
Postižení gastrointestinálního traktu u dědičných chorob ledvin
Gastrointestinal tract involvement in hereditary kidney diseases
Hereditary kidney diseases are an important and specific group of kidney diseases having often systemic and multiorgan characteristics and a tendency to progress to chronic renal failure. The genetic background of the majority of hereditary nephropathies has been reported, an important step has been made to uncover their etiology and pathogenesis, and recently new innovative therapies have been proposed. Gastrointestinal tract (GIT) involvement in hereditary kidney diseases presents in different forms, extents, and intensities. Though only a minor part of hereditary kidney disease, GIT involvement is an integral component of the clinical picture; for example, liver cysts are an important component of autosomal dominant polycystic kidney disease. Even mild symptoms of GIT involvement can be an initial and potentially serious extrarenal manifestation of hereditary nephropathy, which deserves early diagnosis and initiation of treatment.
Keywords:
gastrointestinal tract involvement – hereditary kidney diseases – polycystic kidney disease – renal cysts – liver cysts – congenital hepatic fibrosis – molecular biology methods
The Editorial Board declares that the manuscript met the ICMJE „uniform requirements“ for biomedical papers.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
Submitted: 7. 9. 2018
Accepted: 21. 9. 2018
Authors:
M. Merta
Authors‘ workplace:
Ústav biologie a lékařské genetiky, 1. LF UK a VFN Praha
Published in:
Gastroent Hepatol 2018; 72(5): 441-448
Category:
doi:
https://doi.org/10.14735/amgh2018441
Overview
Dědičná onemocnění ledvin jsou významnou a specifickou skupinou ledvinných onemocnění, vyznačující se často tendencí k progresi do stadia chronického selhání a systémovým a multiorgánovým charakterem onemocnění. Byl odhalen genový podklad většiny dědičných nefropatií, byl učiněn významný pokrok při výzkumu jejich etiologie a patogeneze a recentně byly zavedeny inovativní postupy jejich léčby. Postižení gastrointestinálního traktu (GIT) u dědičných chorob ledvin může mít různou podobu, rozsah a význam. Ačkoli jen v menšině případů tvoří postižení GIT nedílnou součást klinického obrazu dědičného onemocnění ledvin jako např. cystické postižení jater u polycystické choroby ledvin autozomálně dominantního typu, mohou být i méně časté či výrazné projevy postižení GIT prvním či závažným extrarenálním projevem dědičné nefropatie, které vyžaduje včasné stanovení diagnózy a léčbu.
Klíčová slova: postižení zažívacího traktu – dědičné choroby ledvin – polycystická choroba ledvin – ledvinné cysty – jaterní cysty – kongenitální jaterní fibróza – metody molekulární biologie
Úvod
Dědičná onemocnění ledvin tvoří významnou a v řadě ohledů specifickou skupinu onemocnění ledvin. V dětském věku jsou tyto choroby nejčastější příčinou chronického selhání ledvin (CHSL), a ačkoli jejich podíl s věkem klesá, jsou dědičné choroby ledvin v dospělosti příčinou CHSL asi v 10–15 %, přičemž za více než polovinu odpovídá polycystická choroba ledvin autozomálně dominantního typu (PCHLAD). Rozvoj molekulárně genetických metod umožnil definovat velkou většinu známých dědičných onemocnění ledvin z hlediska jejich genového podkladu a přenosu dědičné informace. V současnosti je známo přes 200 nejdůležitějších dědičných onemocnění s monogenním typem dědičnosti, považovaných za primárně ledvinné, u kterých je znám typ dědičnosti, jejich kódující gen a případně genový produkt. Výrazně se rozšířily možnosti stanovení diagnózy pomocí metod DNA diagnostiky, prohloubily se naše poznatky etiologie a patogeneze těchto onemocnění a v některých případech se odkryly možnosti cílené léčby. Postižení gastrointestinálního traktu (GIT) může hrát významnou roli u některých dědičných onemocnění ledvin. Jelikož má značná část dědičných chorob ledvin povahu systémového, případně multi-orgánového onemocnění a nikoli pouze izolovaného postižení ledvin, může být postižení GIT běžnou či méně běžnou součástí dědičné nefropatie. Typickým příkladem pravidelného postižení GIT (konkrétně jater) je tvorba cyst v játrech u PCHLAD, případně postižení jater hepatální fibrózou (HF) u polycystické choroby ledvin autozomálně recesivního typu (PCHLAR), méně časté či závažné postižení GIT lze pozorovat u mnoha dalších dědičných nefropatií (u tuberózní sklerózy (TS), u Fabryho choroby (FCH) aj. Vztah mezi renálním postižením a postižením GIT v rámci dědičného onemocnění může mít také odlišnou a komplikovanější podobu, jako např. u primární hyperoxalurie (PH), jejíž podstatou je defektní enzymatická tvorba v játrech vedoucí k nadměrné hyperoxalurii, která se klinicky projevuje především postižením ledvin, avšak sekundárně i vývojem systémové oxalózy s poškozením dalších orgánů. Možné řešení této situace představuje transplantace obou orgánů (ledvin a jater), která zajistí náhradu funkce selhaných ledvin a jater s defektní tvorbou enzymu [1]. Specifickou roli může hrát postižení GIT u atypického hemolyticko-uremického syndromu (aHUS). Syndrom aHUS představuje vzácnou vrozenou a dědičnou poruchu komplementového systému, vyznačující se poškozením endotelu, hemolýzou, trombotickým poškozením drobných cév a poškozením ledvin. Některé akutní formy GIT (pankreatitida, akutní průjmy) se mohou stát spouštěcím mechanizmem vedoucím k akutní a nekontrolované aktivaci komplementového systému a rozvoje aHUS [2].
Přehled postižení GIT u jednotlivých dědičných onemocnění ledvin
Postižení GIT u polycystické choroby ledvin
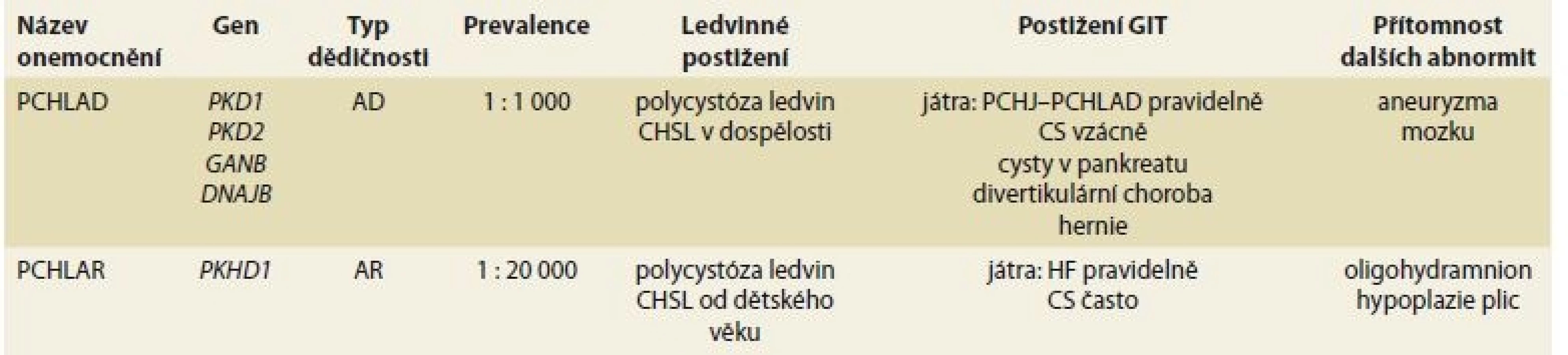
Termín polycystická choroba ledvin zahrnuje dvě nozologické jednotky: PCHLAD a PCHLAR. Obě choroby mají některé společné rysy (především vývoj cyst v ledvinách), avšak v řadě dalších rysů se od sebe liší – typem dědičnosti, typickou dobou vzniku klinických projevů, četností výskytu a v neposlední řadě také typem extrarenálních projevů onemocnění (tab. 1).
Tab. 1. Involvement of GIT in PCHAD and PCHLAR.
Polycystická choroba ledvin autozomálně dominantního typu
PCHLAD je multisystémové onemocnění projevující se obvykle klinicky v dospělém věku a vyznačující se tvorbou cyst v ledvinách, játrech a zvýšeným rizikem vzniku mozkových aneuryzmat. Další méně běžné projevy zahrnují vznik cyst v některých orgánech (v pankreatu, v seminálních veziklech a arachnoidální membráně), kardiovaskulární a cévní abnormity (dilatace kořene aorty, disekce hrudní aorty, prolaps mitrální chlopně) a hernie břišní stěny. K hlavním ledvinným projevů patří především vývoj arteriální hypertenze, bolest v ledvinách a postupný vývoj chronické renální insuficience. Tyto projevy jsou úzce vázány na vývoj a zvětšení ledvinných cest a ledvin. Přibližně u 50 % osob s PCHLAD dojde ke vzniku CHSL do 60. roku věku. S prevalencí přibližně 1 : 1 000 obyvatel se PCHLAD řadí k méně běžným, nikoli však vzácným onemocněním ledvin a představuje nejdůležitější dědičné (monogenní) onemocnění ledvin v dospělosti. Velkou většinu případů PCHLAD kódují dva hlavní geny (PKD1 gen – ze 78 % a PKD2 gen z 15 %), podstatně vzácněji pak dva další geny (GANAB – 0,3 % a DNAJB11 – 0,1 %), u ostatních případů není genotyp znám. Klíčovou roli při vývoji cystického postižení ledvin a dalších orgánů hraje snížená tvorba genových produktů některého z PKD genů – polycystinů, uplatňujících se primárně při snížené funkci řasinek – cilií. Z hlediska renální prognózy byl prokázán významný rozdíl mezi jednotlivými základními genotypy: u pacientů s genotypem PD1 dochází ke vzniku CHSL významně dříve než u pacientů s PD2 genotypem (průměrný věk vzniku CHSL u PKD1 je 58,1 vs. 79,7 let u PKD2 genotypu), u genotypu GANAB dochází ke vzniku CHSL vzácně (avšak u většiny pacientů se vyvíjí cystóza jater) a u genotypu DNAJB se vyvíjí CHSL v pozdním věku. Ačkoli je pro renální i celkovou prognózu onemocnění klíčové, který gen onemocnění kóduje, mohou prognózu významně ovlivnit také typ a závažnost mutační změny (inaktivující vs. neinaktivující mutace), případně některé další genetické i negenetické faktory. Při zachování určitého obdobného průběhu onemocnění v rámci jedné rodiny lze pozorovat významnou intrafamiliální variabilitu [3].
Polycystická choroba jater (PCHJ) je zdaleka nejčastějším extrarenálním projevem PCHLAD (PCHJ-PCHLAD). Za hlavní mechanizmy poruchy vývoje jaterních cyst se v patofyziologii PCHJ-PCHLAD uvádí především malformace zárodečné žlučovodové lišty a následná porucha vývoje žlučovodů a narušená funkce cilií cholangiocytů. Na rozdíl od normální tvorby žlučovodů, které se vytvářejí ze žlučovodové lišty na podkladě komplexních sekvencí růstu a apoptózy, nedochází u PCHJ k adekvátní apoptóze introlobulárních žlučovodů, které zůstávají v podobě von Mayenburgových komplexů a postupně se cysticky dilatují. Cilie cholangiocytů mají mechanosenzorickou kapacitu a modulují intracelulární hladiny cAMP a Ca2+ v reakci na tok žluči. Ciliární defekty vzniklé v důsledku nedostatečné tvorby genového produktu polycystinu způsobují pokles cytoplazmatických hladin Ca2+ a vzestup cytoplazmatických hladin cAMP a v konečném důsledku vedou k hyperproliferaci cholangiocytů a cystogenezi [4,5]. Cysty v játrech jsou vzácné u dětí, ve studii CRISP je jejich přítomnost dle magnetické rezonance 58 % u pacientů ve věku 15–24 let, 85 % u pacientů ve věku 25–34 let a 94 % u pacientů ve věku 34–46 let. PCHJ-PCHLAD se vyvíjí v časnějším věku u žen než u mužů a některá pozorování naznačují, že vývoj PCHJ je závažnější u žen s mnohočetnými těhotenstvími a po menopauze u žen, které byly léčeny suplementací estrogenů. Ve studii HALT PKD, zahrnující soubor 534 pacientů s PCHLAD s normální funkcí ledvin či velmi mírnou chronickou renální insuficiencí, bylo možno pozorovat, že za růst objemu jater s věkem odpovídá jak zmnožení počtu cyst a jejich objemu, tak i zvětšení objemu parenchymu jater mimo cystu [6,7]. Ačkoli ve většině případů zůstávaly laboratorní ukazatele u pacientů s PCHJ v normálním rozmezí, byly závažnější případy PCHJ-PCHLAD spojeny s laboratorními a hematologickými abnormitami a také se sníženými parametry kvality života [6]. Ve studii Chebiba FT et al nebylo možno – poněkud překvapivě – prokázat korelaci mezi závažností PCHJ (mj. rychlost růstu jaterních cyst) a typem mutačních změn u PKD1 a PKD2 genotypů, což naznačuje, že na rozdíl od ledvinného postižení v rámci PCHLAD ovlivňují vývoj PCHJ-PCHLAD významně jiné než genetické změny (konkrétně pohlaví, patrně hormonální a další vlivy) [8]. Pozoruhodné je, že v uvedené studii bylo možno u 58 % žen se závažnou PCHJ-PCHLAD (objem jater vztažený na výšku > 1 800 ml/m) ve věku > 48 let prokázat postupné zmenšení objemu jater proti vstupnímu nálezu. Jaterní cysty jsou obvykle asymptomatické a nezpůsobují selhání jater – pokud se objeví klinické obtíže, bývají to projevy volumového efektu cyst, vývoj komplikací či vzácné asociace [3,7]. Mezi hlavní projevy volumového efektu patří distenze břicha, pocit plnosti, dušnost či bolest v bedrech. Vzácně byly pozorovány komprese dolní duté žíly, jaterních žil či žlučovodů. Jaterní epiteliální buňky produkují a vylučují karbohydrátový antigen 19-9 (CA 19-9), který je tumor markerem pro karcinomy GIT. Koncentrace CA 19-9 v krvi a v tekutině jaterní cysty jsou u pacientů s PCHJ zvýšené, přičemž sérové hladiny korelují s rozsahem PCHJ. PCHJ může být komplikována krvácením do cysty (febrilie, bolesti), infekcí cysty (bolestivost, febrilie, zvýšené zánětlivé parametry, vzestup alkalické fosfatázy, CA 19-9) či rupturou jaterní cysty (prudká abdominální bolest, ascites). Vzácnou a závažnou komplikaci může představovat vrozená dilatace jaterních žlučovodů (Caroliho nemoc), která může predisponovat k zánětům žlučových cest. Častěji byla rovněž u pacientů s PCHJ pozorována dilatace společného žlučovodu. Zobrazovací metody mohou významně přispět k upřesnění diagnózy. U většiny pacientů PCHJ probíhá asymptomaticky a nevyžaduje léčbu [3,7]. Často je důležitá psychologická podpora – pacienty, obvykle ženského pohlaví, je vhodné informovat o tom, že vznik cyst v játrech je běžnou součásti PCHLAD, že PCHJ-PCHLAD nevede k jaternímu selhání, ale mohou vznikat volumové obtíže řešitelné obvykle konzervativně. Je vhodné doporučit omezení exogenních estrogenů u žen, které se mohou potenciálně podílet na vývoji PCHJ-PCHLAD, dále s ohledem na útlakový syndrom GIT přijímat potravu v menších objemech a pro symptomatickou úlevu užívat H2 blokátory či inhibitory protonové pumpy. U pokročilých případů PCHJ se závažnou symptomatologií lze na podkladě individuálního posouzení volit některý z následujících intervenčních zákroků: perkutánní aspiraci a sklerotizaci, laparoskopickou fenestraci, kombinovanou resekci jater a fenestraci cyst, transplantaci jater, případně selektivní embolizaci jaterních žil. Aspirace cyst a sklerotizace (např. alkoholem) je léčebným zákrokem volby v případě bolesti způsobené jednou či několika dominantními cystami. Úspěšnost zákroku je kolem 70 % v případě prvního a podstatně nižší v případě opakovaného zákroku. Méně častou alternativu představuje laparoskopická fenestrace jaterních cyst, která je často komplikována přechodným ascitem a výsledky bývají pouze krátkodobé. U pacientů se zvětšenými játry v důsledku přítomnosti četných malých či středně velkých cyst bývá obvykle určitá část jater zachována, což umožňuje provést kombinovanou resekci jater a fenestraci cyst. Jedná se o náročný zákrok provázený často komplikacemi a také nezanedbatelnou mortalitou, který však může vést k dlouhodobě uspokojivým výsledkům [9]. Transplantace jater je vhodné řešení u pacientů se sníženou funkcí jater a globálním postižení parenchymu jater cystami. Další alternativou je resekce jater a následná transplantace jater ze živého dárce. Selektivní embolizace jaterní tepny může představovat záchovný zákrok u těch pacientů se symptomatickou PCHJ, u kterých nelze uvažovat o chirurgickém zákroku. Novou perspektivu představují aktuálně testované farmakologické postupy mířené na ovlivnění patologických mechanizmů či signálních cest u PCHLAD (např. analoga somatostatinu), v současnosti však nejsou standardní léčebnou možností [3–5].
Z hlediska diferenciální diagnostiky je třeba uvést především polycystickou chorobu jater autozomálně dominantního typu (PCHJAD) – samostatnou nozologickou jednotku, odlišnou od PCHJ-PCHLAD. PCHJAD je v porovnání s PCHJ-PCHLAD podstatně vzácnější onemocnění, které se podobně jako PCHJ-PCHLAD vyznačuje vývojem zřetelného cystického postižení jater. Na rozdíl od postižení PCHJ-PCHLAD je však u PCHJAD cystické postižení ledvin mírné či není přítomno, chybějí další multiorgánové projevy onemocnění a genový podklad je odlišný [3,5]. U PCHJAD je výrazná genová heterogenita (genovým podkladem je více než pět genů), recentně byla částečně definována jejich lokalizace a funkce v endoplazmatickém retikulu a postulována významná funkční interakce mezi těmito genovými produkty a PC1. Klinický průběh a léčba se významně neliší od symptomatologie a léčby u PCHJ-PCHLAD [10].
Další postižení GIT u PCHLAD
Cysty pankreatu
Z řady patologických a zobrazovacích studií vyplývá, že výskyt cyst v pankreatu je proti běžné populaci mírně zvýšený, patrně v závislosti na vyšším věku, a dále že klinický průběh je obvykle asymptomatický, pouze vzácně byly popsány případy pankreatitidy způsobené kompresí pankreatického vývodu [3,11].
Divertikulární choroba
U pacientů s PCHLAD ve stadiu CHSL byl pozorován významně zvýšený výskyt divertikulární choroby střeva a také akutní divertikulitidy ve srovnání s pacienty s jiným základním onemocněním. U pacientů s PCHLAD bez CHSL tento trend prokázán nebyl. Zda se na zvýšeném výskytu divertikulární choroby podílí nadměrná tvorba extracelulární matrice (prokázaná u PCHLAD) či jiné mechanizmy, není známo. Prestižní odborná iniciativa KDIGO (Kidney Disease: Improving Global Outcome) nedoporučuje rutinně vyšetřovat pacienty s PCHLAD ve stadiu CHSL na přítomnost divertikulární choroby [3,11].
Hernie
Prevalence hernií je u pacientů s PCHLAD zvýšená. V jedné retrospektivní studii byla prokázána přítomnost hernií různého typu (inguinální, paraumbilikální aj.) u 45 % (38 z 85) pacientů s PCHLAD ve stadiu CHSL vs. 16 % (7 z 85) ve srovnatelné skupině pacientů s jiným základním onemocněním a ve srovnání se 4 % (3 z 85) v kontrolní skupině obecně chirurgických pacientů. Jako možná příčina byla zvažována nadměrná tvorba extracelulární matrice u PCHLAD a/nebo nadměrný nitrobřišní tlak podmíněný přítomností zvětšených cystických orgánů. Zvláštní pozornost je třeba věnovat pacientům s PCHLAD ve stadiu CHSL, u kterých je zvažováno zavedení peritoneální dialýzy s nutností trvalého napouštění dialyzačního roztoku do břišní dutiny a kde riziko vývoje hernií stoupá mimořádně vysoko [3,11,12].
Polycystické onemocnění ledvin autozomálně recesivního typu
PCHLAR je systémové hepatorenální fibrocystické onemocnění, projevující se obvykle klinicky v novorozeneckém či dětském věku. Frekvence výskytu je odhadována na 1 : 20 000 živých porodů. V ledvinách představuje základní patologii vřetenovitá dilatace sběrných kanálků vedoucí ke zvětšení ledvin. S rostoucím věkem dochází k uzávěru dilatovaných sběrných kanálků do podoby velkých cyst, do určité míry podobných obrazu PCHLAD. K poruše funkce ledvin a progresi do CHSL dochází téměř u všech pacientů, u značné části již v postnatálním či dětském věku, u další části v různém věku až do dospělosti. Od narození je u všech pacientů v játrech přítomna porucha duktálního plátu, postupně vedoucí k vývoji chronické (kongenitální) HF a Caroliho syndromu (CS). Portální hypertenze, hypersplenizmus a jícnové varixy jsou přítomny u většiny pacientů s pokročilou HF. U značné části pacientů je perinatální průběh komplikován přítomností oligohydramnia, zvětšenými ledvinami a „Potterovou sekvencí“ s hypoplazií plic vedoucí k respirační insuficienci a případně k úmrtí až ve 30 % případů [13]. Onemocnění kóduje PKHD1 gen, jeho genový produkt označovaný jako fibrocystin či polyductin představuje nový integrální membránový protein, lokalizovaný v ciliární membráně a ovlivňující komplexním způsobem ciliární vývoj a funkci. Stanovit korelaci genotyp-fenotyp obvykle není snadné, jelikož mutační spektrum u PKHD1 charakterizuje výrazná alelická heterogenita, s četnými unikátními mutacemi, postihující jednu rodinu. Přítomnost dvou inaktivujících mutací bývá spojena s úmrtím v perinatálním či neonatálním období. S ohledem na mimořádně závažný průběh choroby je důležité, že onemocnění lze diagnostikovat často již prenatálně a také že metody DNA lze použít pro preimplantační genetickou diagnostiku (PGD) zamezující přenos patologického genu na potomky [14]. Léčebná péče je zaměřena jednak na náhradu funkce selhaných ledvin dialýzou a/nebo transplantací ledviny a dále na léčbu jaterních a žlučových komplikací. Gastroenterologická, resp. hepatologická dispenzární péče je nezbytná u všech pacientů s pokročilou HF. Ultrazvuk je základní diagnostickou metodou pro stanovení diagnózy, u atypických obrazů či nálezů vyvíjejících se až v dospělém věku je často diagnóza upřesněna biopsií jater a histologickým vyšetřením. U rozvinutých stadií HF dominují projevy portální hypertenze s hypersplenizmem, trombocytopenií nebo krvácením z jícnových varixů. Proteosyntetická funkce jater je obvykle zachována či jen mírně snížena [13,15]. Vhodnou intervencí u rekurujících případů jícnového krvácení je zavedení portosystémového zkratu za účelem snížení portálního tlaku. U řady pacientů je třeba léčit případy cholangitidy, ke kterým často predisponují. Izolované, kombinované či sekvenční transplantace jednotlivých orgánů (ledvin/jater) jsou realizovány na podkladě individuálního posouzení pacienta [13,15]. V rozsáhlé retrospektivní studii z roku 2016 (n = 202) u pacientů s PCHLAR s věkem 19 let či nižším byly provedeny transplantace ve věku s mediánem 9 let. U 163 (80,7 %) pacientů bylo realizována izolovaná transplantace ledviny, u 32 (15,8 %) pacientů byla provedena kombinovaná transplantace ledvin a jater, v ostatních případech nebyly dostatečné údaje k dispozici. V celém souboru bylo 5leté přežívání u pacientů po adjustaci na věk a pohlaví 95,5 % (95% CI; 92,4–98,8 %). Riziko úmrtí u kombinovaných transplantací bylo 6,7× (95% CI; 1,8–25,4) vyšší než u izolovaných transplantací ledviny (p = 0,005) [16].
Vzácná dědičná onemocnění ledvin a hepatální fibróza
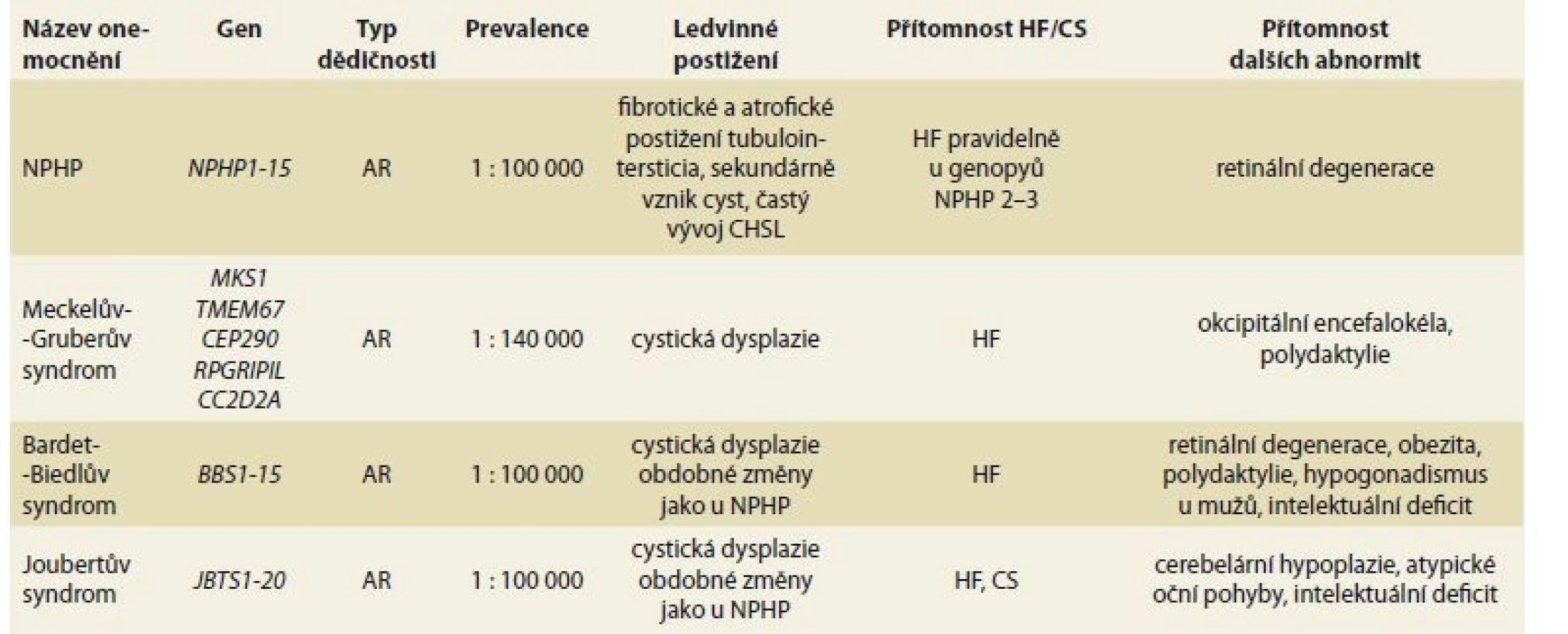
Kromě PCHLAR, v rámci které je HF integrální součástí klinického obrazu onemocnění, je HF asociována s celou řadou vzácných dědičných multisystémových onemocnění, která zahrnují rovněž onemocnění ledvin. Onemocnění ledvin mívá různou podobu a různou klinickou závažnost, často s vývojem CHSL (typicky např. u nefronoftízy). U některých syndromů je HF vždy přítomna (např. u Meckelova-Gruberova syndromu, Joubertova syndromu), u jiných není přesný výskyt znám (tab. 2). Klinický průběh HF není systematicky dokumentován, nicméně z řady kazuistických sdělení vyplývá, že u řady HF se vyvíjí portální hypertenze a CS bývá spojen se záněty žlučových cest [17].
Tab. 2. Hepatal fibrosis in rare hereditary multisystem diseases with renal impairment.
Příznaky postižení GITjako atypické či méně běžné projevy dědičných onemocnění ledvin
Difuzní leiomatóza jícnu u Alportova syndromu
Alportův syndrom (AS) je relativně vzácné dědičné onemocnění ledvin (prevalence 1–9/100 000 obyvatel), způsobené poruchou tvorby alfa-řetězců (A3–5) kolagenu IV (COL4). COL4 je základní stavební součástí glomerulární bazální membrány glomerulů a řady dalších orgánů, což vysvětluje multisystémový charakter AS. Kromě dědičné nefritidy bývá AS provázena abnormitami dalších orgánů – především sluchového aparátu (cca 50 %) a očí (rohovky, čočky, retiny). U většiny pacientů (85 %) je podstatou AS porucha COL4A5 genu s typem přenosu vázaného na chromozom X, mutace COL4A3 a COL4A4 genů způsobují poruchu alfa-řetězců COL4 u AR a velmi vzácně i AD vázaných typů AS. Patologické změny stěny glomerulů způsobují od dětského věku vývoj typického sledu laboratorních a klinických příznaků (erytrocyturie, proteinurie, nefrotický syndrom, pokles glomerulární filtrace), který obvykle vyústí v CHSL. GIT příznaky nejsou běžnou součástí AS, výjimku tvoří difuzní leiomatóza jícnu (asi u 5 % AS). Difuzní leiomatóza jícnu je vzácné onemocnění vyznačující se difuzní benigní hypertrofií svaloviny jícnu a způsobující progredující dysfagii a další klinické obtíže [18]. Onemocnění se izolovaně vyskytuje vzácně, asi u 2/3 pacientů se vyskytuje v asociaci s AS, v některých případech spolu s dalšími vývojovými defekty (bronchu, vulvy, rekta). Metodami molekulární biologie byly u některých pacientů s difuzní leiomatózou jícnu prokázány delece COL4A5–6 genů, přesný mechanizmus vzniku defektu nebyl objasněn [19]. Klinické projevy difuzní leiomatózy jícnu (dysfagie, vomitus, regurgitace) mohou být spolu s výsledky vyšetřovacích metod občas mylně interpretovány jako projevy achalázie, vést k nesprávnému léčebnému postupu a realizaci neúčinných zákroků (myotomie jícnu, dilatace jícnu). Účinnou léčbu představuje úplná ezofagektomie [20].
Hamartomy v GIT u tuberózní sklerózy
Tuberkulózní skleróza (TS) je vzácné dědičné multisystémové onemocnění, které se vyznačuje růstem benigních tumorů v mozku a dalších orgánech, jako jsou ledviny, srdce, játra, oči, plíce či kůže. Příznaky mohou zahrnovat křeče, poruchu intelektu, poruchu růstu aj. Prevalence je 7–12/100 000 obyvatel, typ dědičnosti je AD. TS je způsobena mutací jednoho ze dvou tumor-supresorových genů – TSC1/TSC2, které kódují genové produkty hamartin/tuberin uplatňující se v buněčné proliferaci a diferenciaci. Jejich defektní funkce vede ke vzniku tumorů. Expresivita onemocnění je velmi proměnlivá, což zčásti vysvětluje skutečnost, že příznaky onemocnění často unikají pozornosti [21]. Dědičný charakter nemoci může uniknout pozornosti pro relativně vysoký počet nově vzniklých případů vzniklých v důsledku spontánních mutací TSC1/TSC2 genů. Diagnóza TS se obvykle opírá o průkaz několika typických projevů TS (tzv. velká a malá kritéria nemoci), diagnózu lze potvrdit průkazem mutačních změn v TSC1/TSC2 genech. Postižení ledvin angiomyolipomy, cystami či vzácněji karcinomy patří k běžným projevům TS, které nezřídka vedou k vývoji CHSL. Postižení GIT u TS je méně běžné, vývojové anomálie a zvláště hamartomy se však mohou vyskytnout prakticky v jakékoli části GIT a mít různou podobu. V orální lokalizaci byl popsán výskyt fibromů, fibrózní hyperplazie či papilomů, v jícnu výskyt fibrózních tumorů, v žaludku polypózních hamartomů, v tenkém střevě mukoviscidózy, v tlustém střevě a v rektu výskyt cévních malformací, angiomyolipomů, polypózních hamartomů a adenokarcinomu [22]. U pacientů s TS je třeba počítat s možným výskytem hamartomů v GIT a u pacientů s podezřením na TS může jejich průkaz přispět k včasnému stanovení diagnózy.
Nespecifické obtíže GIT jako projevy Fabryho choroby
Fabryho choroba (FCH) je vzácné dědičné onemocnění způsobené mutacemi alfa-galaktosidázového (GLA) genu a s tím související sníženou či chybějící tvorbou enzymu alfa-galaktosidázy A, a následnému ukládání globotriacylceramidu (GL-3) a dalších glykosfingolipidů do lyzozomů. Intracelulární akumulace GL-3 má za následek závažné poškození řady orgánů vč. mozku, srdce a ledvin s potenciálně fatálním průběhem. Dědičnost FCH je vázána na chromozom X s převážným postižením mužů a celková prevalence se odhaduje na 1 : 120 000. Fenotypická variabilita je značná a odráží patrně další faktory – genetické i negenetické – nezávislé na variantách GLA. Klasický fenotyp u mužů se vyvíjí obvykle od 14. roku věku, mírnější fenotypy – převážně u žen – se obvykle vyvíjejí v pozdějším věku. K hlavním příznakům FCH patří neuropatická bolest, porucha vnímání tepla a chladu či zvýšená potivost. Orgánové postižení představují především kožní změny (angiokeratoma), oční změny (cornea verticillata), renální změny (proteinurie, CHSL) a kardiální změny (hypertrofie levé komory, arytmie). U pacientů s klasickým fenotypem jsou GIT obtíže – především abdominální bolest a průjmy – velmi časté, přítomné asi u 50 % dospělých a 60 % dětí. K dalším příznakům patří zácpa, nauzea a vomitus. Patofyziologický podklad GIT je komplexní a multifaktoriální a zahrnuje především následující tři mechanizmy: dysfunkce autonomního nervového systému odpovědného za střevní motilitu, vaskulopatie postihující cirkulaci GIT a zánětlivé tkáňové změny související s akumulací GL-3. GIT projevy mohou být prvním či vedoucím příznakem u pacientů s FCH a s ohledem na vzácný výskyt FCH a její fenotypickou variabilitu zůstává často příčina obtíží dlouhodobě nerozpoznána a diagnostika FCH oddálena [23]. K úspěšnému průkazu příčiny GIT obtíží při dosud neprokázané FCH je nezbytné, kromě vyšetřovacího postupu zaměřeného na GIT projevy, všímat si případných dalších klinických projevů FCH (často velmi mírných) a údajů o pozitivní rodinné anamnéze ve smyslu výskytu FCH v rodině. Při podezření na FCH lze prokázat různými metodami sníženou tvorbu enzymu alfa-galaktosidázy A (v leukocytech, ve vzorcích krve vyšetřených na testačních proužcích, ve tkáních) a případně mutační analýzou. Včasné stanovení FCH nabylo na významu s tím, že v současné době je k dispozici nová léčba – enzymatická náhradní léčba, nahrazující chybějící enzym pravidelným podáváním enzymu vyráběného rekombinantní cestou [24].
Transplantace jater u primární hyperoxalurie I
Primary hyperoxaluria I (PH I) je vrozená porucha jaterního metabolizmu vzniklá v důsledku deficitu peroxizomálního enzymu alanin-glyoxylát aminotransferázy (AGT), který zprostředkovává konverzi glyoxalátu na glycin. Deficit AGT způsobuje nadprodukci šťavelanů vedoucí k nadměrnému vylučování šťavelanů močí a vzniku krystalové nefropatie s přítomností kalcium oxalátových konkrementů. V další fázi onemocnění dochází k významnému poškození funkce ledvin a schopnost ledvin vylučovat šťavelany je natolik omezena, že dochází k systémovému ukládání šťavelanů do tkání (systémové oxalóze) a poškození funkce řady orgánů. PH I je zdaleka nejčastější a nejvýznamnější ze tří typů PH (PH I–III). Podstatou onemocnění je mutace genu AGXT kódujícího AGT. Dědičnost je AR typu. Výskyt je přibližně 1 : 120 000 živých porodů a odhaduje se, že PH I odpovídá přibližně za 1% všech případů CHSL v dětském věku. Pro podezření na diagnózu PH (I) svědčí přítomnost recidivující kalcium oxalátové urolitiázy, zvláště v koincidenci s recidivující kalcium oxalátovou urolitiázou u dalších členů rodiny, průkaz hyperoxalurie, případně nadměrné hladiny šťavelanů v krvi. Průkaz onemocnění lze podpořit průkazem snížené enzymové aktivity enzymu AGT a průkazem mutačních změn AGXT genu. Symptomatická léčba PH I spočívá především ve zvýšeném příjmu tekutin, u menší části pacientů s PH I (5–30 %) může být přínosná suplementace pyridoxinem. U většiny pacientů s PH I je klíčovým a účinným řešením transplantace jater, jejichž funkce je sice jinak intaktní, avšak neumožňuje vytvořit dostatečné množství AGT k metabolizaci glyoxalátů [25]. Izolovaná transplantace ledvin představuje v případě vzniku CHSL pouze přechodné řešení [26]. Transplantace jater – ať již preventivní, kombinovaná s transplantací ledvin či sekvenční (po transplantaci ledvin) – bývá indikována pouze v případě nepříznivého vývoje klinického průběhu PH I při dodržení konzervativních prostředků léčby [1,25].
Infekční a zánětlivé projevy onemocnění GIT – možný spouštěcí mechanizmus aHUS
Hemolyticko-uremický syndrom (HUS) definuje triáda hemolytické anémie, trombocytopenie a renálního poškození, k dalším typickým rysům patří porucha endotelu, vývoj trombotické mikroangiopatie (TMA) či přítomnost závažné hypertenze. HUS je vzácné onemocnění, často velmi závažné s potenciálním poškozením ledvin a dalších orgánů. HUS lze rozdělit na dvě základní podskupiny: získanou formu HUS představuje tzv. STEC-HUS, vyvolaný Shiga-toxinem produkovaným některými kmeny Escherichia coli, a vrozenou či dědičnou formu představuje aHUS, způsobený vrozenými, resp. dědičnými poruchami alternativní cesty komplementu či bílkovin, které tuto cestu regulují. STEC-HUS tvoří převážnou většinu případů HUS v dětském věku a malou část HUS v dospělosti a charakterizuje jej obvykle přítomnost akutních (krvavých) průjmů v úvodu onemocnění. aHUS tvoří jen malou část HUS v dětském věku, avšak převážnou část HUS v dospělosti, jeho prevalence se odhaduje na 1–9 : 1 mil. obyvatel. U pacientů s aHUS dochází v důsledku vrozené poruchy alternativní cesty komplementu za určitých okolností k nekontrolované aktivaci komplementového systému a následnému vývoji klinických projevů aHUS s poškozením ledvin, mozku, srdce, GIT a dalších tkání a orgánů. GIT postižení (kolitida, bolesti břicha, pankreatitida, vomitus, gastroenteritida, jaterní nekróza či průjem) bylo dokumentováno téměř u 40 % pacientů s aHUS [2]. U pacientů s aHUS bývají prokázány mutační změny v genech, které kódují proteiny alternativní cesty komplementu či molekuly regulující tuto cestu (C3, CD46 (MCP), CFB, CFH, CFHR1, CFHR3, CFHR4, CFI, DGKE, a THBD). Pokud je dědičný přenos dokumentován, má obvykle podobu AR či AD přenosu. V případě abnormální genetické predispozice může u pacienta dojít k aktivaci komplementového systému a vývoji další kaskády klinických dějů působením spouštěcích mechanizmů, které mohou mít různou podobu či intenzitu. Aktivace komplementového systému a vývoj aHUS/TMA byly kromě jiných „triggerů“ (těhotenství, léky, maligní hypertenze atd.) pozorovány velmi často při infekcích, např. horních cest dýchacích, ale také při průjmovitém onemocnění/gastroenteritidě. Pokud se po průjmovitém onemocnění/gastroenteritidě, případně jiném infekčním onemocnění GIT rozvíjejí známky hemolýzy, trombocytopenie a poškození ledvin, je potřeba uvažovat o možnosti vzniku HUS (STEC-HUS, aHUS). Jsou-li v rámci vyšetřovacího programu vyloučeny další možnosti TMA (STEC-HUS, trombotická trombocytopenická purpura), je možno podezření na diagnózu aHUS podpořit laboratorním průkazem aktivace komplementu (snížené hladiny vybraných složek komplementového systému v plazmě, případně přítomnost specifických protilátek proti těmto složkám – konkrétně anti-CFH), provedením biopsie ledviny a histologickým průkazem typických změn TMA v ledvinné tkáni a konečně genetickým vyšetřením a průkazem mutačních změn v genech, které kódují proteiny alternativní cesty komplementu [27,28]. Včasné stanovení diagnózy je naprosto klíčové, jelikož na jedné straně aktivace, ev. rekurence aHUS může velmi rychle vést k ireverzibilním poškozením ledvin a dalších orgánů, na straně druhé je v současné době v léčebném arzenálu aHUS k dispozici velmi účinná látka – eculizumab. Eculizumab je humanizovaná monoklonální protilátka, která váže složku komplementu C5 odpovědnou za aktivaci membránu atakujícího komplexu, a tím způsobuje inhibici nekontrolované terminální aktivity komplementu a umožňuje dosáhnout remise onemocnění [29].
Závěr
Pokrok, jehož jsme svědky v oblasti dědičných chorob ledvin, byl do značné míry podmíněn rozvojem vědních poznatků na poli molekulární biologie, odráží však také změny v dalších oborech, které s péčí o pacienty a rodiny s dědičnými chorobami ledvin souvisejí. Od identifikace genového lokusu pro PCHLAD v polovině 80. let minulého století se do současnosti podařilo odhalit genový podklad všech významnějších monogenních onemocnění ledvin [30]. U dědičných chorob ledvin byla přijata jednotná nomenklatura a terminologie založená na poznatcích o příslušném genovém podkladu. Použití metod molekulární biologie a mutační analýza se staly integrální součástí genetického vyšetření a podařilo se odhalit některé základní poznatky v etiologii a patogenezi těchto onemocnění. Rozvoj nových metod (jako např. PGD) a zavedení inovativních léčebných látek (např. náhradní enzymatické léčby u vybraných metabolických onemocnění či monoklonální protilátky eculizumab pro léčbu aHUS) byly dalšími impulzy, které iniciovaly současný mimořádný zájem o dědičné choroby ledvin. Stejně tak jako se nové poznatky z oblasti genetiky zřetelně promítly do našich přesnějších znalostí o genotypu dědičných chorob ledvin, je možné poznamenat, že klinický průběh a klinický obraz řady dědičných onemocnění ledvin (fenotyp) byl výrazně ovlivněn a modifikován rozvojem některých lékařských disciplín jako např. neonatologie či transplantologie, umožňující dlouhodobě přežívat jedincům se závažnými vrozenými orgánovými poruchami. Stále častěji se péče o pacienty s vrozenými a dědičnými chorobami ledvin a také průvodním postižením GIT přesouvá z dětského věku i do dospělosti. Klinický význam a závažnost extrarenálních projevů dědičných chorob ledvin vč. postižení GIT tak může v budoucnu relativně narůstat. Přítomnost příznaků postižení GIT může být v některých případech prvním projevem nerozpoznaného dědičného onemocnění ledvin, a proto by tato možnost měla být zvažována v diferenciálně diagnostických úvahách.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Doručeno: 7. 9. 2018
Přijato: 21. 9. 2018
prof. MUDr. Miroslav Merta, CSc.
Ústav biologie a lékařské genetiky 1. LF UK a VFN Praha
Purkyňův ústav
Albertov 4
128 00 Praha 2
Sources
1. Van Hoeve K, Mekahli D, Morava E et al. Liver involvement in kidney disease and vice versa. Pediatr Nephrol 2018; 33 (6): 957–971. doi: 10.1007/s00467-017-3715-3.
2. Hofer J, Rosales A, Fischer C et al. Extra-renal manifestations of complement-mediated thrombotic microangiopathies. Front Pediatr 2014; 2 : 97. doi: 10.3389/fped.2014.00097.
3. Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Adam MP, Ardinger HH, Pagon RA et al. (eds). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle: 1993–2018.
4. Abu-Wasel B, Walsh C, Keough V et al. Pathophysiology, epidemiology, classification and treatment options for polycystic liver diseases. World J Gastroenterol 2013; 19 (35): 5775–5786. doi: 10.3748/wjg.v19.i35.5775.
5. Cnossen WR, Drenth JP. Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet J Rare Dis 2014; 9 : 69. doi: 10.1186/1750-1172-9-69.
6. Hogan MC, Abebe K, Torres VE et al. Liver involvement in early autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol 2015; 13 (1): 155–164. doi: 10.1016/j.cgh.2014.07.051.
7. Kim H, Park HC, Ryu H et al. Clinical correlates of mass effect in autosomal dominant polycystic kidney disease. PLoS One 2015; 10 (12): e0144526. doi: 10.1371/journal.pone.0144526.
8. Chebib FT, Jung Y, Heyer CM et al. Effect of genotype on the severity and volume progression of polycystic liver disease in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2016; 31 (6): 952–960. doi: 10.1093/ndt/gfw008.
9. Chebib FT, Harmon A, Irazabal Mira MV et al. Outcomes and durability of hepatic reduction after combined partial hepatectomy and cyst fenestration for massive polycystic liver disease. J Am Coll Surg 2016; 223 (1): 118–126.e1. doi: 10.1016/j.jamcollsurg.2015.12.051.
10. Besse W, Dong K, Choi J et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest 2017; 127 (9): 3558. doi: 10.1172/JCI96729.
11. Mikolajczyk AE, Te HS, Chapman AB. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol 2017; 15 (1): 17–24. doi: 10.1016/j.cgh.2016.06.017.
12. Morris-Stiff G, Coles G, Moore R et al. Abdominal wall hernia in autosomal dominant polycystic kidney disease. Br J Surg 1997; 84 (5): 615–617.
13. Wehrman A, Kriegermeier A, Wen J. Diagnosis and management of hepatobiliary complications in autosomal recessive polycystic kidney disease. Front Pediatr 2017; 5 : 124. doi: 10.3389/fped.2017.00124.
14. Bergmann C. Genetics of autosomal recessive polycystic kidney disease and its differential diagnoses. Front Pediatr 2018; 5 : 221. doi: 10.3389/fped.2017.00221.
15. Gunay-Aygun M, Font-Montgomery E, Lukose L et al. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology 2013; 144 (1): 112–121. e2. doi: 10.1053/j.gastro.2012.09.056.
16. Mekahli D, van Stralen KJ, Bonthuis M et al. Kidney versus combined kidney and liver transplantation in young people with autosomal recessive polycystic kidney disease: data from the European Society for Pediatric Nephrology/European Renal Association-European Dialysis and Transplant (ESPN/ERA-EDTA) registry. Am J Kidney Dis 2016; 68 (5): 782–788. doi: 10.1053/j.ajkd.2016.06.019.
17. Hartung EA, Guay-Woodford LM. Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics 2014; 134 (3): e833–845. doi: 10.1542/peds.2013-3646.
18. Abbes K, Ayadi L, Makni S et al. Esophageal leiomyomatosis revealing an Alport syndrome. Rev Med Interne 2009; 30 (1): 88–90. doi: 10.1016/j.revmed.2008.02.025.
19. Nozu K, Minamikawa S, Yamada S et al. Characterization of contiguous gene deletions in COL4A6 and COL4A5 in Alport syndrome-diffuse leiomyomatosis. J Hum Genet 2017; 62 (7): 733–735. doi: 10.1038/jhg.2017.28.
20. Burgos R, Muñiz E, Rosa ER et al. Comprehensive management of diffuse leiomyomatosis in a patient with Alport syndrome. P R Health Sci J 2013; 32 (4): 200–202.
21. Staley BA, Vail EA, Thiele EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics 2011; 127 (1): e117–125. doi: 10.1542/peds.2010-0192.
22. Santos L, Brcic I, Unterweger G et al. Hamartomatous polyposis in tuberous sclerosis complex: Case report and review of the literature. Pathol Res Pract 2015; 211 (12): 1025–1029. doi: 10.1016/j.prp.2015.09.016.
23. Hilz MJ, Arbustini E, Dagna L et al. Non-specific gastrointestinal features: Could it be Fabry disease? Dig Liver Dis 2018; 50 (5): 429–437. doi: 10.1016/j.dld.2018.02.011.
24. Ortiz A, Germain DP, Desnick RJ et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab 2018; 123 (4): 416–427. doi: 10.1016/j.ymgme.2018.02.014.
25. Bhasin B, Ürekli HM, Atta MG. Primary and secondary hyperoxaluria: Understanding the enigma. World J Nephrol 2015; 4 (2): 235–244. doi: 10.5527/wjn.v4.i2.235.
26. Bollée G, Cochat P, Daudon M. Recurrence of crystalline nephropathy after kidney transplantation in APRT deficiency and primary hyperoxaluria. Can J Kidney Health Dis 2015; 2 : 31. doi: 10.1186/s40697-015-0069-2.
27. Sakari Jokiranta T, Viklicky O, Al Shorafa S et al. Differential diagnosis of thrombotic microangiopathy in nephrology. BMC Nephrol 2017; 18 (1): 324. doi: 10.1186/s12882-017-0727-y.
28. Loirat C, Frémeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis 2011; 6 : 60. doi: 10.1186/1750-1172-6-60.
29. Kim SH, Kim HY, Kim SY. Atypical hemolytic uremic syndrome and eculizumab therapy in children. Korean J Pediatr 2018; 61 (2): 37–42. doi: 10.3345/kjp.2018.61.2.37.
30. Reeders ST, Breuning MH, Davies KE et al. A highly polymorphic DNA marker linked to adult polycystic kidney disease on chromo-some 16. Nature 1985; 317 (6037): 542–544.
Labels
Paediatric gastroenterology Gastroenterology and hepatology SurgeryArticle was published in
Gastroenterology and Hepatology

2018 Issue 5
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Inhibítory protónovej pumpy vo svetle klinických štúdií a bezpečnostný profil ich dlhodobého užívania
- Duodenálne stenty, nechirurgické riešenie poruchy evakuácie žalúdka pri malígnych ochoreniach
- Laparoskopická hemipankreatoduodenektomie u ampulárního adenokarcinomu – kazuistika
- Karcinom pankreatu z pohledu pacienta