů výrobce [8].
Celogenomová sekvenace a úprava dat
Celogenomová sekvenace izolátů S. pneumoniae proběhla na pracovišti EMBL (European Molecular Biology Laboratory, Heidelberg, Německo) s využitím platformy Illumina MiSeq. Výsledné překrývající se sekvence o délce cca 300 bp byly následně v naší laboratoři zpracovány pomocí softwaru Velvet de novo Assembler [9]. Tento proces byl optimalizován pomocí skriptu Velvet-Optimiser script. Průměrná hodnota parametru “K-mer lenght“ se pohybovala okolo 147 (minimum = 97, maximum = 173). Sestavené genomy byly vloženy do PubMLST databáze [7], která využívá platformy BIGSdb (Bacterial Isolate Genome Sequence Database) [10], pod následujícími IDs: 51320 – 51354.
Analýza a vizualizace WGS dat
Nahrané genomové kontigy jednotlivých izolátů byly v PubMLST skenovány automaticky a následně charakterizovány alelovým profilem 53 ribozomálních genů (rpsA – rpsU, rplA – rplF, rplI – rplX, rpmA – rpmJ) a 7 MLST genů (aroE, ddl, gdh, gki, recP, spi, xpt). Celkový počet ribozomálních alel je 55, jelikož gen rpmG se u S. pneumoniae vyskytuje ve třech kopiích. Na základě alelových variant MLST genů byla u jednotlivých izolátů určena příslušnost k sekvenačnímu typu (ST) [11]. Profil alelových variant ribozomálních genů (rMLST) následně poskytl ribozomální profil izolátů (rST) [12]. Nové alely byly skenovány manuálně a nahrány do databáze PubMLST. Po anotaci jim bylo díky automatickému vkládacímu nástroji platformy BIGSdb přiděleno číselné označení.
Tvorba fylogenetických sítí proběhla pomocí programu Genome Comparator, který je součástí PubMLST databáze. Izoláty byly vzájemně porovnány na třech úrovních rozlišení: na úrovni MLST genů (7 lokusů), rMLST genů (53 lokusů) a s využitím schématu “all loci“ (1 420 lokusů). “Distance matrix“, založené na počtu a variabilitě alel všech analyzovaných lokusů, byly vygenerovány automaticky programem Genome Comparator. Vlastní fylogenetické sítě byly vytvořeny a editovány v programu SplitsTree4, který využívá algoritmus NeighborNet. Konečná grafická úprava proběhla programem Inkscape.
VÝSLEDKY A DISKUSE
Ribozomální profil, který je v PubMLST databázi popsaný a tedy již spojený s příslušným číselným označením, mělo 22 z 35 izolátů našeho výběru (62,9 %). Ostatní izoláty nesly buď neznámou kombinaci již popsaných alelových variant (n = 6), nebo obsahovaly jednu či více mutovaných alel, které nebyly dosud popsány (n = 4). Ve třech případech se pak jednalo o kombinaci obou těchto jevů. Nové alelové varianty byly zjištěny celkem u osmi různých ribozomálních genů (rpsC, rpsD, rpsT, rplB, rplD, rplE, rplW, rpmC). Do PubMLST databáze bylo zařazeno 12 nepopsaných ribozomálních profilů rST, zjištěných v námi studovaném souboru (tab. 1).
Table 1. Molecular characterization of 35 S. pneumoniae isolates from IPD, 2017–2019, Czech Republic
*newly described ribosomal profile
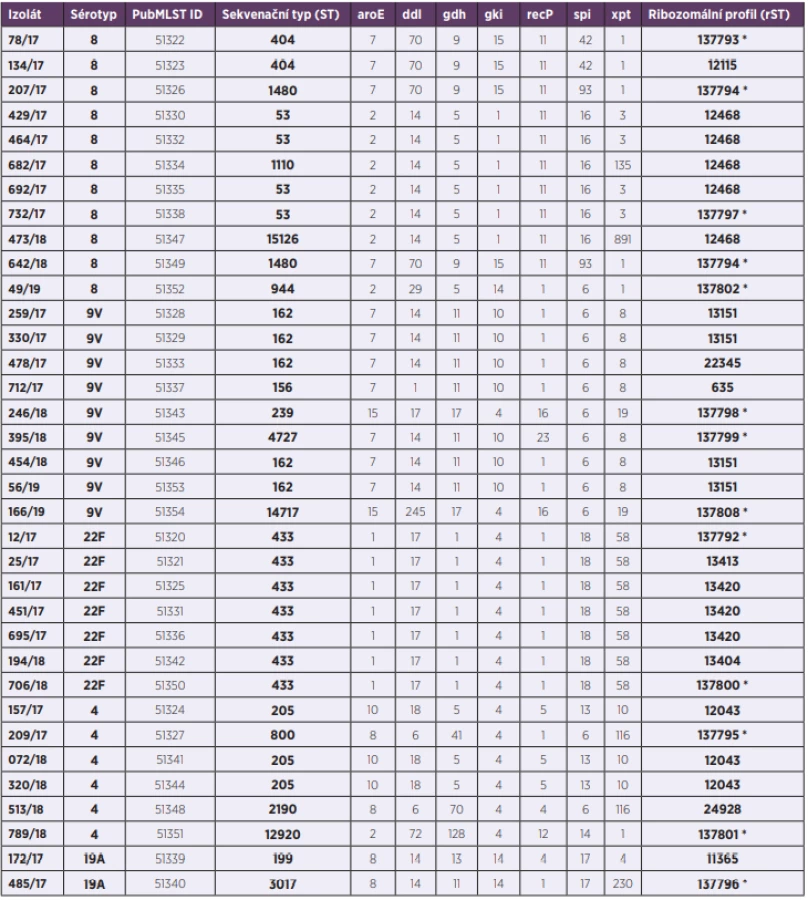
MLST (obr. 1)
Figure 1. Visualisation of genetic diversity according to the MLST scheme of 35 S. pneumoniae isolates from IPD, 2017–2019, Czech Republic
První fylogenetická síť znázorňuje genetické vztahy izolátů S. pneumoniae na základě genetické variability MLST genů.
Izoláty nejpočetnějšího sérotypu 8 jsou zastoupeny dvěma hlavními, geneticky odlišnými, liniemi. Do první patří čtyři izoláty sekvenačního typu ST-53, spolu s dvěma izoláty ST-1110 a ST-15126, které se liší od ST-53 pouze v jednom MLST genu (xpt). Druhou genetickou linii tvoří dva izoláty ST-404 a dva izoláty ST-1480. Tato ST se od sebe odlišují opět změnou alely v jednom MLST genu (spi). Ačkoliv obě tyto skupiny izolátů patří ke stejnému sérotypu, jejich MLST profily jsou značně odlišné. Jedinou alelou MLST genu, kterou tyto dvě skupiny sdílí, je alela 11 genu recP. Posledním izolátem, patřícím k sérotypu 8, je izolát 49/19 (ST-944) ležící samostatně a jeho MLST profil je unikátní. Můžeme zde pozorovat tři alelové varianty, které se objevují u izolátů dvou hlavních genetických skupin sérotypu 8 – aroE alela 2, gdh alela 5 (skupina izolátů ST-53) a xpt alela 1 (skupina izolátů ST-404 a ST-1480). V alelových variantách ostatních MLST genů (ddl, gki, recP a spi) se tento izolát liší.
Sedm z devíti izolátů sérotypu 9V tvoří na fylogenetické síti zřetelně ohraničenou, geneticky blízkou, hlavní skupinu. Jedná se o pět izolátů ST-162, spolu s izolátem 712/17 (ST-156) a izolátem 395/18 (ST-4727). Izoláty ST-156 a ST-4727 se liší od izolátů ST-162 v jednom MLST genu. V případě izolátu ST-156 je to gen ddl, izolát ST-4727 nese odlišnou alelovou variantu genu recP. Zbylé dva izoláty sérotypu 9V (246/18 – ST-239 a 166/19 – ST-14717) se nachází zcela odděleně od hlavní skupiny. Tomu odpovídá i jejich MLST profil, který je ve většině genů odlišný. Vzájemně se tyto dva izoláty mezi sebou liší v alelové variantě genu ddl. Jediným genem, který se u všech izolátů sérotypu 9V vyskytuje ve stejné alelové variantě, je gen spi (alela 6).
Naprostou homogenitu vykazují izoláty sérotypu 22F. Jelikož všechny tyto izoláty patří k sekvenačnímu typu ST-433, jsou na fylogenetické síti znázorněny jedním společným bodem.
Izoláty sérotypu 4 tvoří dvě hlavní geneticky odlišné skupiny a jeden samostatně ležící izolát. První skupinou jsou tři izoláty ST-205, druhou tvoří izoláty 209/17 (ST-800) a 513/18 (ST-2190). Tyto dva izoláty se vzájemně liší ve dvou MLST genech (gdh a recP) a vykazují vzdálenou příbuznost k izolátům sérotypů 9V (246/18 a 166/19) a 22F. Izolát 789/18 (ST-12920) má zcela unikátní kombinaci MLST alel a leží zcela samostatně. Jedinou společnou alelou MLST genů u izolátů sérotypu 4 je alela 4 genu gki.
Izoláty sérotypu 19A (ST-199 a ST-3017) leží na fylogenetické síti v blízkosti hlavní skupiny izolátů sérotypu 9V (ST-162; ST-156; ST-4727), a vykazují tak vyšší příbuznost k těmto izolátů než například k izolátům sérotypu 8 nebo 22F. MLST profily izolátů sérotypu 19A se vzájemně shodují ve 4 MLST genech (aroE, ddl, gki a spi).
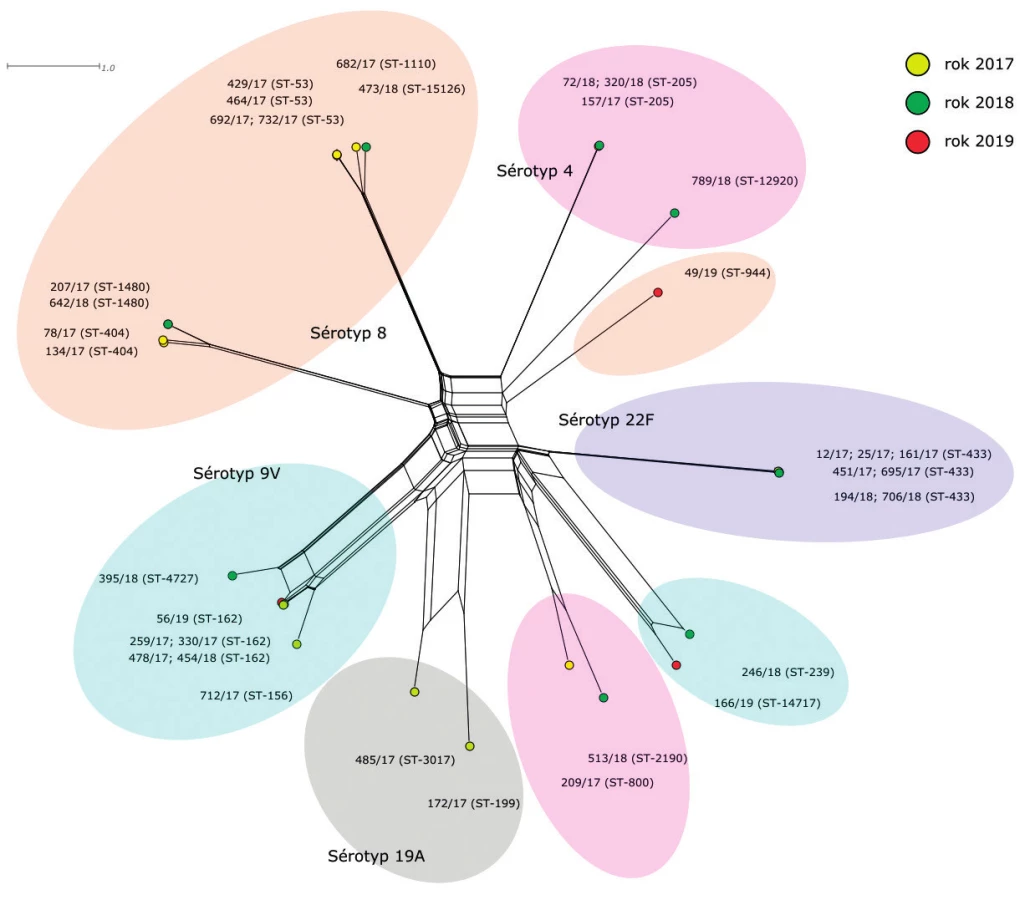
rMLST (obr. 2)
Figure 2. Visualisation of genetic diversity according to the rMLST scheme of 35 S. pneumoniae isolates from IPD, 2017–2019, Czech
Republic
Druhá fylogenetická síť znázorňuje genetické vztahy izolátů S. pneumoniae na základě srovnání variability jejich ribozomálních genů (rMLST).
Pozice izolátů sérotypu 8 je podobná jako na předchozí fylogenetické síti. Opět jsou zjištěny dvě hlavní genetické linie sérotypu 8 a izolát ST-944, který je geneticky vysoce vzdálený ostatním. První linie opět obsahuje 6 izolátů sekvenačních typů ST-53, ST-1110 a S T-15126, které s jednou výjimkou sdílí stejné alely všech 53 rMLST genů a jejich ribozomální profil je tedy identický rST-12468. Zmíněnou výjimku tvoří izolát 732/17 (ST-53), který se od ostatních liší v alele genu rpsA a jeho ribozomální profil je nově popsaný rST-137797. Zatímco izoláty ribozomálního profilu rST-12468 nesou v genu rpsA alelu 113, u izolátu 732/17 se nachází alela 2506, která se od původní alely 113 liší jednonukleotidovou substitucí. Druhá hlavní genetická linie sérotypu 8 obsahuje dvě sublinie, které se vzájemně liší v alelách sedmi rMLST genů (rpsD, rplL, rplJ, rplS, rplW, rpmE, rpmG). Sublinie ST-404 je zastoupena dvěma izoláty. Izolát 134/17 s ribozomálním profilem rST-12115 a izolát 78/17, který má nově popsanou kombinaci alel ribozomálních genů (rST-137793). Rozdíl tvoří jednonukleotidová substituce v genu rpsP, která mění původní alelu 26 (rST-12115) na alelu 749. Druhou sublinii tvoří dva izoláty ST-1480 se shodným, nově popsaným, ribozomálním profilem rST-137794. Oba izoláty nesou novou alelu genu rplW, která vznikla identickou jednonukletidovou substitucí z alely 54. Poslední izolát sérotypu 8 (49/19, ST-944) je svým profilem ribozomálních genů zcela odlišný od ostatních izolátů stejného sérotypu a jeho pozice na fylogenetické síti značí příbuznost ke dvěma izolátům sérotypu 9V (246/18 a 166/19). Jeho nově popsaný ribozomální profil rST-137802 nese 14 alel, které se u ostatních izolátů sérotypu 8 nevyskytují, a novou alelu genu rplE.
Pozice izolátů sérotypu 9V je podobná jako na předchozí fylogenetické síti. Hlavní linii tvoří sedm izolátů ST-162, ST-156 a ST-4727. Čtyři z nich (ST-162) sdílí stejný ribozomální profil rST-13151 a tvoří proto jediný bod. K těmto izolátům vykazují vysokou příbuznost izoláty 712/17 (rST-635) a 478/17 (rST-22345), které se od ribozomálního profilu rST-13151 shodně liší v alelách 4 genů (rpsA, rplJ, rplL, rplS). Izolát 478/17 nese navíc odlišnou alelu genu rpmA. Posledním izolátem hlavní linie sérotypu 9V je izolát 395/18 (ST-4727), který se od ostatních liší už podstatně více. Jeho ribozomální profil rST-137799 je velmi unikátní, podle dat PubMLST databáze se liší od nejpodobnějšího profilu v devíti různých alelách ribozomálních genů, což je neobvykle vysoké číslo. Zbylé dva izoláty sérotypu 9V (246/18 a 166/19) leží samostatně v geneticky specifické linii (spolu s izolátem 49/19 sérotypu 8). Oba nesou nově popsané ribozomální profily (rST-137798 a rST-137808) a vzájemně se liší v alelách 4 ribozomálních genů (rpsA, rpsP, rplI, rpmG).
Izoláty sérotypu 22F (ST-433) se na základě alelových variant ribozomálních genů rozdělily do dvou geneticky odlišných klastrů. První klastr obsahuje tři izoláty s identickými ribozomálními profily rST-13420 (161/17, 451/17, 695/17) a izolát 706/18 (rST-137800), který se od nich liší novou alelovou variantou genu rpsD. Tato alela se liší od původní alely 2042 (rST-13420) jednonukleotidovou substitucí. Druhý klastr obsahuje tři izoláty (12/17, 25/17, 194/18), jejichž ribozomální profily se mezi sebou liší vždy jednou alelou. Izolát 25/17 (rST-13413) se od izolátu 194/18 (rST-13404) liší alelou 49 genu rpsI. Izolát 12/17, který má nově popsaný ribozomální profil rST-137792, nese alelu 6333 v genu rplS.
Izoláty sérotypu 4 tvoří dvě geneticky nepříbuzné linie. Změna nastala u izolátu 789/18 (ST-12920), který vykazuje příbuznost s izoláty sekvenačních typů ST-800 a ST-2190 a jeho pozice na fylogenetické síti již není zcela samostatná. Příbuznost izolátů této linie sérotypu 4 je nicméně mnohem nižší než mezi většinou ostatních linií, což se obrazí i v ribozomálních profilech těchto izolátů. Společně sdílí pouze 29 alel ribozomálních genů. Izoláty 209/17 a 789/18 nesou nově popsané ribozomální profily, izolát 209/17 (rST-137795) se liší od nejpodobnějšího známého profilu třemi alelami (rpsL, rpsP, rplA) a izolát 789/18 (rST-137801) sedmi alelami (rpsF, rpsG, rpsL, rpsT, rplA, rplC, rplD). V případě genů rpsT a rplD se pak jedná o nově identifikované alely. Izolát 513/18 nese ribozomální profil rST-24928. Druhou linii sérotypu 4 tvoří tři identické izoláty ST-205, rST-12043.
Izoláty sérotypu 19A leží na ribozomální fylogenetické síti již zcela samostatně a jsou vzájemně blízce příbuzné. Izolát 485/17 (rST-137796) se od izolátu 172/17 (rST-11365) liší na úrovni ribozomálních genů pouze novou alelou genu rpmC, která vznikla z původní alely 14 jednonukleotidovou substitucí.
“All loci“ (obr. 3)
Figure 3. Visualisation of genetic diversity according to the all loci scheme of 35 S. pneumoniae isolates from IPD, 2017–2019, Czech Republic
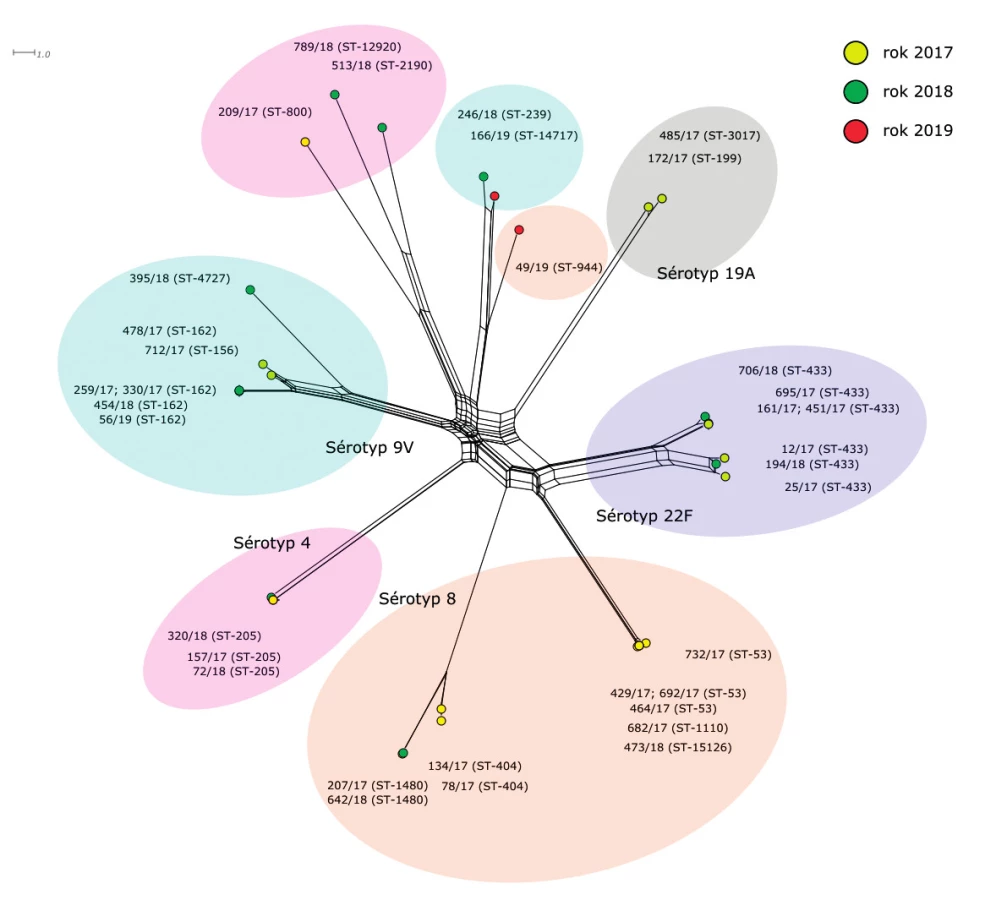
Třetí fylogenetická síť je zkonstruována na základě srovnání 1420 genů. Schéma “all loci“ zahrnuje MLST geny, ribozomální geny a geny, tvořící schéma core genom MLST (cgMLST) pro S. pneumoniae.
Izoláty sérotypu 8 tvoří na fylogenetické síti tři linie, které jsou vzájemně geneticky vysoce vzdálené. První linie je tvořena dvěma téměř identickými izoláty sekvenačního typu ST-1480 a dvěma izoláty ST-404, které jsou méně příbuzné. Druhá linie je tvořena šesti geneticky blízkými izoláty (ST-53, ST-1110, ST-15126). Mezi nimi lze pozorovat klastr tří vysoce příbuzných izolátů 692/17 (ST-53), 732/17 (ST-53) a 682/17 (ST-1110), izolát 473/18 (ST-15126), který patří do stejného klastru, ale jeho příbuznost je nižší, a dva variabilnější izoláty ST-53 (429/17 a 464/17), které tvoří druhý klastr. Třetí linii tvoří samostatně ležící izolát 49/19 (ST-944).
Sérotyp 9V se rovněž skládá ze dvou geneticky nepříbuzných linií. V první linii je pět izolátů sekvenačního typu ST-162. Čtyři z nich jsou prakticky identické (259/17, 330/17, 454/18 a 56/19), pátý je pak, navzdory stejnému sekvenačnímu typu, geneticky významně vzdálenější (478/17). Do první linie se dále řadí izoláty 712/17 (ST-156) a 395/18 (ST-4727), mezi kterými je vzájemná příbuznost zhruba na stejné úrovni jako jejich příbuznost k ostatním izolátům této linie. Zajímavý je fakt, že dva izoláty sérotypu 19A vykazují k izolátům první linie sérotypu 9V vyšší příbuznost než izoláty druhé linie stejného sérotypu. Tato příbuznost je nicméně velice vzdálená. Dva izoláty druhé linie 9V leží na fylogenetické síti samostatně a vzájemně se liší v cca 100 genech.
Navzdory příslušnosti ke stejnému sekvenačnímu typu (ST-433) zůstaly izoláty sérotypu 22F i při srovnání na základě 1420 genů rozděleny do dvou zřetelně oddělených klastrů, které mezi sebou mají rozdíl zhruba 150 genů. První klastr je tvořen čtyřmi vysoce příbuznými izoláty 161/17, 451/17, 695/17, 706/18 a druhý klastr obsahuje tři podobně příbuzné izoláty 12/17, 25/17, 194/18.
Rovněž sérotyp 4 tvoří dvě geneticky vzdálené linie. První je tvořena třemi izoláty ST-205, které jsou geneticky prakticky stejné. Uvnitř druhé linie můžeme pozorovat výjimečně vysokou variabilitu. Je tvořena dvěma vzdáleně příbuznými izoláty 209/17 (ST-800), 513/18 (ST-2190) a ještě méně příbuzným 789/18 (ST-12920).
Výčet izolátů uzavírají dva izoláty sérotypu 19A (172/17, 485/17), které tvoří samostatně stojící linii a vykazující poměrně značnou genetickou blízkost.
Při celkovém pohledu na fylogenetické sítě lze říci, že genetické linie vybraných izolátů uvnitř jednotlivých sérotypů (4, 8, 9V, 19A a 22F) jsou u S. pneumoniae vysoce nepříbuzné, a to v míře stejné, jako by se jednalo o izoláty rozdílných sérotypů. Izoláty S. pneumonie stejného sérotypu, ač shodných nebo rozdílných sekvenačních typů, můžeme podle výsledků popsat jako nehomogenní skupinu o určitém počtu nepříbuzných genetických klastrů, které společně sdílí geny zodpovědné za jejich zařazení ke konkrétnímu sérotypu. Kromě genů určujících sérotypy byla u jednotlivých genetických linií izolátů stejných sérotypů nalezena velice nízká příbuznost. Genetické linie, které pozorujeme uvnitř jednotlivých sérotypů, jsou tvořeny buď izoláty stejného ST (př. sérotyp 4 = ST-205) nebo skupinou několika příbuzných ST (př. sérotyp 8 = ST-404 a ST-1480). Díky WGS se podařilo oddělit dva zřetelné klastry izolátů sérotypu 22F o totožném ST-433. Liší se již na úrovni ribozomálních genů, kde klastrují a vytváří dvě vzájemně bližší skupiny. Toto potvrdila i analýza na úrovni schématu “all loci“.
Klonální charakterizaci pneumokoků je možno provádět kromě MLST a WGS i metodou MLVA (Multiple-Locus Variable number tandem repeat Analysis). Tato metoda není rutinně používána, ale lze ji například použít jako doplňkovou pro zrychlené zjištění klonální příbuznosti izolátů ze situací lokálního charakteru [13]. Určování sérotypů S. pneumoniae je prováděno metodou Quellung, která je celosvětově považována za zlatý standard pro typizaci pneumokoků. Tato metoda je však nákladná finančně i časově, a jsou proto hledány molekulární metody, které by ji mohly nahradit. Srovnání metod sekvenační multiplexové PCR a WGS ukázalo, že pro určení sérotypů S. pneumoniae je úspěšnější metoda WGS [14].
Metoda WGS umožňuje detailnější fylogenetickou analýzu pneumokoků, která je velmi důležitá zejména v postvakcinačním období a umožňuje zjišťovat, zda nedochází vlivem selekčního tlaku k rozšíření nových fylogenetických linií nevakcinačních sérotypů. Například v Kanadě byl po implementaci PCV13 pneumokokové vakcíny zjištěn vzestup nevakcinačního sérotypu 22F, který byl metodou WGS charakterizován jako homogenní klonální komplex ST-433 [15]. Stejnou homogenitu S. pneumoniae ST-433 jsme prokázali v souboru námi studovaných izolátů sérotypu 22F. Důležitost metody WGS v detekci nových sekvenačních typů v postvakcinačním období prokázala nosičská studie v UK, kde byly díky této metodě zjištěny dosud nepopsané sekvenační typy u nevakcinačních sérotypů S. pneumoniae. Data WGS ukázala, že replacement sérotypů byl nejčastěji způsoben změnou genotypu [16]. Implementace WGS do molekulární surveillance IPO dává detailní podklady pro zhodnocení situace v postvakcinačním období a následnou aktualizaci vakcinační strategie.
ZÁVĚR
Tento článek prezentuje první české izoláty S. pneumoniae, které byly podrobeny sekvenaci celého genomu. Metoda WGS přinesla doposud nejdetailnější charakterizaci izolátů S. pneumoniae z využívaných metod v České republice. Velice obohacuje dosavadní charakterizaci izolátů S. pneumoniae z rutinně získávaných 7 genů až na 1420 genů. Je velice žádoucí zařazení WGS do molekulární surveillance IPO v České republice, stejně jak probíhá již v některých jiných státech.
Poděkování
Podpořeno z programového projektu Ministerstva zdravotnictví ČR s reg. č. 17-29256A, 2017-2020. Veškerá práva podle předpisů na ochranu duševního vlastnictví jsou vyhrazena.
Do redakce došlo dne 26. 5. 2020.
Adresa pro korespondenci:
MUDr. Jana Kozáková
Státní zdravotní ústav Praha
Šrobárova 48
100 42 Praha 10
e-mail: jana.kozakova@szu.cz
Sources
1. Vyhláška č. 275/2010 Sb., kterou se mění vyhláška č. 473/2008 Sb., o systému epidemiologické bdělosti pro vybrané infekce, příloha č. 21 invazivní pneumokoková onemocnění.
2. Vacková Z, Klímová M, Kozáková J. Nová metoda a schéma typi-zace Streptococcus pneumoniae. Epidemiol Mikrobiol Imunol, 2013;62(2):50–58.
3. Enright MC, Spratt BG. A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology, 1998;144 : 3049–3060.
4. Revez J, Espinosa L, Albiger B, et al. Survey on the Use of Whole-Genome Sequencing for Infectious Diseases Surveillance: Rapid Expansion of European National Capacities, 2015–2016. Front Public Health, 2017;5 : 347. DOI: 10.3389/fpubh.2017.00347.
5. Kozáková J, Okonji Z, Šebestová H, Klímová M, Křížová P. Invazivní pneumokokové onemocnění v České republice v roce 2018. Zprávy CEM (SZÚ, Praha), 2019;28(7):277–282.
6. https://tessy.ecdc.europa.eu/tessyweb.
7. https://pubmlst.org/spneumoniae/.
8. Manuál QIAamp DNA Mini Kit. Dostupné na www: http://www.qiagen.com/Products/Catalog/Sample-Technologies/DNA-Sample-Technologies/Genomic-DNA/QIAamp-DNA-Mini-Kit#technicalspecification
9. Zerbino DR. Using the Velvet de novo assembler for short-read sequencing technologies. Curr Protoc Bioinformatics, 2010;11(11.5). DOI: 10.1002/0471250953.bi1105s31.
10. Jolley KA, Maiden MCJ. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics, 2010;11(595). DOI: 10.1186/1471-2105-11-595.
11. Maiden MCJ, Bygraves JA, Feil E, et al. Multilocus sequence ty-ping: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A, 1998;95(6):3140–3145.
12. Jolley KA, Bliss CM, Bennett JS, et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology, 2012;158(4):1005-15. DOI: 10.1099/mic.0.055459-0.
13. Kozáková J, Okonji Z, Musílek M. Klonální charakterizace kmenů Streptococcus pneumoniae metodami MLST a MLVA – může metoda MLVA charakterizaci zkvalitnit? Epidemiol Mikrobiol Imunol, 2020;69(1):20–28.
14. Mauffrey F, Fournier É, Demczuk W, et al. Comparison of sequential multiplex PCR, sequetyping and whole genome sequencing for serotyping of Streptococcus pneumoniae. PLoS One, 2017;12(12):e0189163. DOI: 10.1371/journal.pone.0189163.
15. Demczuk WHB, Martin I, Hoang L, Van Caeseele P, et al. Phylogenetic analysis of emergent Streptococcus pneumoniae serotype 22F causing invasive pneumococcal disease using whole genome sequencing. PLoS One, 2017;12(5):e0178040. DOI: 10.1371/journal.pone.0178040.
16. Sheppard CL, Groves N, Andrews N, et al. The Genomics of Streptococcus Pneumoniae Carriage Isolates from UK Children and Their Household Contacts, Pre-PCV7 to Post-PCV13. Genes (Basel), 2019;10(9):687. DOI: 10.3390/genes1009068
Labels
Hygiene and epidemiology Medical virology Clinical microbiologyArticle was published in
Epidemiology, Microbiology, Immunology

2020 Issue 3
Most read in this issue
- Epidemiologie vankomycin-rezistentních enterokoků ve Fakultní nemocnici Hradec Králové v roce 2017
- Koutule skvrnitá – Clogmia albipunctata (Diptera: Psychodidae) – muška s epidemiologickým potenciálem a rizikem myiáz
- Délka vylučování SARS-CoV-2 u pacientů uzdravujících se z COVID-19
- Séroprevalence IgG protilátek proti spalničkám u zaměstnanců Nemocnice Strakonice, a.s.