
Fenotypová a genotypová analýza vrodenej hypofibrinogenémie a dysfibrinogenémie
Phenotype and genotype analysis of hereditary hypofibrinogenaemia and dysfibrinogenaemia
Inherited fibrinogen disorders, either quantitative (afibrinogenemia, hypofibrinogenemia) or qualitative (dysfibrinogenemia) represent a rare blood coagulation disorders. Clinical manifestation is variable and the bleeding may paradoxically be associated with the thrombotic events. Recently the function of all components of the complex fibrinogen molecule as well as the molecular basis of fibrinogen disorders were defined. Currently the attention is focused upon the relationship between the clinical phenotype and the type of molecular defect with the aim to predict the clinical picture of disease. The phenotype analysis in 67 patients with inherited hypofibrinogenemia and dysfibrinogenemia registered in the National Registry of Inherited Bleeding Disorders in Bratislava has confirmed a wide variation in bleeding manifestation and unpredictable association of bleeding with the invasive procedures in patients with fibrinogen disorders. Genetic analysis in 24 patients has revealed three heterozygous novel mutations: in hypofibrinogenemia the exon 1 FGG gene mutation (Trp3→Stop) and exon 7 FGG gene mutations (Trp253→Cys), in dysfibrinogenemia the exon 2 FGA gene mutation (Aα Gly13→Glu). The results of our study, although limited, suggest an importance of the mutation identification in patients with inherited disorders of fibrinogen. However, further studies are needed to improve the predictive value of the mutational type for the clinical phenotype in particular patient.
Key words:
hypofibrinogenemia – dysfibrinogenemia – bleeding – thrombosis – gene mutations
Authors:
A. Bátorová 1; D. Horváthová 1; P. De Moerloose 2; M. Neerman-Arbez 3; M. Mistrík 1
Authors‘ workplace:
Národné hemofilické centrum Kliniky hematológie a transfuziológie LF UK, Bratislava, Slovenská republika, prednosta doc. MUDr. Martin Mistrík, Ph. D.
1; Division of Angiology and Hemostasis, University Hospital, Ženeva, Švajčiarsko, director prof. Dr. H. Bounameaux
2; Departement of Genetic Medicine and Development, University Medical School, Ženeva, Švajčiarsko, director prof. Dr. Stylianos Antonarakis
3
Published in:
Vnitř Lék 2005; 91(7 a 8): 802-808
Category:
128th Internal Medicine Day - 21rd Vanysek's Day Brno 2005
Overview
Vrodené poruchy fibrinogénu, či už kvantitatívne (afibrinogenémia a hypofibrinogenémia) alebo kvalitatívne (dysfibrinogenémia) patria medzi zriedkavé poruchy zrážania krvi. Klinické prejavy sú značne variabilné a popri krvácaní môžu byť paradoxne spojené aj s výskytom trombózy. V posledných rokoch bola definovaná funkcia jednotlivých častí zložitej molekuly fibrinogénu a odhalená genetická podstata porúch fibrinogénu. V súčasnosti sa intenzívne skúma nielen súvislosť medzi klinickým fenotypom a typom genetickej poruchy ale aj možnosti predvídania priebehu choroby na základe konkrétneho molekulového defektu. Analýza fenotypu u 67 pacientov s vrodenou hypofibrinogenémiou a dysfibrinogenémiou, evidovaných v Národnom registri vrodených koagulopatií v Bratislave, potvrdila variabilnosť klinických prejavov a nepredvídateľnosť krvácavých komplikácií pri invazívnych výkonoch. Genetické vyšetrenie u 24 jedincov odhalilo nové, doteraz neopísané mutácie v heterozygotnej forme: pri hypofibrinogenémii mutáciu FGG génu na exóne 1 (Trp3→Stop) a mutáciu FGG na exóne 7 (Trp253→Cys), pri dysfibrinogenémii mutáciu na exóne 2 FGA génu (Aα Gly13→Glu). Naše, i keď limitované výsledky naznačujú význam vyšetrenia genotypu pri poruchách fibrinogénu, jeho možné využitie ako prediktívneho markera klinického fenotypu si vyžiada ďalšie štúdium na väčších súboroch chorých.
Kľúčové slová:
hypofibrinogenémia – dysfibrinogenémia – krvácanie – trombóza – génové mutácie
Vrodené poruchy fibrinogénu patria medzi zriedkavé abnormality zrážania krvi. Od prvého opisu vrodenej afibrinogenémie v roku 1920 [1] a hypofibrinogenémie v roku 1935 [2] bolo zaznamenaných viac jako 200 prípadov kvantitatívnych porúch fibrinogénu. Častejšími sa ukázali byť kvalitatívne abnormality, známe až od 60. rokov minulého storočia [3], s dodnes opísanými viac jako 400 prípadmi dysfibrinogenémií. S postupným vývojom sofistikovaných metód analýzy sa menila aj definícia a klasifikácia abnormalít fibrinogénu. Afibrinogenémia je definovaná ako imunologickou metódou potvrdená absencia fibrinogénu v cirkulujúcej krvi, hypofibrinogenémia ako zníženie hladiny normálneho fibrinogénu. Vrodenú dysfibrinogenémiu charakterizujú štrukturálne abnormality molekuly fibrinogénu, ktoré vedú k zmene jeho funkčných vlastností. Klasickým nálezom je diskrepancia medzi nízkou hladinou fibrinogénu vyšetrenou Claussovou koagulačnou metódou (FBG:koag) a jeho normálnou hladinou pri stanovení imunologickou metódou (FBG:Ag). Pri hypodysfibrinogenémii je znížená aj imunologická hladina abnormálneho fibrinogénu pod 1,5 g/l v dôsledku jeho zníženej sekrécie alebo zvýšenej clearance. Prevalencia afibrinogenémie sa odhaduje na 1 : 1 000 000 [4], pričom geografické rozdiely výskytu odrážajú vysokú incidenciu u detí rodičov z pokrvných príbuzenských vzťahov v moslimských krajinách [5,6]. Incidencia dysfibrinogenémie nie je známa, možno však predpokladať, že súčasné údaje o skutočnom výskyte sú podhodnotené. Na Slovensku v Národnom registri vrodených koagulopatií evidujeme takmer 160 jedincov s rôznymi poruchami fibrinogénu. V práci predkladáme najnovšie poznatky o patofyziológii týchto porúch a výsledky fenotypovej a genotypovej analýzy v našom súbore chorých s hypo - a dysfibrinogenémiou.
Štruktúra fibrinogénu a jeho úloha v hemostáze
Fibrinogén (FBG) je glykoproteín o veľkosti 340 kDa s homodimérickou štruktúrou, pričom každá podjednotka pozostáva z troch polypeptidových reťazcov, Aα, Bβ a γ. Podľa elektrónmikroskopického obrazu má molekula fibrinogénu trinodulárnu štruktúru: centrálna globulárna doména E obsahuje aminoterminálne konce všetkých 6 polypeptidových reťazcov a je prostredníctvom dvoch helikálnych špirálovitých domén spojená s karboxyterminálnymi globulárnymi doménami D. Súčasné radiologické kryštalografické štúdie objasnili štruktúru viacerých častí molekuly fibrinogénu a poskytli bázu pre detailnejšie modelovanie protofibríl fibrínu [7]. Poznanie presnej úlohy jednotlivých častí molekuly FBG v hemostatických reakciách je kľúčom k pochopeniu funkčných porúch při rôznych typoch dysfibrinogenémií.
Fibrinogén je syntetizovaný prevažne v hepatocytoch, nachádza sa v plazme a v α-granulách trombocytov, ktoré ho získavajú z plazmy endocytózou. Fibrinogén je reaktantom akútnej fázy, mediátorom jeho zvýšenej syntézy je interleukín-6, ktorý zvyšuje koncentráciu m-RNA fibrinogénu v hepatocytoch. Silný zápalový stimulus môže viesť až k 20násobnému zvýšeniu koncentrácie fibrinogénu [8]. Biologický polčas fibrinogénu je 3–5 dní.
Hlavnou fyziologickou funkciou fibrinogénu je jeho účasť na tvorbe fibrínového koagula. Na začiatku sa fibrinogén mení na fibrín účinkom trombínu, ktorý sa v mieste Arg130 viaže na Glu11 Aα reťazca fibrinogénu. Trombín štiepi väzbu Arg16–Gly17 na aminoterminálnom konci reťazca Aα a väzbu Arg14–Gly15 na reťazci Bβ, čo vedie k uvoľneniu fibrinopeptidov A (FPA; Aα 1–16) a B (FPB; Bβ 1–14) a vzniku monomérov fibrínu. V doméne E sa po uvoľnení fibrinopeptidov exponujú väzobné „miesto A“ (tzv. A gombík, tvorený aminoterminálnou sekvenciou α Gly17–Pro18–Arg19) a „miesto B“ (sekvencia β Gly14–His15–Arg16) pre komplementárne väzobné miesta na D doménach iných monomérov fibrínu. V prvej fáze polymerizácie fibrínu dochádza k agregácii desA fibrínu, tvorbe oligomérov a dvojreťazcových protofibríl, ktoré vznikajú spájaním reťazcov end-to-end systémom „gombík-dierka“. Väzobné „miesto A“ α reťazca (gombík) sa spája s korešpondujúcim „miestom a“ γ reťazca (dierka), reprezentovaným karboxylovými skupinami γ Gln329, Asp330 a Asp364. Podobne „miesto B“ sa spája s „jamkou b“, tvorenou rezíduom Glu132 γ reťazca. Takto vznikajú reťazce obsahujúce striedavo domény D-E-D. V tejto fáze dochádza aj k asociácii D domén susedných fibrín monomérov, nachádzajúcich sa v jednom zväzku, bez vytvorenia špecifickej väzby (tzv. D:D interface). Špecifická väzba F XIIIa v mieste γ Gln398 zabezpečuje recipročné väzby medzi D doménami γ reťazcov (D xD) a stabilizáciu fibrínovej siete [7,9]. Laterálne spájanie reťazcov v mieste C-terminálnych segmentov α reťazcov (αC–αC doména) priľahlých protofibríl vedie k tvorbe stočených vlákien a hrubých zväzkov a vytvoreniu hustej fibrínovej siete.
Vznik fibrínu je stimulom pre uvoľnenie tkanivového aktivátora plazminogénu (t–PA) z endotelových buniek. V procese fibrinoformácie dochádza k inkorporácii zložiek fibrinolytického systému do vznikajúceho koagula, ktoré zabezpečia jeho následné rozpustenie. Miestom väzby plazminogénu je Arg554 na Cterminálnom konci α reťazca polymerizujúceho fibrínu, pre väzbu t-PA je potrebná prítomnosť dvojreťazcových protofibríl a normálna štruktúra oblastí reťazcov α 148–160, γ 311–336 a N–terminálnej časti β 9–72 [9].
N–terminálna časť β reťazca je dôležitá aj pre väzbu trombínu na fibrín, ktorá limituje jeho ďalšie prokoagulačné pôsobenie, čo má regulačný význam pre ohraničenie procesu hemostázy. Chýbanie tejto funkcie podporuje pretrvávanie trombinémie a jej aktivačného pôsobenia na trombocyty. Významnou úlohou fibrinogénu je aj jeho väzba na bunkové integríny: väzba na endotelové bunky podporuje normálny proces hojenia a väzba na GP IIbIIIa trombocytov ich agregáciu a proces primárnej hemostázy.
Klinický obraz vrodených porúch fibrinogénu
Klinické prejavy vyplývajú z chýbania alebo poruchy niektorej z fyziologických funkcií fibrinogénu a fibrínu. Charakteristiky klinického obrazu sa väčšinou opierajú len o publikované kazuistiky [10,11,12,13,14,15,16, 17,18] a dva početnejšie súbory chorých s afibrinogenémiou [5,6]. Pre afibrinogenémiu je typická zvýšená náchylnosť na krvácanie s variabilnou manifestáciou a zhoršené hojenie rán. K najčastejším prejavom patrí krvácanie z pupočníka, slizničné krvácanie a krvácanie do kĺbov. Svalové, CNS s GIT krvácania sú zriedkavé, boli opísané aj prípady intraabdominálneho krvácania v dôsledku spontánnej ruptúry sleziny [5,10,11,12,13]. Pri hypofibrinogenémii sú prejavy krvácania mierne, vyskytujú sa najmä pri invazívnych procedúrach. Pri oboch poruchách sa u viacerých chorých popri krvácaní paradoxne vyskytli aj prejavy trombózy, a to nielen v súvislosti so substitučnou liečbou [12,16,17], ale aj bez nej [6]. U žien fertilného veku sa okrem hypermenorrhoe pozorovali aj komplikácie gravidity, krvácanie v gravidite a po pôrode [4] ale aj spontánne potraty. Výskyt opakovaných abortov v 6.–8. týždni gravidity, teda v čase, keď trofoblast infiltruje myometrium, poukazuje na význam fibrinogénu pre implantáciu embrya [19].
Dysfibrinogenémia je charakterizovaná variabilným klinickým fenotypom. Prehľad 250 publikovaných prípadov dysfibrinogenémie spracovaný SSC komisiou ISTH (International Society of Hemostasis and Thrombosis) demonštroval asymptomatický priebeh u 133 (53 %) chorých, 65 (26 %) chorých malo prejavy krvácania a až u 52 (21 %) chorých sa manifestovala trombóza, 6 chorých malo kombinovaný krvácavý a trombotický fenotyp [15]. Krvácanie, väčšinou len mierne, sa vyskytuje najčastejšie po provokácii (extrakcia zubov, operácia, pôrod), umbilikálne a CNS krvácania sú zriedkavé, vznikajú len pri veľmi nízkej hladine fibrinogénu [20]. Prehľad 26 pacientov s familiárnou dysfibrinogenémiou a trombózou v databáze ISTH a analýza ich príbuzných s a bez vrodenej dysfibrinogenémie [15], i ďalšie publikované prípady potvrdzujú spojenie medzi dysfibrinogenémiou a trombofíliou [14,18]. Dysfibrinogenémia sa v súčasnosti považuje za nezávislý, i keď menej frekventovaný rizikový faktor venóznej trombózy s prevalenciou 0,8 % v súboroch chorých s venóznou trombózou [21]. Komplikácie tehotenstva zahŕňajú spontánne aborty a intrauterínne odumretie plodu, pôrodné a popôrodné krvácanie a puerperálne trombózy.
Diagnostika vrodených porúch fibrinogénu
Diagnostika sa v súčasnosti opiera o štandardné metódy vyšetrenia koagulačnej a imunologickej hladiny fibrinogénu. Pri afibrinogenémii sa zisťujú nemerateľné hodnoty trombínového času, protrombínového času a aktivovaného parciálneho tromboplastínového času. Pri hypofibrinogenémii a dysfibrinogenémii sú uvedené testy mierne predĺžené. Dysfibrinogenémiu charakterizuje diskrepancia medzi hladinou FBG:koag a FBG:Ag, odrazom funkčnej poruchy je pravidelný nález pozitivity fibrín degradačných produktov (FDP) a u časti chorých aj D-dimérov. Diferenciálne diagnosticky je potrebné vylúčiť získané poruchy fibrinogénu pri hepatopatii, DIC a malignitách, po liekoch (L-asparagináza, anti-thymocytový globulín) a inhibítory fibrinogénu pri autoimúnnych a iných ochoreniach (heparin-like molekuly, paraproteín, makroglobulinémia, FDP). V súčasnosti sa obracia pozornosť na molekulovú analýzu defektov fibrinogénu a jej diagnostický a diferenciálne diagnostický význam.
Molekulová báza vrodených porúch fibrinogénu
Syntéza každého reťazca fibrinogénu je kódovaná vlastným génom: reťazec Aα génom FGA (5,4 kb, 6 exónov), Bβ génom FGB (8,2 kb, 8 exónov) a reťazec γ génom FGG (8,4 kb, 10 exónov). Všetky tri gény sú klustrované na dlhom ramienku chromozómu 4 q23–q32.
Vrodená afibrinogenémia je autozomálne recesívne ochorenie. Po odhalení prvej mutácie spôsobujúcej defekt syntézy fibrinogénu (delécia 11 kb na géne FGA) [22] boli identifikované ďalšie mutácie lokalizované na všetkých troch génoch fibrinogénu. S výnimkou troch „missense“ mutácií na géne FGB išlo vždy o „nulové“ mutácie (frameshift, nonsense alebo splice–site) spôsobujúce chýbanie korešpondujúceho reťazca [12,23,24,25].
Keďže sa vrodená dysfibrinogenémia dedí autozomálne dominantne, pozitívna rodinná anamnéza sa považuje za dôležité diagnostické kritérium poruchy. V negatívnom prípade sa za dôkaz vrodenej dysfibrinogenémie považuje potvrdenie a lokalizácia defektu na molekulovej úrovni. Doteraz sa identifikovalo už viac ako 300 abnormálnych fibrinogénov a viac ako 100 štrukturálnych defektov, podmienených väčšinou „missense“ mutáciami. Podľa databázy mutácií je väčšina z nich (180) lokalizovaná na géne FGA (z nich 74 mutácií v mieste Aα 16), 27 mutácií sa našlo na FGB a 75 mutácií na FGG (z nich 25 v mieste γ 275) [23]. Prítomnosť abnormálnej molekuly fibrinogénu sa dotýka všetkých aspektov jeho fyziologických funkcií [9,15].
Porucha väzby trombínu na fibrinogén pri dysfibrinogenémii Lille (Aα Asp7→Asn), Mitaka II (Aα Glu11→Gly) a Rouen (Aα Gly12→ Val), sprevádzaná opozdeným uvoľňovaním FPA, prebieha väčšinou asymptomaticky.
Poruchu štiepenia FPA a FPB podmieňujú mutácie v mieste Aα Arg16–Gly17 a Bβ Arg14–Gly15. Dysfibrinogenémie Aα Arg16→His (Giesen I, Chapell Hill II, Bern IV), Aα Arg16→Cys (Osaka I) a Bβ Arg14→Cys (Christchurch) prebiehajú asymptomaticky alebo s miernym krvácaním, ale môže sa vyskytnúť aj trombóza.
Porucha polymerizácie fibrínu je podmienená mutáciami reťazcov Aα a γ. Kritickými sú mutácie v polohe γ 268–280 (γArg275→His; Haifa) a mutácie väzobného „a miesta“ na reťazci γ: Gln 329→Val (Nagoya), Asp 330→Val (Milano I), Asp 330→Tyr (Kyoto III) a Asp 364→Val (Melun I). Dysfibrinogenémia Melun I a Tokyo V (γ Asp 327→Thr) boli spojené s rekurentnými trombotickými prejavmi v dôsledku tvorby fragilného, ale na fibrinolýzu rezistentného fibrínového koagula [18]. Aj mutácia Aα Arg 554→Cys (Chapell Hill III, Paris V) s poruchou laterálneho spájania protofibríl a vznikom kompaktnejšieho koagula so zníženou permeabilitou, sa manifestovala trombózou.
Porucha väzby trombínu na fibrín sprevádzajúca deléciu Bβ7–92 (New York I) a mutáciu BβAla68→Thr (Neapol) spôsobuje perzistenciu trombinémie v cirkulácii s pokračujúcou aktiváciou trombocytov a koagulácie. Tieto poruchy majú vzťah k výskytu rekurentnej arteriálnej aj venóznej trombózy [9,15].
Poruchu fibrinolýzy spôsobuje znížená väzba plazminogénu na patologický fibrinogén Aα Arg 554→Cys (Chapell Hill III, Paris V) a znížená väzba t–PA pri poruche väzobných miest Aα 148–160 alebo γ 311–336. Zatiaľ čo pri fibrinogéne Kyoto III (Asp 330→Tyr) sa trombóza nepozorovala, tromboembolická choroba môže sprevádzať deléciu N-terminálneho konca Bβ7–92 (New York I) a zámenu Bβ Arg44→Cys (Nijmegen). Patologická molekula fibrínu Marburg (Aα 461Lys→Stop) podlieha kritickým štrukturálnym zmenám pod vplyvom faktora XIIIa, ktorých výsledkom je rezistencia na plazmín. Porucha sa prejavila závažným post-partum krvácaním a rekurentnou tromboembolickou chorobou.
Vzhľadom na skutočnosť, že v niektorých prípadoch vrodenej dysfibrinogenémie existuje korelácia medzi molekulovým defektom a klinickým fenotypom [9,15,23], zvyšuje sa v súčasnosti záujem o diagnostiku dysfibrinogenémií na molekulovej úrovni s cieľom predpovede klinického priebehu choroby a možného genetického poradenstva.
Liečba porúch fibrinogénu
Liečba hypofibrinogenémie a dysfibrinogenémie spočíva v substitúcii fibrinogénu vo forme bezpečného koncentrátu fibrinogénu alebo kryoprecipitátu, ktorý však nie je antivírusovo opracovaný. Za hemostatické minimum sa považuje hladina 0,5 až 1,0 g/l funkčne normálneho fibrinogénu. Zvýšenie hladiny fibrinogénu v plazme príjemcu približne o 1 g/l sa dosiahne podaním 2–3 g fibrinogénu alebo 10 TU kryoprecipitátu. Spontánne prejavy krvácania a zvyčajne mierne krvácanie po extrakciách zubov nevyžadujú substitučnú liečbu. V literatúre však chýbajú relevantné informácie o reálnom stupni rizika a frekvencii krvácania po invazívnych výkonoch pri dysfibrinogenémii a ich prevencii. Príprava na operácie a manažment tehotenstva a perinatálnej starostlivosti u žien by mali byť individualizované podľa predchádzajúceho klinického priebehu [4]. U pacientov s anamnézou krvácavosti sa na zabezpečenie adekvátnej hemostázy odporúča pred operáciou zvýšiť hladinu fibrinogénu o 1 g/l nad bazálnu hodnotu fibrinogénu pacienta, neskôr udržiavať zvýšenie hladiny o 0,5 g/l nad bazálnou hodnotou až do zhojenia. Súčasná aplikácia antifibrinolytík nepatrí k paušálnej medikácii vzhľadom na možnosť zvýšenia rizika trombózy. U predtým symptomatických pacientov sa v prípade operácie odporúča len pozorovanie a substitúcia len pri krvácaní, naopak u pacientov s anamnézou trombózy by mali byť operačné výkony clonené aj adekvátnou tromboprofylaxiou.
Fenotypová a genotypová analýza vrodenej hypofibrinogenémie a dysfibrinogenémie na Slovensku
V Národnom registri vrodených koagulopatií Slovenskej republiky v súčasnosti evidujeme 118 jedincov s dysfibrinogenémiou (dysFBG), 40 jedincov s hypofibrinogenémiou (hypoFBG) a 1 pacienta s afibrinogenémiou. V roku 2004 sme vykonali analýzu fenotypu u 67 chorých s poruchami fibrinogénu (tab. 1), a v spolupráci so zahraničným pracoviskom vyšetrenie genotypu u 24 jedincov. V súbore 51 chorých s dysfibrinogenémiou (16 M/35 Ž), pochádzajúcich z 20 nepríbuzných rodín s mediánom hladiny FBG:koag a FBG:Ag 0,76 g/l a 2,8 g/l prebieha porucha asymptomaticky u 25 (49 %) jedincov a 24 (47 %) chorých má anamnézu miernych prejavov krvácania (hematómy po úderoch, epistaxy a krvácanie z ďasien). Horšie hojenie rán udáva 7 (14 %) jedincov. Trombóza sa manifestovala len u dvoch (4 %) chorých v neprítomnosti iných markerov trombofílie. 33 chorých s dysFBG sa podrobilo celkom 107 invazívnym výkonom bez substitučnej clony: krvácaním bolo komplikovaných 6 z 38 extrakcií zubov (16 %) a len 3 zo 69 operácií (4,3 %), z nich 1 z 21 (5 %) tonzilektómií/adenotómií. V 2 prípadoch pooperačného krvácania bola nutná substitúcia fibrinogénu. U 5 pacientov boli niektoré operácie bez krvácania, kým iné výkony boli komplikované krvácaním. 20 operácií u 13 chorých pod clonou fibrinogénu prebehlo bez krvácavých a trombotických komplikácií. Hypermenorrhoea sa vyskytla u 7 z 24 (29 %) žien vo fertilnom veku, všetky ženy mali úspešné tehotenstvá, jedna z nich mala 1 spontánny abort a dve ženy pôrod mŕtveho plodu. Z celkového počtu 51 pôrodov bez substitučnej clony boli len 4 (8 %) komplikované krvácaním.
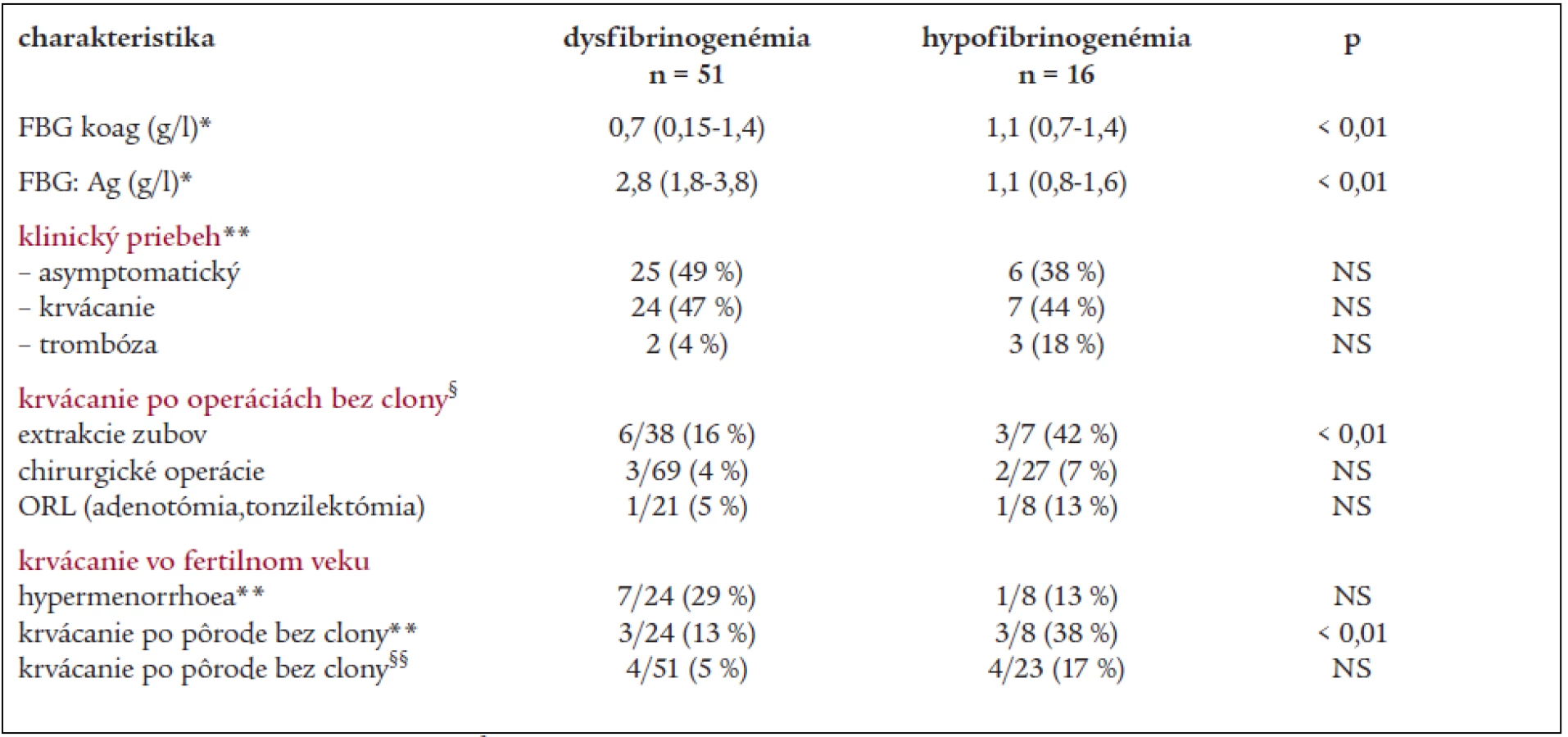
V skupine 16 chorých s hypofibrinogenémiou (6 M/10 Ž) zo 6 rodín s mediánom hladiny FBG:koag 1,1 g/l (rozptyl 0,7–1,4 g/l) a FBG:Ag 1,3 g/l (rozptyl 0,8–1,57 g/l) má mierne prejavy krvácania 7 (44 %) chorých (tab. 1). U troch chorých bez krvácavých prejavov došlo k manifestácii trombózy, 2 príbuzné ženy s recidivujúcimi trombózami majú súčasne heterozygotnú formu mutácie F V Leiden a MTHFR C677T, u jednej z nich bolo 7 gravidít ukončených pôrodom mŕtveho plodu. Z 27 operácií vykonaných u 13 chorých bez hemostatickej prípravy boli 2 operácie (8 %) komplikované krvácaním pri hladine FBG 0,7 a 0,8 g/l (tonzilektómia a laparoskopia), po extrakcii zubov sa krvácanie vyskytlo u 3 z 7 pacientov (42 %). Hypermenorrhoe mala len 1 z 8 žien fertilného veku, 7 žien malo úspešné tehotenstvá, u 3 z nich sa vyskytol aj 1 abort. Krvácanie po pôrode sa vyskytlo u 3/8 žien, z 23 pôrodov bez substitučnej clony boli krvácaním komplikované 3 spontánne pôrody a 1 sekcia (17,4 %).
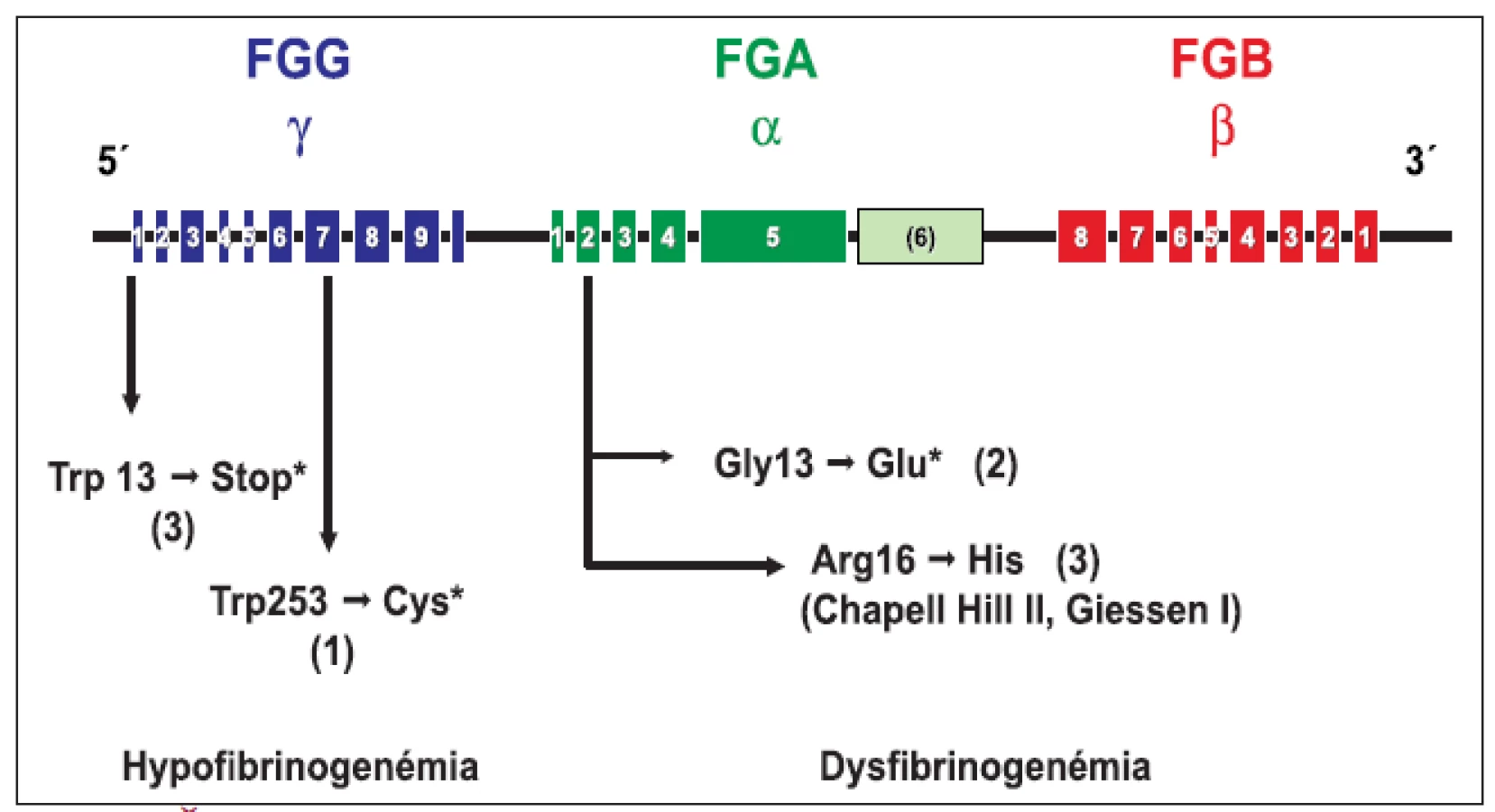
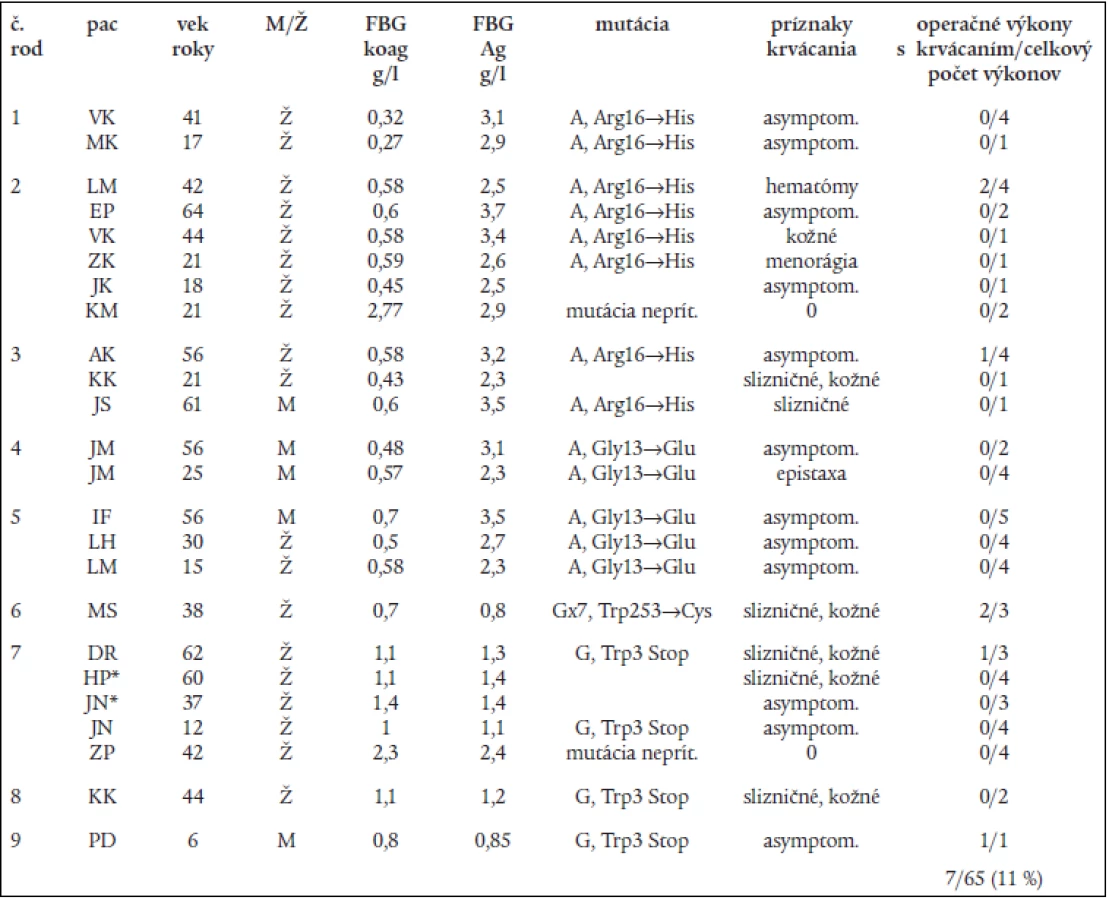
Genotypová analýza (obr. 1 a tab. 2) u 16 jedincov z 5 nepríbuzných rodín s familiárnou dysfibrinogenémiou potvrdila heterozygotnú formu mutácií: v 3 rodinách častú mutáciu génu FGA Arg16→His, a v 2 rodinách novú, zatiaľ neopísanú mutáciu génu FGA Gly13→Glu. Vyšetrenie 8 jedincov s hypofibrinogenémiou zo 4 nepríbuzných rodín odhalilo dve nové, zatiaľ neopísané mutácie. V 3 rodinách sa vyskytla hetrozygotná mutácia génu FGG Trp3→Stop, podmieňujúca zníženú syntézu fibrinogénu. Túto mutáciu mala aj 1 pacientka s anamnézou tromboembolickej choroby a opakovanými mŕtvymi plodmi. Vo štvrtej rodine sa u 1 pacientky s hladinou FGB:koag 0,7 g/l a anamnézou ťažkého popôrodného krvácania zistila bodová mutácia FGG Trp253→Cys v heterozygotnej forme. Ďalšia analýza potvrdila, že uvedená mutácia spôsobuje poruchu sekrécie fibrinogénu [26].
Diskusia
Dostupné literárne prehľady a rovnako aj analýza klinického fenotypu v našom súbore chorých s hypo - a dysfibribogenémiou potvrdzujú veľkú variabilitu klinických prejavov nielen medzi jedincami s porovnateľnými hodnotami fibrinogénu, ale aj v priebehu života jednotlivcov. Prejavy krvácania sú väčšinou mierne a ani krvácania po invazívnych výkonoch, zaznamenané u malého počtu chorých, neboli život ohrozujúce. Otázka správnej indikácie profylaktického podania fibrinogénu ostáva problematickou, keďže nie vždy sa dajú komplikácie predvídať na základe predchádzajúceho klinického priebehu. V našich súboroch 5 z 8 pacientov s krvácaním po invazívnych výkonoch boli predtým asymptomatickí, naopak, mnohí pacienti, ktorí udávali výskyt miernych krvácaní, absolvovali aj veľké operácie bez komplikácií. Krvácanie bolo pritom menej časté pri dysfibrinogenémii, napriek nižším hodnotám fibrinogénu, ako pri hypofibrinogenémii. V našom súbore chorých s dysFBG sme zistili nižší výskyt trombózy (4 %) ako udáva medzinárodný register, kde sa táto komplikácia vyskytla až u 21 % jedincov [15]. Vznik trombózy vysvetľujú 2 hlavné mechanizmy: a) defektná väzba trombínu na fibrín; b) defektná stimulácia fibrinolýzy sprostredkovanej prostredníctvom t-PA. U našich geneticky vyšetrených pacientov sme nezistili poruchu lokalizovanú v miestach podmieňujúcich poruchu uvedených funkcií molekuly fibrinogénu a fibrínu. Paradoxne vyšší výskyt trombózy u chorých s hypofibrinogenémiou bol ovplyvnený malou početnosťou súboru a skutočnosťou, že u 2 z 3 chorých boli súčasne prítomné ďalšie genetické faktory trombofílie.
Rozbor klinického fenotypu u pacientov s dysfibrinogenémiou a definovanou genetickou poruchou ukázal variabilné prejavy krvácania při mutácii FGA Arg16→His, spôsobujúcej opozdené štiepenie FPA trombínom a známej z literatúry ako fibrinogén Chapell Hill II a Giessen I [9]. Pacienti s novo zistenou mutáciou FGA Gly13→Glu mali asymptomatický priebeh. To je v zhode s literárnymi údajmi, podľa ktorých mutácie v blízkosti väzobných miest trombínu prebiehajú väčšinou asymptomaticky [9,15]. Pri hypofibrinogenémii sa mutácia FGG Trp3→Stop prejavila variabilným klinickým fenotypom ale mutácia FGG Trp253→Cys viedla k významnému krvácaniu po provokácii. Táto mutácia spôsobuje poruchu sekrécie fibrinogénu a v homozygotnej forme pravdepodobne môže spôsobiť afibrinogenémiu [26].
Naše, i keď zatiaľ limitované pozorovania naznačujú význam vyšetrenia genotypu pre diagnózu a diferenciálnu diagnózu porúch fibrinogénu, jeho využitie ako spoľahlivého prediktívneho markera klinického fenotypu si vyžaduje ďalšie štúdium väčších súborov pacientov s identickými mutáciami.
doc. MUDr. Angelika Bátorová, Ph.D.
www.fmed.uniba.sk
e-mail: batorova@hotmail.com
Doručeno do redakce: 18. 4. 2005
Přijato k otištění: 18. 4. 2005
Sources
1. Rabe F, Salomon E. Ueber-faserstoffmangel im Blute bei einem Falle von Haemophilie. Arch Klin Med 1920; 95 : 2–14.
2. Risak E. Die Fibrinopenie. Ztsch Klin Med 1935; 128 : 605–629.
3. Mammen EF, Prasad AS, Barnhart M et al. Congenital dysfibrinogenemia: fibrinogen Detroit. J Clin Invest 1969; 48 : 235–249.
4. Bolton-Maggs PHB, Perry DJ, Chalmers EA et al. Guidelines. The rare coagulation disorders – review with
guidelines for management from the United Kingdom Haemophilia Centre Doctor’s Organization. Haemophilia 2004; 10 : 593–628.
5. Lak M, Keihani M, Elahi F at al. Bleeding and thrombosis in 55 patients with inherited afibrinogenemia. Br J Haematol 1999; 107 : 204–206.
6. Fried K, Kaufmann S. Congenital afibrinogenemia in 10 of spring of uncle-niece marriages. Clin Genet 1980; 17 : 223–227.
7. Doolittle RF. X-ray crystallographic studies in fibrinogen and fibrin. J Thromb Haemost 2003; 1 : 1559–1565.
8. Roberts HR, Monroe DM, Hoffman M. Molecular biology and biochemistry of the coagulation factors and pathways of hemostasis. In: Beutler E et al (eds). Williams Haematology. 6th ed. New York – Chicago: The McGraw-Hill Comp, Inc. 2001 : 1409–1434.
9. Matsuda M, Sugo T, Yoshida N et al. Structure and function of fibrinogen: Insights from dysfibrinogens. Thromb Haemost 1999; 82 : 283–290.
10. Henselmans JM, Meijer K, Haaxma R et al. Recurrent spontaneous intracerebral heamorrhage in a congenital afibrinogenemic patient: diagnostic pittfals and therapeutic options. Stroke 1999; 30 : 2479–2482.
11. Paramaswaran R, Dickinson JP, De-Lord S et al. Spontaneous intracranial bleeding in two patients with congenital afibrinogenaemia and the role of replacement therapy. Haemophilia 2000; 6 : 705–708.
12. Vu D, Bolton-Maggs HB, Parr JR et al. Congenital afibrinogenemia: identification and expression of a missense mutation in FGB impairing fibrinogen secretion. Blood 2003; 102 : 4413–4415.
13. Shima M, Tanaka I, Sawamoto Y et al. Succesful treatment of two brothers with congenital afibrinogenemia for splenic rupture using heat – and solvent detergent–treated fibrinogen concentrates.
J Pediatr Hematol Oncol 1997; 19 : 462–465.
14. Lefebvre P, Velasco PT, Dear A et al. Severe hypodysfibrinogenemia in compound heterozygotes of the fibrinogen AaIVS4+ 1G>T mutation and an AaGLN328 truncation (fibrinogen Keokuk). Blood 2004; 103 : 2571–2576.
15. Haverkate F, Samama M. Familial dysfibrinogenemia and thrombophilia. Report on a study of the SSC Subcommittee on Fibrinogen. Thromb Haemost 1995; 73 : 151–161.
16. Chafa O, Chellali T, Sternberg C et al. Severe hypofibrinogenemia associated with bilateral ischemic necrosis of toes and fingers. Blood Coag Fibrinol 1995; 6 : 549–552.
17. Dupuy F, Soria C, Mohol P et al. Embolized ischemic lesions of toes in an afibrinogenemic patient: possible relevance toin vivo circulating thrombin. Thromb Res 2001; 102 : 211–219.
18. Hamano A, Mimuro J, Aoshima M et al. Thrombophilic dysfibrinogen Tokyo V with the amino acid substitution of Ala327Thr: formation of fragile but fibrinolysis–resistant finrin clots and its relevance to arterial thromboembolism. Blood 2004; 103 : 3045–3050.
19. Suh TT, Holmback K, Jensen NJ et al. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen - deficient mice. Genes Dev 1995; 9 : 2020–2033.
20. Peyvandi F, Duga S, Akhavan S et al. Rare coagulation disorders. Haemophilia 2002; 8 : 308–321.
21. Rosendaal FR. Risk factors for venous thrombotic disease. Thromb Haemost 1999; 82 : 610–619.
22. Neerman-Arbez M, Honsberger A, Antonarakis SE et al. Deletion of fibrinogen (correction of fibrinogen) alphachain gene (FGA) causes congenital afibrinogenemia. J Clin Invest 1999; 103 : 215–218.
23. Hanss M, Biot F. A database for human fibrinogen variants. Ann N Y Acad Sci 2001; 936 : 89–90.
24. Neerman-Arbez M, de Moerloose P, Honsberger A et al. Molecular analysis of the fibrinogen gene cluster in 16 patients with congenital afibrinogenemia: novel truncating mutations inthe FGA and FGG genes. Hum Genet 2001; 108 : 237–240.
25. Neerman-Arbez M. The molecular basis of inherited afibrinogenemia. Thromb Haemost 2001; 86 : 154–163.
26. Vu D, de Moerloose P, Batorova A et al. Identification and expression of a missense mutation in FGG impairing fibrinogen secretion. J Med Gen; v tlači.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2005 Issue 7 a 8
Most read in this issue
- Potransfuzní reakce
- Trombocytózy a trombocytemie
- Antifosfolipidový syndrom – diagnostika a léčba
- Protidestičková léčba