
Protidestičková léčba
Antiaggregant therapy
Antiaggregant treatment is suitable for all at risk patients with atherothrombosis and its cardiovascular complications. Antiaggregant treatment decreases severe vascular events in at risk patients, myocardial infarctions, non-fatal strokes, transient ischaemic attacks, unstable angina, obstructive peripheral vascular disease. It decreases a risk of embolism caused by atrial fibrillation and a risk of vascular occlusion in other patients at high risk. Antiaggregant treatment should be administered long-term. The basic antiaggregant drug is acetylsalicylic acid (ASA) which is commonly used in doses of 75–150 mg daily. Acetylsalicylic acid is an effective antiaggregant drug with clearly demonstrated beneficial effect on atherothrombotic complications in cardiovascular diseases. Other substances and drugs with antiaggregant effect including their modes of action and indications are reviewed.
Keywords:
cardiovascular diseases – antiaggregant therapy
Authors:
J. Malý; M. Pecka; P. Ďulíček; M. Blažek; L. Smolej
Authors‘ workplace:
II. interní klinika Lékařské fakulty UK a FN, Hradec Králové, přednosta prof. MUDr. Jaroslav Malý, CSc.
Published in:
Vnitř Lék 2005; 91(7 a 8): 826-832
Category:
128th Internal Medicine Day - 21rd Vanysek's Day Brno 2005
Overview
Protidestičková léčka je vhodná u všech rizikových nemocných s aterotrombózou a jejími kardiovaskulárními komplikacemi. Protidestičková léčba snižuje závažné cévní příhody u rizikových nemocných, srdečního infarktu, nefatálních iktů, transientních ischemických atak, nestabilní anginy pectoris, obliterující aterosklerózy tepen dolních končetin. Dále snižuje rizika embolie při fibrilaci síní a rizika cévních uzávěrů u jiných nemocných s vysokým rizikem. Protidestičková léčba se má podávat dlouhodobě. Základním protidestičkovým lékem je kyselina acetylsalicylová (ASA), která se obvykle podává v dávkách 75–150 mg/denně. Kyselina acetylsalicylová je účinným protidestičkovým lékem, u kterého byl jasně prokázán příznivý vliv na aterotrombotické komplikace u kardiovaskulárních onemocnění. V přehledu jsou uvedeny další látky a léky s protidestičkovým účinkem, včetně mechanizmů působení a indikací.
Klíčová slova:
kardiovaskulární choroby – protidestičková léčba
Úvod
V primární hemostáze mají trombocyty více úloh:
- přispívají k zachování integrity cévní stěny uzavíráním drobných defektů endoteliální výstelky;
- tvorbou primitivní hemostatické zátky zastavují v iniciální fázi krvácení;
- stabilizují a zpevňují primární koagulum uvolňováním koagulačně aktivních látek během release reakce a poskytnutím povrchu, na němž některé koagulační rekce proběhnou [20].
Krevní destičky v primární hemostáze se aktivují ve 4 krocích:
- adherují na poškozený endotel a umělé povrchy;
- uvolňují ze zásobních granulí aktivační působky, především ADP;
- agregují spolu a vytvářejí hemostatickou zátku;
- vytvářejí prokoagulační povrch pro aktivní koagulační proteiny [11].
Destičky jsou stimulovány poraněním endotelu a adherují k subendoteliální matrix a absorbují plazmatické proteiny včetně von Willebrandova faktoru a fibrinogenu. Destičky adherují na von Willebrandův faktor pomocí receptoru destičkových glykoproteinů (GP) Ib/IX/V a prostřednictvím fibrinogenu pomocí glykoproteinu (GP) IIb/IIIa. Adherující destičky mění po kontaktu s kolagenem svůj tvar. Vytvářejí pseudopodie a rozprostírají se po exponovaném povrchu. Současně s tím dochází ke změnám povrchových vlastností membrány destiček a k uvolňovací reakci. Destičková adheze stimuluje intracelulární signál, který vede k uvolnění destičkového faktoru 4 (PF4) a β-tromboglobulinu (β-TG), růstového destičkového faktoru, faktoru V a dále trombospondinu z α-granulí. Zároveň se uvolňují fosfolipidy, které dovolují zahájení koagulační reakce na povrchu destiček a vedou k tvorbě fibrinu. Adenozindifosfát (ADP) se uvolňuje z denzních granulí, aktivuje GP IIb/IIIa receptor a podmiňuje agregaci krevních destiček. Spolu s ním se uvolňuje z denzních granulí ATP, serotonin a Ca++ ionty [20].
Hlavními fyziologickými induktory agregace je ADP a tromboxan A2 (TXA2), který vzniká metabolickým pochodem zahájeným aktivací destičkové fosfolipázy. Na agregaci se též podílí trombin vzniklý v malém množství na povrchu destiček. Agregace trombocytů je závislá na přítomnosti dvou dalších glykoproteinů (GP IIb a GP IIIa) v membráně destiček, fibrinogenu a Ca++. Fibrinogen spojuje destičky a podmiňuje tím tvorbu agregátů. Zatímco TXA2 podporuje uvolňování ADP, druhý prostaglandin – prostacyklin PGI2, který vzniká v cévních endoteliích, uvolňování ADP brzdí, inhibuje agregaci destiček a zabraňuje tvorbě destičkových trombů mimo místo poranění. Vede navíc k lokální vazodilataci [11].
Vyšetřovací metody destiček se v minulých desetiletích soustředily především na zjišťování krvácivých stavů destičkového původu. Metod jak prokázat destičkovou hyperaktivitu a z ní usuzovat na větší schopnost trombogeneze je poměrně málo a jejich interpretace je pro komplexnost změn a interakce s endotelem a krevními elementy stále problematická. Vyšetření destičkových funkcí se často omezuje pouze na vyšetření agregace krevních destiček a z tohoto vyšetření se dělají zásadní klinické závěry. Vyšetření agregace destiček je jen jedna laboratorní metoda se svými metodickými zásadami a specifickou interpretací. Při pátrání po chorobách destiček zůstává základním přístupem k vyšetření destičkových funkcí vyšetření anamnézy a fyzikální vyšetření. Vyšetřovací metody jsou uvedeny v tab. 1.

Přijdou-li krevní destičky během cirkulace do styku s obnaženým kolagenem v subendoteliální vrstvě cévní stěny v důsledku ruptury aterosklerotického plátu, dochází v tomto místě k jejich adhezi. Záleží na rovnováze proagregačních a antiagregačních mechanizmů, zda tento děj přestoupí do tvorby destičkového trombu [24].
Aktivity destiček v primární hemostáze jsou vzájemně provázány a agregace je pouze jednou nedílnou součástí primární hemostázy.
V současnosti dostupné protidestičkové léky interferují s různými kroky aktivačních procesů, včetně adheze, uvolňovací reakce nebo agregace, a snižují riziko arteriální trombózy, ale mohou zvýšit riziko krvácení při jejich předávkování. Indikace protidestičkové léčby vyplývá především z přítomnosti rizikových faktorů tepenné okluze.
Antiagregační léčba je zaměřena na omezení shlukovací schopnosti krevních destiček [4]. Aktivace krevních destiček nabízí vzhledem k počtu zúčastněných faktorů několik úrovní, ve kterých je možno farmakologicky zasáhnout a léčbu je možné kombinovat. Trombocyty je možné inhibovat ve fázi adheze, aktivace nebo agregace, popřípadě můžeme potencovat jejich stabilitu (tab. 2) [19].
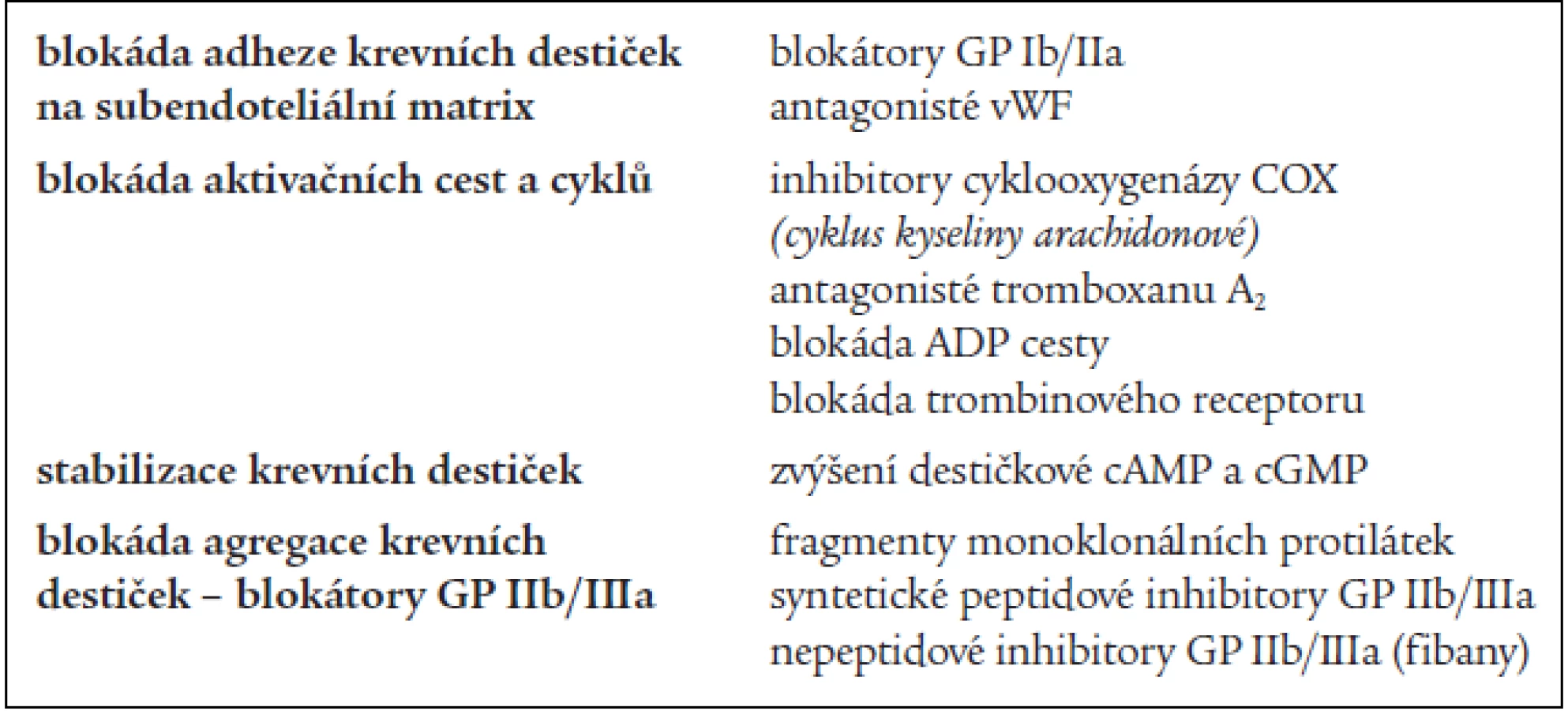
Existuje celá řada chemických látek, včetně léků, které prokazatelní snižují destičkové funkce. Jenom některé z nich je možné využít léčebně (tab. 3).

V tab. 4 jsou uvedeny léky užívané v léčbě a prevenci arteriálních tromboembolizmů, hlavní farmakologické působení spočívá v ovlivnění destičkových funkcí [5].

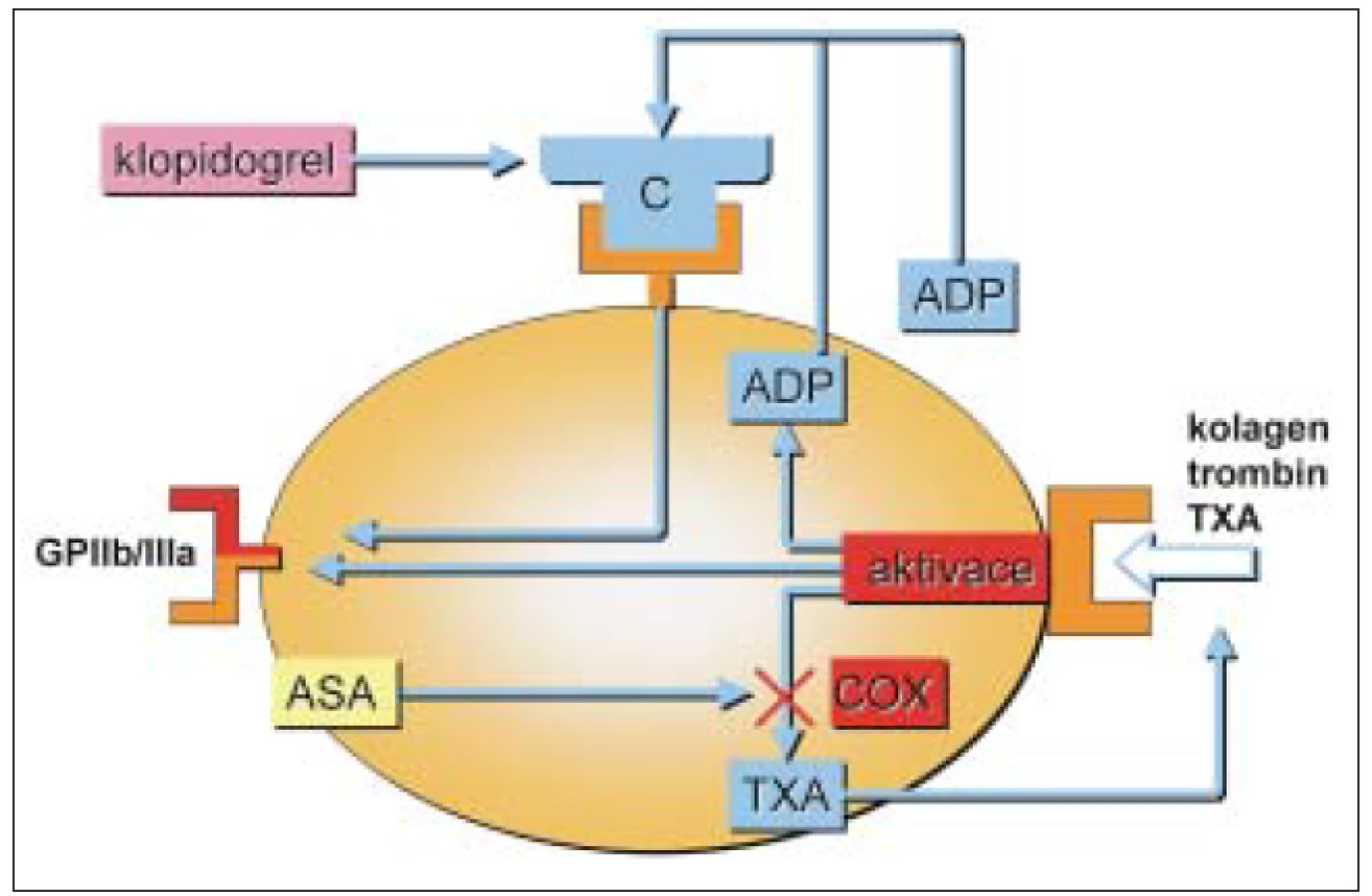
Mechanizmus působení protidestičkových léků je uveden na obr. 1.
Blokáda adheze krevních destiček na subendoteliální matrix
Ve fázi adheze lze inhibovat vazbu na obnažená kolagenní vlákna buď na úrovni destičkových receptorů, nebo blokádou vWF, který tuto vazbu zprostředkuje.
Blokátory GP Ib/IIa: zkouší se blokáda kyselinou aurinkarboxylovou, heterogenní směsí polykarboxylových polymerů nebo proteiny izolovanými z jedu Bothrops jararaca.
Antagonisté vWF: používají se nefunkční fragmenty vWF, které obsazují vazebná místa. Blokáda na tomto místě zabrzdí vazbu trombocytů na obnažená kolagenní vlákna, zabrání jejich adhezi, aktivaci a pozdější agregaci [23].
Blokáda enzymů cyklu kyseliny arachidonové
Blokace acetylsalicylovou kyselinou (ASA)
Kyselina acetylsalicylová je bezpochyby nejstarším a nejužívanějším lékem, který má nepochybný protidestičkový efekt. Již Hippokrates používal 400 let před Kristem jako analgetikum a antipyretikum odvar z vrbové kůry, která obsahuje salicin – název odvozen od latinského názvu vrby – salix [6]. Italský chemik Rafael Piria vyluhoval z extraktu vrbové kůry kyselinu salicylovou a v roce 1859 ji syntetizoval Herman Kolb [15]. V roce
1899 syntetizoval Felix Hoffmann kyselinu acetylsalicylovou [21]. Teprve v roce 1950 bylo prokázáno prodloužení krvácivosti po podání ASA. Vliv acetylsalicylové kyseliny na krevní destičky publikoval v roce 1967 Morris. Antiagregační účinek ASA je dán interferencí s biosyntézou cyklických prostanoidů v cyklu kyseliny arachidonové [3]. ASA vyvolá nevratnou inhibici cyklooxygenázy a následné potlačení syntézy tromboxanu A2 a dalších prostaglandinů [9]. ASA je poměrně rychle deacetylována játry – plazmatický poločas je asi 20 minut.
V roce 1991 byly zjištěny 2 izoformy cyklooxygenázy: COX-1 a COX-2 [21]. COX-1 se nachází v krevních destičkách, kdežto COX-2 hlavně v monocytech a makrofázích, ve kterých se tvoří „de novo“. Obě izoformy cyklooxygenázy jsou membránově vázané enzymy, které tvoří úzký a dlouhý hydrofobní kanál, do kterého vstupuje kyselina arachidonová uvolněná z poškozené membrány. Enzymy katalyzují přeměnu této kyseliny na prostaglandiny. COX-1 vzniká expresí genu o velikosti 22 kB na chromozomu 2 nebo 9, kdežto COX-2 je kódován genem o velikosti 8,3 kB lokalizovaným na chromozomu 1. Bílkovinné řetězce mají stejnou molekulovou hmotnost (72 kDa) a vykazují 63% homologii v sekvenci aminokyselin [22]. ASA je 50–100krát
účinnějším inhibitorem destičkové COX-1 než monocytární COX-2. Přestože plazmatický poločas ASA je poměrně krátký, stačí za tuto dobu inhibovat prakticky všechny destičky. Navíc se předpokládá, že ASA inaktivuje COX i v relativně zralých megakaryocytech, ve kterých jsou již přítomny zárodky krevních destiček [3]. Inhibice je nevratná a vzhledem k tomu, že destičky nedokáží COX syntetizovat, přetrvává po celou dobu biologického života trombocytu (10–12 dní). V současné době se používají terapeutické dávky ASA v rozmezí 50–100 mg za den. Tato léčba by měla zajistit účinnou blokaci syntézy tromboxanu A2, aniž by současně blokovala endoteliální produkci PGI2.
Účinek ASA na krevní destičky a endotelové buňky
ASA ireverzibilně acetyluje cyklooxygenázu (COX-1) v cyklu kyseliny arachidonové. Molekulární mechanizmus této inhibice spočívá v blokádě COX kanálu v důsledku acetylace hydroxylové skupiny serinového rezidua v místě Ser529. Tím je zabráněno přístupu substrátu ke katalytickému místu enzymu [19].
ASA dále ireverzibilně inaktivuje COX-1 v krevních destičkách, tím zamezuje arachidonové kyselině se vázat částí své molekuly ke COX-1 [21]. Konečně ASA acetyluje COX-1 v endotelových buňkách a zamezuje tak následné konverzi arachidonové kyseliny na výrazné vazodilatátory, zejména na prostacyklin PGI2.
Pro acetylaci serinového rezidua COX-1 platí následující:
- a) COX-1 (prostaglandin H syntáza: PGHS-1) je exprimována mnoha tkáněmi a buňkami, zahrnujícími krevní destičky, endotelové buňky, buňky střevní sliznice a jiné.
- b) Inhibiční vliv ASA k COX-1 je závislý na dávce. Jednorázová dávka 160 mg ASA nebo denní dávka 30–50 mg ASA po dobu 7–10 dnů by měla plně inhibovat destičkovou COX-1 [14].
Pro léčbu inhibitory cyklooxygenáz je tedy nejčastěji využívána ASA. Inhibice cyklooxygenáz po ASA je ireverzibilní, nebo reverzibilní, například po indomethacinu a sulfinpyrazonu. Kyselina acetylsalicylová inaktivuje cyklooxygenázu acetylací jejich aktivních míst po celou dobu života destiček, které jako bezjaderné elementy nejsou schopné syntézy nového enzymu. Nežádoucím výsledkem působení acetylsalicylové kyseliny je i inhibice cyklooxygenázy v endoteliálních buňkách, a tím i inhibice prostacyklinu. Malé dávky acetylsalicylové kyseliny by měly selektivně inhibovat tvorbu tromboxanu v destičkách bez významnější inhibice prostacyklinu ve stěně cévní. Disociace obou účinků je pouze relativní, protože již jen velmi nízká dávka ASA (20 mg) může blokovat syntézu prostacyklinu. ASA blokádou cyklooxygenáz tedy zabraňuje tvorbě obou základních metabolitů kyseliny arachidonové s opačnými biologickými účinky tromboxanu i prostacyklinu. Látky, které specificky blokují tromboxansyntetázu, by teoreticky měly tento problém vyřešit, protože tromboxansyntetáza je v hojné míře přítomna v destičkách, zatímco zcela chybí v endotelu [12]. Při selektivní blokádě jedné cesty kyseliny arachidonové dochází nahromadění ostatních produktů, v tomto případě účinného agregačního faktoru PHG2. Vzhledem k tomu, že k aktivaci fosfolipáz je nezbytná zvýšená hladina cytoplazmatického vápníku, vedou k redukci syntézy prostaglandinů i blokátory vápníkových kanálů. Jinou cestou k ovlivnění produkce eikosanoidů je manipulace s nenasycenými mastnými kyselinami, které jsou jejich zdrojem. Dihomo-gama-linolenová kyselina (DHLA) je 20uhlíková kyselina s 3 dvojnými vazbami, která dává vzniknout sérii prostaglandinů, finálním produktem je PGE2, účinný inhibitor destičkových funkcí, účinkující stimulace adenylcyklázy, zatímco tromboxan a prostacyklin jsou tvořeny jen velmi málo a mají zanedbatelný biologický efekt [14, 22,24].
V některých zemích se využívá inhibice triflusalem, který podobně jako ASA blokuje cyklooxygenázu. Výhodou je, že inhibice je omezena na cyklooxygenázu krevních destiček a cyklooxygenáza endotelií není ovlivněna. Syntéza prostacyklinu je tak plně zachována.
Další látkou zasahující do syntézy prostaglandinů, je eikosapentaneová kyselina (EPA). Je to také 20uhlíková kyselina s 5 dvojnými vazbami a je také prekurzorem jiné série prostaglandinů včetně PGI3, jehož antiagregační účinek je stejně silný jako efekt prostacyklinu, zatímco takto vznikající TXA3 má ve srovnání s tromboxanem A2 velmi slabý antiagregační a vazokonstrikční účinek. Variace ve vzniku prostaglandinů lze ovlivnit dieteticky zvýšeným přísunem například ryb s vysokým obsahem EPA.
Inhibitory tromboxansyntetázy
Využívá se imidazolových přípravků. Jedná se o kombinované blokátory receptorů TXA2 a tromboxansyntetázy. Inhibitory tromboxansyntetázy se ukázaly v klinické prevenci arteriální trombózy jako nedostatečné, protože zvýšená produkce prostacyklinu je provázena zvýšenou aktivitou prostaglandinu D2 (PGD2), a tím se kompenzuje nedostatek tromboxanu. Používají se léky, ve kterých se kombinuje inhibice tromboxanu a endoperoxidů (ipetrpofan, vapiprost, daltroban).
Stabilní analoga prostacyklinu, např. iloprost, mají významný antiagregační efekt, protože zvyšují cyklický AMP a mají vliv na cytosolický vápník [16].
Blokace membránových receptorů krevních destiček
Selektivní inhibitory receptorů ADP
ADP je vylučovaný z denzních granul destiček a z aktivovaného cévního subendotelu.
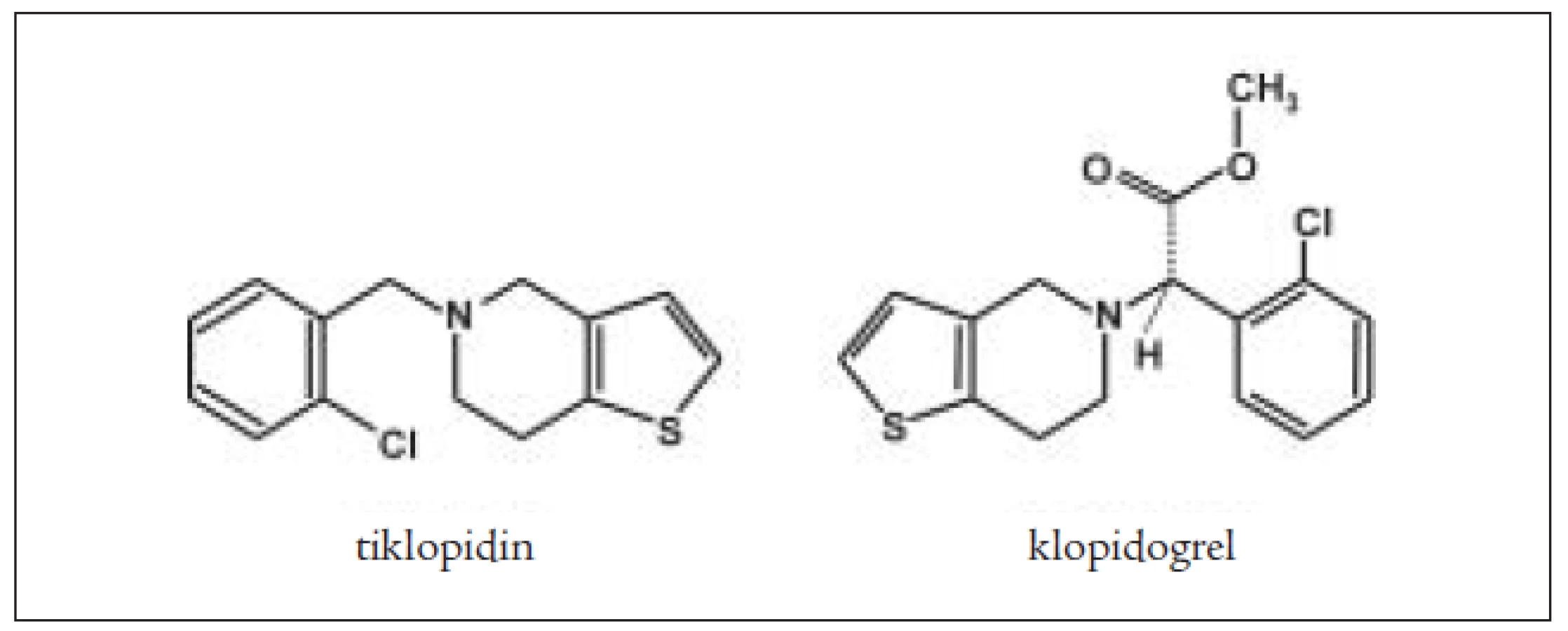
Tiklopidin a klopidogrel jsou thienopyrimidinové deriváty (obr. 2), které ireverzibilně blokují receptory aktivace destiček pro ADP typu P2Y1. Jiné cesty aktivace zůstávají neovlivněny. Bylo prokázáno, že prodlužují přežívání destiček, nepřímo ovlivňují vazbu fibrinogenu na GPIIb/IIIa. Neovlivňují agregaci provokovanou kolagenem a trombinem a neovlivňují syntézu prostaglandinů. Blokováním ADP thienopyridiny inhibují vazbu na fibrinogen pomocí receptoru GPIIb/IIIa, ale na GPIIb/IIIa přímo nepůsobí. Inhibice ADP indukované agregace je 50–60% po 4–6 dnech. Blokují uvolňování ADP z denzních granul, a tím blokují sekundární agregaci vyvolanou ionty Ca++ a serotoninem a velmi denzních granulí (α) (trombospodin a fibrinogen). Klopidogrel je indikován v prevenci ischemických příznaků u nemocných s aterosklerózou. Mechanizmus účinku je stejný, působí jako inhibitor funkce destiček pomocí ireverzibilní inhibice ADP indukované agregace destiček. Dávkování je 75 mg/denně p.o., 1krát denně. Absorpce klopidogrelu je rychlá a nezávislá na jídle a antacidech. Metabolicky se aktivuje v játrech a jeho plazmatická koncentrace má vrchol po 1 hodině po podání. Lze prokázat jeho neaktivní metabolit SR 26334, jeho aktivní metabolit není znám. Eliminuje se do 7 hodin. Z vedlejších účinků jsou některé častější jako průjem a vyrážka, nebývá cytopenie a u některých nemocných může vzniknout gastrointestinální krvácení. Klopidogrel při současném podání s trombolytiky (tPa) zvyšuje riziko krvácení, stejně tak zvyšuje riziko krvácení i současné podávání nesteroidních antiflogistik [8]. Klopidogrel se metabolizuje cytochromem P 450 v játrech. U starších osob není nutné měnit dávkování. Klopidogrel zvyšuje krvácení u nemocných s renální nedostatečností a u jaterního selhání [8]. Současné podávání warfarinu vede ke výšení rizika krvácení. Destičky nemocných, kteří užívají tyto léky, nejsou schopné agregovat po stimulaci ADP, ale zachovávají si schopnost změny tvaru po přidání ADP. Blokáda receptorů P2Y1 není schopna inhibovat všechny ADP receptory aktivace destiček. Je snaha najít inhibitory receptorů P2YAD. Po tiklopidinu jsou popisovány častější aplazie kostní dřeně [16,19].
Receptory trombinu jsou tyto:
- blokátory trombinových destičkových receptorů PAR–1: jejich účinek je založen na principu protilátek proti vlastnímu receptoru
- trombostatin: jedná se o degradační oligopeptidový produkt bradykininu o 5 aminokyselinách, který se váže přímo na aktivní místo receptoru.
Komplexní inhibitory jsou léčiva s komplexním působením. Patří sem anagrelid, který inhibuje aktivaci po řadě podnětů (ADP, trombin, kyselina arachidonová, kolagen aj). Po dlouhodobém podávání se může objevit trombocytopenie.
Blokáda agregace krevních destiček – blokátory GP IIb/III
Inhibitory glykoproteinových receptorů typu IIb/IIIa zasahují v místě konečného a společného bodu všech cest vedoucích k aktivaci krevních destiček, v důsledku toho blokují agregaci destiček, která je vyvolána všemi agonisty. Blokátory GP IIb/IIIa blokují spojování trombocytů přes můstky Ca++ a fibrinogen. Inhibitory blokují připojení fibrinogenu k receptoru IIb/IIIa na krevních destičkách tím, že obsadí receptorové místo na konformačně změněné molekule GP IIb/IIIa. Antagonisté GP IIb/IIIa tím, že ovlivňují fibrinogenový receptor, také inhibují adhezivitu krevních destiček svým působením na von Willebrandův faktor a fibronektin. Antagonisté GP Ib brání interakci von Willebrandova faktoru s endotelem [17,18].
Přehled blokátorů GP IIb/III
Inhibitory na bázi monoklonální protilátky
Abciximab – chimerická myší/lidská monoklonální protilátka blokující nespecificky sterickou zábranou destičkový receptor GP IIb/IIIa. Abciximab blokuje i vitronektinový receptor (αvβ3) endotelií, buněk hladkého svalu a destiček a váže se i na αuβ2 (Mac-1) receptor monocytů, neutrofilů a NK buněk (natural killer cells).
Inhibitory s malou molekulou
Tyto látky napodobují RGD sekvenci aminokyselin fibrinogenu a váží se specificky na receptor krevních destiček IIb/IIIa.
K dispozici jsou tyto nízkomolekulární látky:
Peptidové inhibitory
- eptifibatid: cyklický heptapeptid odvozený z hadího jedu pygmejového chřestýše. Má nahrazen Lys za Arg. Jedná se tedy o sekvenci KGD, která napodobuje RGD sekvenci a váže se s vysokou afinitou na GP IIb/IIIa.
Nepeptidové inhibitory
- tirofiban: nepeptidový derivát tyrozinu
- lamifiban [23].
Blokátory aktivátoru agregace krevních destiček – trombinu
Nízkomolekulární hepariny – LMWH v přítomnosti AT potencují jeho účinek na aktivovaný faktor Xa, a tím snižují produkci aktivátoru krevních destiček – trombinu. Použití LMWH umožňuje snížit destičkovou agregaci vyvolanou trombinem, což má význam zejména při endovaskulárních výkonech na koronárním řečišti. Aktivace destiček ostatními mechanizmy zůstává zachována [1,23].
Ostatní možné inhibitory destičkových funkcí
Sulodexid je látka se zajímavým účinkem, má 2 frakce. Iduronyl-glykosaminoglykan sulfát se váže na antitrombin a dermatan sulfát se váže na heparin kofaktor II. Významným způsobem ovlivňuje endoteliální dysfunkci, čímž upravuje i funkce krevních destiček u hyperreaktivních stavů.
Statiny se také počítají mezi látky s protidestičkových efektem a jsou nazývány buněčnými antitrombotiky. Statiny inhibují 3-hydroxy-3-methylglutaryl-CoA reduktázu, která produkuje kyselinu mevalonovou ve 3. kroku v cyklu izoprenoidů. Inhibicí 3-hydroxy-3-methylglutaryl-CoA reduktázy zastavují tvorbu a účinky kyseliny mevalonové, a tím zastavují tvorbu izoprenů. Touto cestou dochází k inhibici tvorby a aktivity trombinu a inhibici proliferace hladkých svalových vláken intimy vyvolané trombinem. Statiny se podílejí na úpravě destičkové dysfunkce tím, ze inhibují růstový destičkový faktor (PDGF) a endoteliální růstový faktor (ERGF).
Snahou objektivizovat vliv protidestičkové léčby u nemocných s vysokým rizikem arteriálních uzávěrů se zabývá řada prací. Výsledky prací na toto téma jsou shrnuty v analýze 287 studií, které referovaly o výsledcích 135 000 pacientů s protidestičkovou léčbou proti kontrolám. Bylo porovnáno 77 000 pacientů v různých režimech protidestičkové léčby. Hlavním sledovaným výsledkem byl: výskyt závažných cévních uzávěrů, dále nefatální srdeční infarkty, nefatální ikty nebo úmrtí na cévní onemocnění. Výsledky jsou shrnuty v publikaci Antithrombotic Trialists’ Collaboration [2].
Výsledky svědčí pro tyto závěry:
- protidestičková léčka je vhodná u všech rizikových nemocných
- protidestičková léčba snižuje závažné cévní příhody u rizikových nemocných, srdeční infarkt, nefatální ikty, tranzientní ischemické ataky, nestabilní anginu pectoris, obliterující aterosklerózu tepen dolních končetin. Dále snižují rizika embolie při fibrilaci síní a rizika cévních uzávěrů u jiných nemocných s vysokým rizikem.
- protidestičková léčba se má podávat dlouhodobě
- nízkodávkovaná kyselina acetylsalicylová (ASA) (75–150 mg/den) je stejně účinná jako vysoké dávky ASA
- antagonisté ADP-receptorů jsou jediné, které se ukazují jako efektivnější než ASA (% odds reduction)
- kombinace ASA s jiným protidestičkovým lékem (klopidogrel, antagonisté GPIIb/IIa) je pro nemocné výhodná.
Existuje fenomén, který se nazývá rezistence na ASA. O tomto jevu lze hovořit buď z klinického pohledu – jako o selhání protektivního účinku ASA před trombotickou komplikací, nebo ji lze definovat laboratorně – jako neschopnost způsobit in vitro prokazatelnou inhibici destičkových funkcí [7,10,13].
Práce byla podpořena grantem IGA MZ NR8036-3/04 a výzkumným záměrem MZO 001179906.
prof. MUDr. Jaroslav Malý, CSc.
www.fnhk.cz
e-mail: maly@fnhk.cz
Doručeno do redakce: 11. 3. 2005
Přijato k otištění: 11. 3. 2005
Sources
1. Andersson LO, Borrowcliffe TW, Holmer E et al. Molecular weight dependency of the heparin potentiated inhibition of thrombin and activated factor X. Effect of heparin neutralization in plasma. Thromb Res 1979; 115 : 531–541.
2. Antithrombotics Trialist’s Collaboration. Collaborative metaanalysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction and stroke in high risk patiens. BMJ 1999; 318 : 759–764.
3. Awtry HA, Loscalzo J. Aspirin. Circulation 2000; 101 : 1206–1218.
4. Badimon L, Badimon JJ. Interaction of platelet activation and coagulation. In Fuster V, Topol EJ (Eds.) Atherosklerosis and coronary artery disease. Philadelphia: Lippincott – Raven Publishers 1996 : 639–656.
5. Bultas J, Karetová D. Léčba trombotických stavů – kde jsme a kam se ubíráme. Remedia 2004; 14 : 182–200.
6. Dalen JE. An apple a day or an aspirin a day. Arch Intern Med 1991; 151 : 1066–1069.
7. Gum PA, Kottke Marchant K, Poggio ED et al. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol 2001; 88 : 230–235.
8. Harrington RA, Becker RC, Ezekowitz Met al. Antithrombotic Therapy for Coronary Artery Disease: The 7th ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 : 513S–548S.
9. Hirmerová J, Filipovský J. Klinický význam aspirinové rezistence. Vnitř Lék 2004; 50 : 462–469.
10. Karetová D, Bultas J. Rezistence na aspirin – laboratorní odchylka nebo klinický problém. Interní medicína pro praxi 2005; 1 : 10–13.
11. Kottke-Marchant K, Corcoran G. The Laboratory Diagnosis of Platelet Disorders. Arch Pathol Lab Med 2001; 126 : 133–146.
12. Kurth T, Glynn RJ, Walker AM et al. Inhibition of clinical benefits of aspirin on first myocardial infarction by nonsteroidal intiinflamatory drugs. Circulation 2003; 108 : 1191–1195.
13. Malý J. Vyšetření aktivity destičkových funkcí se vztahem k rezistenci na kyselinu acetylsalicylovou. Vnitř Lék 2005; 51(2): 157–162.
14. Maree AO, Fitzgerald DJ. Aspirin and coronary artery disease. Thromb Haemost 2004; 92 : 1175–1181.
15. McKee SA, Sane DC, Dellargyris EN. Aspirin resistance in cardiovascular diseases: A review of prevalence, mechanisms and clinical significance. Thromb Haemost 2002; 88 : 711–715.
16. Patrono C, Coller B, FitzGerald GA et al. Platelet-Active Drugs: The Relationships Among Dose, Effectiveness, and Side Effects: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 : 234S–264S.
17. Popma JJ, Berger P, Ohman EM et al. Antithrombotic Therapy During Percutaneous Coronary Intervention: The 7th ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 : 576S–599S.
18. Patrono C, Bachmann F, Baigent C et al. European Society of Cardiology Expert consensus document on the use of antiplatelet agents. The task force on the use of antiplatelet agents in patients with atherosclerotic cardiovascular disease of the European society of cardiology. Eur Heart J 2004; 25 : 166–181.
19. Patrono C, Coller B, Dalen JE et al. Platelet – Active Drugs. The relationship among dose, effectiveness and side effects. Chest 2001; 119 : 39–63.
20. Pecka M. Laboratorní hematologie v přehledu. Fyziologie a patofyziologie hemostázy. Český Těšín: FINIDR 2004.
21. Samama MM, Elalamy I. Aspirine et hémostase. Rev Méd Interne 2000; 21(Suppl 1): 27–34.
22. Vane JR, Bakhle YS, Botting RM. Cyklooxygenases 1 a 2. Ann Rev Pharmacol Toxicol 1998; 38 : 97–120.
23. Vojáček J. Inhibitory destičkových glykoproteinových receptorů typu Iib/IIIa. Remedia 2003; 13 : 84–92.
24. Vojáček J, Malý M, Hraboš V et al. Hladina tkáňového faktoru, inhibitoru tkáňového faktoru a solubilního P–selektinu u nemocných s akutním koronárním syndromem. Cor Vasa 2002; 44 : 148–151.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2005 Issue 7 a 8
Most read in this issue
- Potransfuzní reakce
- Trombocytózy a trombocytemie
- Antifosfolipidový syndrom – diagnostika a léčba
- Protidestičková léčba