
Některé vzácnější formy hereditárních anémií vyskytující se v dospělé populaci v ČR - β-talasemie a nestabilní hemoglobinové varianty
Rare forms of hereditary anaemia in the Czech and Slovak populations - β - and δβ-thalassaemia and unstable haemoglobin variants
The authors present a review of the spectrum and frequency of mutations of the β-globin gene in the Czech and Slovak patients with clinical symptoms of β-thalassaemia or δβ-thalassaemia and of Heinz body haemolytic anaemia associated with unstable haemoglobinopathies. In the Czech and Slovak populations, β-thalassaemia appears to be an uncommon disorder, which, however, must be considered as the prevailing cause of congenital hypochromic microcytic anaemia. All β-thalassaemia patients were heterozygous, manifesting thalassaemia minor, with rare exceptions of dominantly inherited β-thalassaemia with phenotype that ranged from severe thalassaemia minor to thalassaemia intermedia. We hypothesize that genetic drift and migration in the past are responsible for introduction of the Mediterranean alleles, while several mutations, described in single families, originated locally in the Czech Republic and Slovakia.
Key words:
β-thalassaemia - Heinz body haemolytic anaemia - unstable haemoglobin variants - Czech and Slovak populations
Authors:
V. Divoký 1,2; S. Walczysková 2; D. Pospíšilová 3; I. Kostelecká 2; M. Divoká 1; Š. Vlachová 1; M. Jarošová 1; J. Čermák 4; K. Indrák 1; Česko-Slovenská Kooperativní Skupina Pro Diagnostiku Hemoglobinopatií
Authors‘ workplace:
Hematoonkologická klinika Lékařské fakulty UP a FN, Olomouc, přednosta prof. MUDr. Karel Indrák, DrSc.
1; Ústav biologie Lékařské fakulty UP, Olomouc, přednosta prof. RNDr. Karel Lenhart, DrSc.
2; Dětská klinika Lékařské fakulty UP a FN, Olomouc, přednosta prof. MUDr. Vladimír Mihál, CSc.
3; Ústav hematologie a krevní transfuze, Praha, ředitel prof. MUDr. Pavel Klener, DrSc.
4
Published in:
Vnitř Lék 2005; 91(7 a 8): 886-893
Category:
128th Internal Medicine Day - 21rd Vanysek's Day Brno 2005
Overview
β - a δβ-talasemické alely jsou ve střední Evropě relativně vzácné, přesto jsou v České republice i na Slovensku nejčastější příčinou vrozené mikrocytární anémie. V průběhu 15 let jsme na molekulární úrovni identifikovali 19 různých β-talasemických alel a 2 δβ-talasemické alely u 230 členů 109 rodin českého a slovenského původu. Většina těchto alel k nám byla importována ze Středomoří. Všichni nemocní byli heterozygotní nosiči mutace s klinickými projevy talasemia minor, výjimečně talasemia intermedia. Hemoglobinové varianty jsou v České republice a na Slovensku vzácné, klinicky i laboratorně jsou nejvýznamnější nestabilní hemoglobinopatie, asociované s vrozenou anémií s Heinzovými tělísky. Molekulární příčiny mikrocytární anémie s talasemickými rysy u některých rodin nejsou vázány na globinové geny a jejich odhalování přispívá ke zpřesnění diagnostiky i léčby nemocných.
Klíčová slova:
β-talasemie - vrozené anémie s Heinzovými tělísky - nestabilní hemoglobinové varianty - česká a slovenská populace
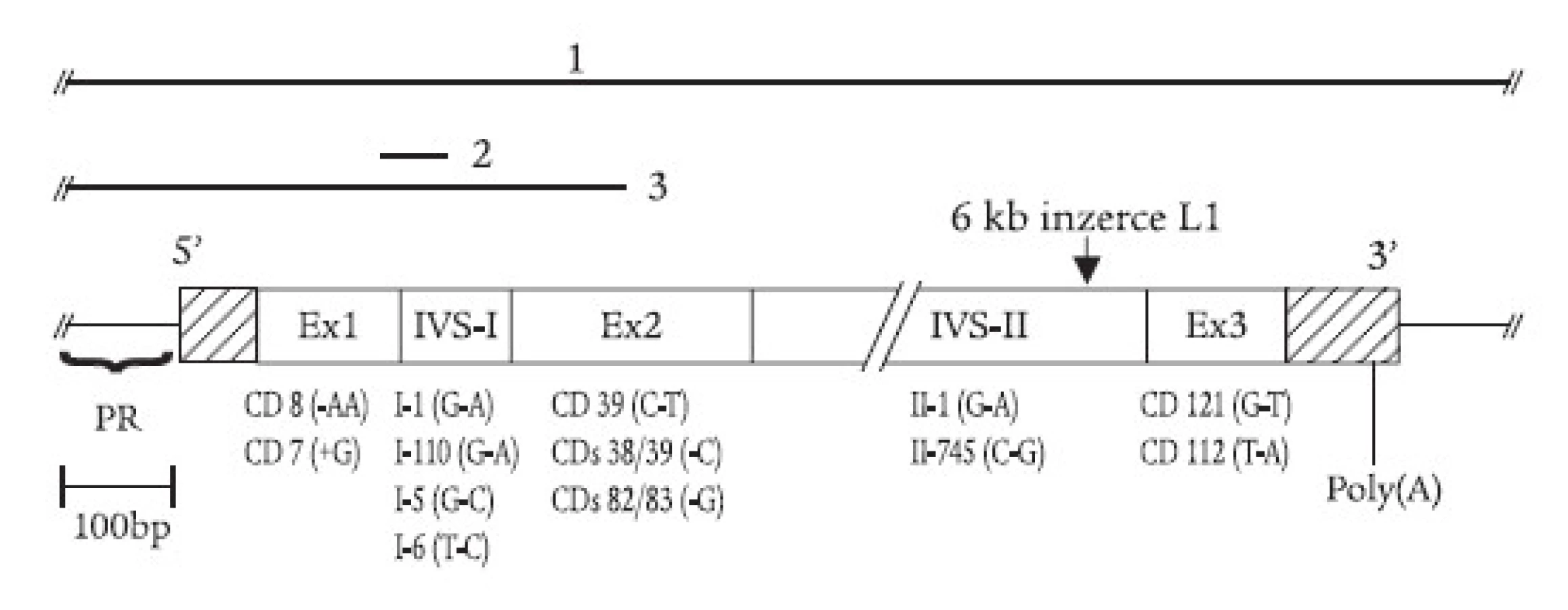
Hemoglobin (Hb) je tvořen bílkovinou globinem a prostetickou skupinou, hemem. Všechny lidské Hb molekuly jsou tetramery sestávající ze 2 různých párů globinových řetězců. Jeden pár globinových řetězců je produkován geny α-globinového lokusu a druhý pár řetězců je produkován geny β-globinového lokusu (obr. 1). Hemoglobinové přepínání je postupná kvalitativní náhrada červených krvinek v krevním řečišti, během které jsou červené krvinky obsahující převážně jeden typ hemoglobinu nahrazeny červenými krvinkami obsahujícími jiný typ hemoglobinu [1,2]. Jednotlivé globinové řetězce syntetizované ve žloutkovém váčku, játrech, slezině a kostní dřeni během ontogenetického vývoje, vyjádřené v procentu celkového globinu, jsou znázorněny na obr. 1. Přepnutí z embryonálních hemoglobinů žloutkového váčku na fetální Hb (HbF) fetálních jater je ukončeno v 10. týdnu; přepnutí z HbF na Hb dospělého typu probíhá v období těsně po narození. V dospělosti jsou v kostní dřeni produkovány dva typy Hb: HbA (tvoří asi 98 %) a minoritní, jen 1-3 % zastoupený HbA2. Zbytkový HbF bývá u normálního dospělého člověka zastoupen méně než 1 % celkového Hb. Bílkovinná složka Hb dospělého typu (HbA) se skládá ze 2 řetězců α a 2 řetězců β. Každý řetězec váže 1 hemovou skupinu uloženou v kapsových dutinách. Krystalografické studie odhalily, že se hem nachází v hydrofobní hemové kapse, kterou vytváří každý ze 4 globinových řetězců a která brání průniku vody a chrání ionty železa před autooxidací. Nabité aminokyseliny tvoří vnější povrch Hb molekuly. Stabilní α1/β1 kontakty jsou zabezpečeny celkem 40 aminokyselinami a řadou vodíkových můstků; α1/β2 vazebné kontakty nejsou tak stabilní a podléhají změnám v oxy - a deoxy-hemoglobinové konformaci.
![Struktura globinových lokusů a syntéza globinů. Schematické znázornění α globinového lokusu na chromozomu 16 (A) a β globinového lokusu na chromosomu 11 (C). HS-40 a β-LCR jsou regulační oblasti globinových genových rodin, působící v poloze cis. Funkční geny na lokusech jsou vyznačeny černě, nevybarvené obdélníky jsou pseudogeny (ψ). (B) Jednotlivé globinové řetězce syntetizované ve žloutkovém váčku, játrech, slezině a kostní dřeni během ontogenetického vývoje, vyjádřené v procentu celkového Hb. Přepnutí z embryonálních Hb na HbF je ukončeno v 10. týdnu; přepnutí z HbF na Hb dospělého typu probíhá v období těsně po narození. Upraveno dle [1].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/20cbbd635cefdbae19dcfeb2f527818b.jpg)
Vrozené poruchy syntézy či struktury vlastního Hb - hemoglobinopatie - jsou nejrozšířenější monogenní dědičné nemoci na světě. Postihují asi 7 % světové populace [1,3]. Jsou způsobeny buď poruchou tvorby některého z globinů, jejímž důsledkem je nerovnováha v poměru α - a ne-α-globinových řetězců v erytrocytech (kvantitativní globinové poruchy, talasemie), a nebo syntézou strukturálně abnormálních globinových řetězců (kvalitativní globinové defekty, hemoglobinové varianty). Kvalitativní porucha, tj. syntéza některých abnormálních globinových řetězců, může být provázena redukcí tvorby abnormálních řetězců, tak vznikají hemoglobinové varianty s nerovnováhou α - a β-řetězců a s talasemickým klinickým obrazem. Jiná skupina dědičných globinových poruch je způsobena poruchou fyziologického přepínání exprese globinových řetězců, ke kterému dochází v průběhu vývoje organizmu. Jedná se o poruchy přechodu ze syntézy HbF na HbA, kdy hovoříme o patologickém dědičném přetrvávání produkce fetálního hemoglobinu (Hereditary Persistence of Fetal Hemoglobin - HPFH). Vzhledem k rozšíření hemoglobinových defektů je v některých populacích běžné, že se vyskytují v kombinacích, tj. jedinec zdědí gen pro talasemii i gen pro hemoglobinovou variantu nebo HPFH.
Výskyt většiny hemoglobinopatií, ale i např. deficitu glukózo-6-fosfát-dehydrogenázy (G6PD), další široce rozšířené dědičné poruchy červených krvinek, je soustředěn především do malarických oblastí. Obrovská heterogenita talasemických alel svědčí pro navzájem nezávislý, lokální původ těchto mutací [1,4]. Také mutace pro srpkovitou anémii vznikla nezávisle v několika oblastech Afriky a Asie. Proč jsou hemoglobinopatie tak časté a proč se vyskytují převážně v malarických oblastech? Globinové mutace poskytují heterozygotním nosičům ochranu před malárií. To v průběhu evoluce převážilo nad cenou, kterou platí homozygoti, jejichž postižení je letální. Již před 50 lety vyslovil J. B. S. Haldane hypotézu, že důvodem rozšíření talasemie a srpkovité anémie v malarických oblastech je selektivní zvýhodnění těchto alel v průběhu evoluce (základem teorie byly epidemiologické studie prováděné v 50. letech minulého století v Africe). Erytrocytární parazité způsobující malárii, především Plasmodium falciparum, degradují hemoglobin a vedou k rozpadu červených krvinek. Existuje řada mechanizmů, kterými jsou heterozygotní nosiči hemoglobinopatie chráněni proti malárii: zvýšená hladina HbF (resp. zpomalené zastavení syntézy HbF) chrání heterozygoty v kojeneckém období; HbF je rezistentní k natrávení malarickými hemoglobinázámi, a brání tak růstu parazita [5]. Parazitem infikované srpkovité erytrocyty jsou intenzivněji vychytávány makrofágy a jejich přežití je zkráceno, a tak je zabráněno množení parazita [6]. Heterozygoti pro α-talasemii jsou náchylnější k infekci méně virulentním parazitem Plasmodium vivax, což vede k jejich přirozené imunizaci proti závažnějším infekcím Plasmodium falciparum [7].
Hemoglobinopatie se však mohou vyskytnout v každé etnické skupině, včetně zemí střední Evropy. I zde dříve byla malárie, a navíc byly mutované alely do střední Evropy importovány z oblasti Středomoří, ale také z Mongolska a jiných oblastí Asie [3]. Některé mutace vznikly ve střední Evropě de novo, avšak díky nepřítomnosti selekčního tlaku malárie je jejich rozšíření izolované, postihující jen několik rodin a vesměs se jedná o heterozygoty, nosiče talasemické nebo strukturní hemoglobinové mutace.
β-talasemie
Poměr α - a β-globinových řetězců v molekule HbA je fyziologicky 1 : 1. Nerovnováha vzájemného poměru globinových řetězců je charakteristická pro talasemie. Talasemie jsou definovány podle typu postiženého řetězce (α - nebo β-talasemie, případně δβ-talasemie). Tíži talasemie kvantitativně charakterizuje buď snížení produkce globinového řetězce (u α+ - nebo β+-talasemií), nebo úplná ztráta syntézy globinového proteinu (α0 - nebo β0-talasemie).
Patofyziologie a klinické projevy talasemií obecně závisí na míře nerovnováhy α - a ne-α-globinových řetězců. Mezi α - a β-talasemiemi je však zásadní rozdíl: při srovnatelné nerovnováze syntézy řetězců jsou β-talasemie vždy klinicky a morfologicky závažnější (větší mikrocytóza).
α-talasemie jsou u nás vzácné, většina nemocných s talasemickým obrazem jsou nosiči β-talasemické, příp. δβ-talasemické alely. β-talasemické a δβ-talasemické mutace detekované v české a slovenské populaci jsou shrnuty v tab. 1 a na obr. 2. První molekulárně-genetickou charakteristiku talasemií u rodin českého a slovenského původu publikovali Indrák et al [8]. Na tuto práci navazovalo několik dalších studií [9,10]. Tab. 1 shrnuje výsledky těchto studií a je rozšířena o dosud nepublikované výsledky molekulárně-genetické diagnostiky u více než 100 nemocných. Molekulárními mechanizmy zodpovědnými za β-talasemie v Čechách a na Slovensku jsou většinou bodové mutace, tj. jednoduché záměny bází v sekvenci β-globinového genu nebo jeho promotoru (obr. 2). Výjimku tvoří tzv. „česká“ delece u české rodiny žijící v Kanadě [11] a inzerce retrotranspozonu do β-globinového genu u matky a dcery ukrajinského původu [12,13] (obr. 2). U několika námi sledovaných rodin segreguje fenotyp nezávisle na β-globinovém lokusu, tzn. že mutace jiných genů, kódujících (neznámé) regulační faktory globinové exprese, mohou vést k vrozené mikrocytární anémii a talasemii-podobnému fenotypu.
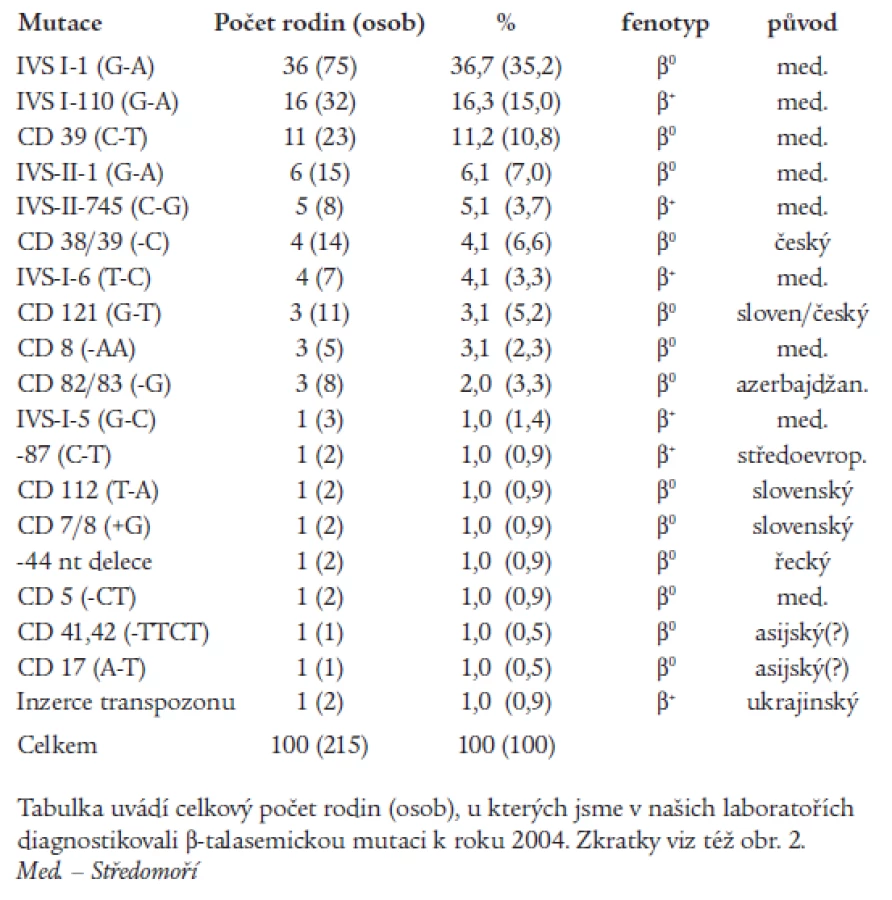
Jak již bylo uvedeno, uvedené typy mutací vedou k autozomálně-recesivnímu onemocnění. Heterozygoti pro β+ - i β0-talasemii jsou klinicky asymptomatičtí jedinci s mírně nižší hladinou Hb (hodnoty kolem 100 g/l), s mikrocytózou (MCV 55-70 fl) a hypochromazií a často i nápadnou erytrocytózu (až 6 x 1012/l), tj. s formou talasemia minor. Malá skupina β-talasemických mutací vykazuje dominantní dědičnost, s fenotypem β-talasemia intermedia u heterozygotů. V jejich erytroblastech nacházíme výrazné morfologické změny s intracytoplazmatickými inkluzemi. Proto tuto formu talasemií označujeme také jako β-talasemie s inkluzními tělísky (Heinz-body-β-thalassemia) [14,15]. Typická je středně těžká až těžká hemolytická anémie s inefektivní erytropoézou. Při fyzikálním vyšetření nemocných bývá nápadný subikterus až ikterus a mírná splenomegalie. V krevních nátěrech bývají patrné výrazné morfologické změny červené krevní řady: terčovité buňky, bazofilní tečkování erytrocytů, mikro-, anizo-, poikilo-, schisto - a echinocyty, a zvláště nápadné jsou zmíněné intracytoplazmatické inkluze po barvení brilantkresylovou modří. Na molekulární úrovni jsou způsobeny nesmyslnými mutacemi nebo mutacemi měnícími smysl kodonu ve 3. exonu β-globinového genu (obr. 2). Na rozdíl od recesivních alel produkují dominantní talasemické alely buď zkrácený řetězec (tzv. nesmyslné mutace), nebo vysoce nestabilní Hb variantu (tj. důsledek mutací měnících smysl kodonu, u kterých produkované β-globinové řetězce nemají schopnost vazby s α-globinovými řetězci, tj. nejsou schopné tvořit α1/β1 dimery). V obou případech vznikají hypernestabilní polypeptidy, které nejsou ani přechodně schopny vytvářet Hb tetramery (na rozdíl od nestabilních Hb variant, viz dále), a precipitují spolu s nadbytečnými α-globinovými řetězci [15]. Tím dominantní β-talasemie připomínají inefektivní erytropoézu u talasemia intermedia, ale současně mají znaky kongenitální hemolytické anémie nestabilních hemoglobinopatií.
Postižení vedoucí k dominantní β-talasemii bylo dosud zjištěno u 5 nepříbuzných rodin českého a slovenského původu [16], z toho 2 mutace byly nesmyslné, 1 mutace (v kodonu 115 β-globinového genu) vede ke vzniku Hb Hradec Králové [CD 115 (GCC-GAC) a je dále zmíněna v části věnované nestabilním hemoglobinovým variantám [17]. V případě mutace v CD 112 (T-A) odpovídal fenotyp pacienta spíše talasemia minor, přestože jsme podle typu mutace očekávali těžší klinické projevy [18].
Homozygoti nebo dvojití heterozygoti pro β0-talasemické mutace mají závažný fenotyp známý jako β-talasemia major nebo Cooley’s anaemia [19]. U nás je zatím výskyt homozygotů nebo dvojitých heterozygotů nepravděpodobný (vzhledem k relativní vzácnosti mutantních alel), ale není vyloučený. Nemocní z malarických oblastí mívají těžkou chudokrevnost (při diagnostikování 20-30 g/l), která se vyvíjí během několika měsíců po narození, progresivně se zhoršuje a vyžaduje pravidelnou transfuzní léčbu. U neléčených pacientů bývá β-talasemia major provázena deformitami kostí vznikajícími v důsledku expanze krvetvorby z dřeňových prostor, častou retardací růstu a mongoloidním vzezřením. Nemocní mohou mít výraznou hepatosplenomegalii. Jsou trvale závislí na transfuzní léčbě s postupně vznikajícím obrazem přetížení organizmu železem. To vede u neléčených nemocných k úmrtí již ve 2. až 3. dekádě života. U β-talasemia major nacházíme výrazně zvýšenou hladinu HbF, který může tvořit téměř všechen (> 90 %) Hb.
Diagnostika
Většina nosičů β-talasemické alely (heterozygoti), s výjimkou nemocných se současným nedostatkem železa a se vzácnější δβ-talasemií, má přibližně 2násobně zvýšenou hladinu HbA2 (α2δ2) oproti normálu (tj. na 3-5 %) a asi 1/3 nosičů má zvýšenou i hladinu HbF. Proto je nejrozšířenějším diagnostickým testem chromatografické nebo elektroforetické stanovení hladiny HbA2 [20]. HbF stanovujeme s dostatečnou přesností alkalickou denaturací [21]. U homozygotů pro β0-talasemii detekujeme jen HbF a normální nebo jen mírně zvýšený HbA2; absolutní hladina HbF je dána mírou proliferace a přežití F-buněk, které je velmi variabilní. Přímá detekce mutací na DNA úrovni využívá molekulárně-genetických metod: štěpení restrikčními enzymy, PCR a sekvenování nebo hybridizace DNA se specifickými sondami. Tyto metody jsou rychlé a spolehlivé a jsou využívány v prenatální diagnostice v malarických oblastech. Všechny uvedené metody také rutinně provádíme v naší laboratoři.
Diferenciální diagnostika
Zvýšenou hladinu HbA2 mají i nemocní se srpkovitou anémií (HbSS) v kombinaci s homozygotním postižením pro α-talasemii-2 (genotyp -α/-α). Mírně zvýšený HbA2 bývá někdy detekován i u megaloblastických anémií a hypertyroidizmu. Vysoké zvýšení HbF je typické u juvenilní myeloidní leukémie; mírně zvýšený HbF je nacházen u Fanconiho anémie, Diamond-Blackfanovy anémie a u řady dalších myeloproliferativních chorob.
δβ-talasemie
δβ-talasemie jsou většinou způsobeny delecemi příslušných úseků DNA. Specifickou skupinou delečních forem δβ-talasemií jsou delece vznikající v meióze nerovnoměrným překřížením mezi δ - a β-globinovými geny a vedoucí ke genové fúzi těchto genů, přičemž fúzní gen zachovává čtecí rámec, ale je pod vlivem slabého δ-globinového promotoru. Produktem jsou chimérické δβ-řetězce, které se kombinují s α-řetězci a dávají vzniknout skupině Hb Lepore s talasemickým fenotypem. Hb Lepore jsme prokázali u 8 členů 5 různých českých rodin. Z delecí vedoucích k δβ0-talasemii je v Evropě častá tzv. sicilská 13 kb dlouhá delece, vyskytující se vzácně i v ČR (obr. 2; u 7 členů 4 rodin). Homozygoti produkují pouze HbF, heterozygoti mají talasemický krevní obraz s 5-15 % HbF.
Strukturní hemoglobinové varianty
Strukturní hemoglobinové varianty neboli abnormální hemoglobiny vznikají v důsledku strukturní změny v globinové složce Hb. Ta je výsledkem aminokyselinové záměny v globinovém řetězci. Na genové úrovni jsou nejčastěji příčinou bodové mutace v kódujících oblastech globinových genů, ale mohou to být i krátké delece, elongace či genové fúze. Záměna aminokyseliny většinou změní molekulový náboj řetězce. Toho se využívá u detekčních a separačních elektroforetických nebo chromatografických metod. Funkce a stabilita, a tím i klinické projevy mutantních hemoglobinů, představují široké spektrum fenotypických projevů, od „normálních“ bez klinické manifestace až po výrazně pozměněné, způsobující typické hematologické syndromy. Dosud bylo popsáno více než 800 hemoglobinových variant (aktuálně na Globin Gene Server, http://globin.cse.psu.edu); 270 z nich jsou varianty α-globinového řetězce, 450 varianty β-globinového řetězce, vzácnější jsou varianty δ - a γ-globinových řetězců a hybridní globiny (produkty fúzních genů). Z klinického hlediska jsou významné hemoglobiny vedoucí k srpkovatění erytrocytů, nestabilní Hb varianty a Hb varianty s abnormální vazbou ke kyslíku. Několik málo hemoglobinových variant (HbS, HbC, HbD, HbE) způsobuje světově nejrozšířenější dědičné choroby. V České republice jsou hemoglobinové varianty vzácné [22], jejich přehled je uveden v tab. 2.

Nestabilní Hb varianty v české a slovenské populaci: vrozené anémie s Heinzovými tělísky
Více než 100 různých globinových mutací vede ke vzniku nestabilní hemoglobinové varianty. Hemolytická anémie způsobená nestabilním Hb bývá autozomálně dominantně dědičná; pacienti jsou většinou heterozygoti, výskyt homozygotního postižení je vzácný. Molekulární mechanizmy nestabilních Hb variant zjištěných v české a slovenské populaci je možné shrnout do 3 skupin: (i) záměny aminokyselin v blízkosti hemové kapsy vedou k zeslabení vazby hemu na globin, a tím ke snížení stability celé podjednotky; (ii) mutace histidinu (proximálního nebo distálního), tj. hem-kontaktní aminokyseliny za aminokyselinu, která nemůže vázat hem; (iii) mutace narušující α1/β1 nebo α1/β2 vazby (jako vodíkové můstky mezi aminokyselinami, nejčastěji jsou to záměny za prolin).
Patofyziologie
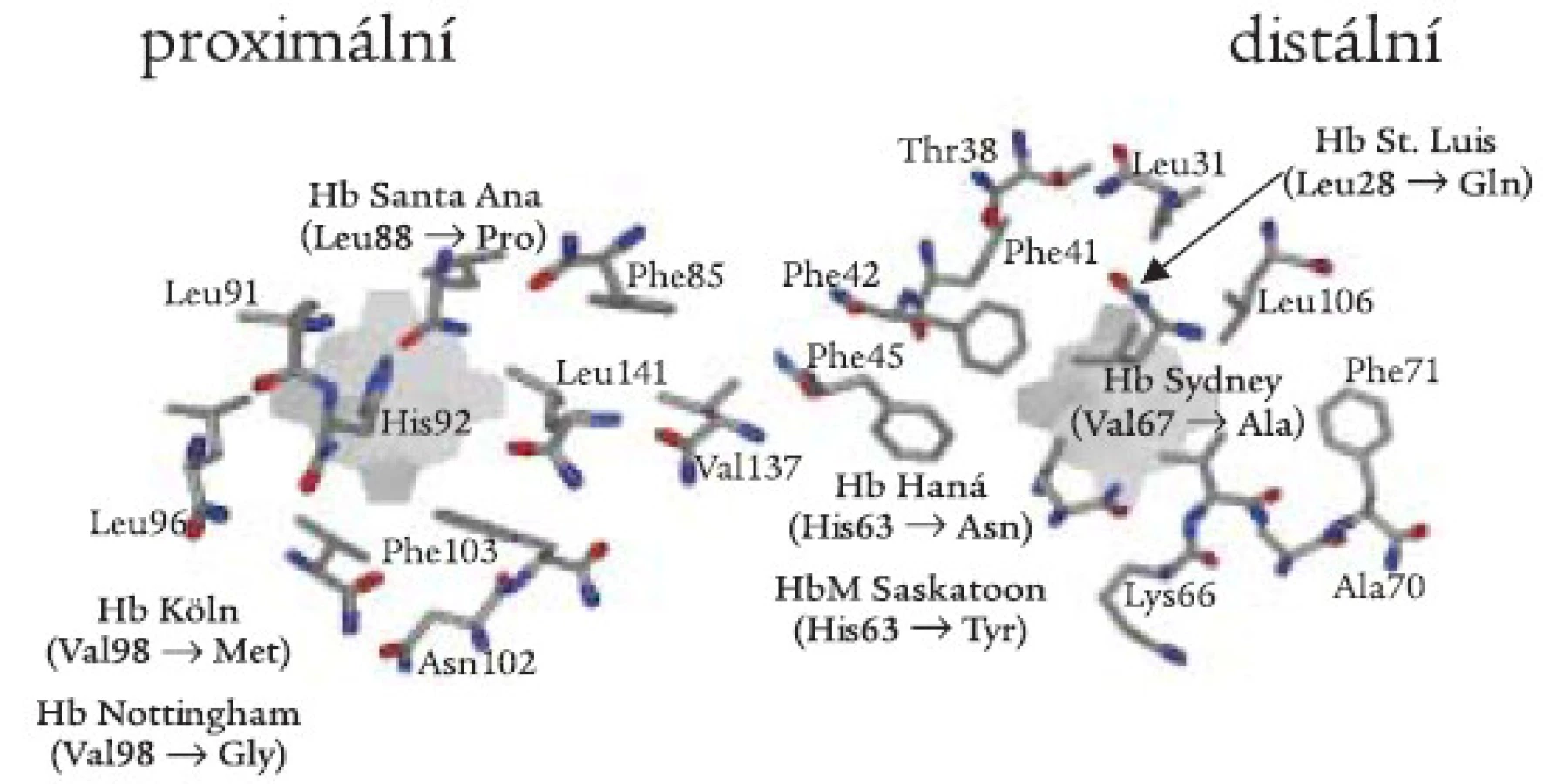
Důležitým patofyziologickým projevem nestabilního Hb je jeho precipitace. Mechanizmus precipitace hemoglobinu za vniku mikroskopicky patrných Heinzových tělísek a následné hemolýzy je znázorněn na schématu 1. Inkluzní tělísko narušuje plasticitu erytrocytární membrány a zhoršuje průchodnost erytrocytů mikrocirkulací zvláště ve slezině, což vede k deformaci a rozpadu červené krvinky. Rozpadem nestabilního Hb dochází i k autooxidaci hemoglobinu za vzniku methemoglobinu a uvolnění kyslíkových radikálů (HbFe2+ + O2 → HbFe3+ + O2-), které dále poškozují hemoglobinovou molekulu. Proto zde také lékové oxidanty zvyšují hemolýzu a mohou způsobit akutní hemolytickou krizi. Prohloubení hemolýzy v době infekce je způsobeno tím, že neutrofily během infekce ve zvýšené míře produkují kyslíkové radikály.
Některé Hb varianty jsou vysoce nestabilní a není je možné identifikovat ani pomocí vysokotlaké kapalinové chromatografie (HPLC), ani elektroforézou, ba ani testy stability, protože se rozpadnou během vyšetření. Jak bylo uvedeno v části věnované dominantním β-talasemiím, takovéto Hb varianty (hypernestabilní globiny) mohou vést v důsledku nerovnováhy α - a β-řetězců k talasemickému fenotypu. Do této skupiny patří i Hb Hradec Králové (β 115 Ala-Asp) [17].
(i) Nepolární aminokyseliny, jako např. leucin nebo valin, pomáhají udržet hydrofobní vlastnosti hemové kapsy. Záměny těchto aminokyselin mohou mít za následek vniknutí molekuly vody do hemové kapsy a následnou oxidativní denaturaci Hb (obr. 3). Hb Köln (β 98 Val-Met) je nejrozšířenější nestabilní Hb variantou, postihující všechny etnické skupiny, včetně české [23]. Hemolytická anémie u nemocných je důsledkem uvolnění hemu a vzniku vysoce nestabilních tetramerů s následnou tvorbou hemichromů a Heinzových tělísek (schéma 1). Další Hb varianty této skupiny byly v České republice nebo Slovenské republice diagnostikovány ojediněle jako „de novo“ mutace u dětí: Hb Saint Louis (β 28 Leu-Glu) [24], Hb Nottingham (β 98 Val-Gly) [25], a Hb Santa Ana (β 88 Leu-Pro) [26]. U Hb Santa Ana vede navíc záměna leucinu za prolin k narušení sekundární struktury globinového řetězce. Leucin β 88 je důležitý pro tvorbu α-helikální struktury; aminokyselina prolin není schopna participovat na tvorbě α-helixu. Záměna valinu v pozici β 67 za alanin vede ke vzniku nestabilního Hb Sydney, který byl v ČR diagnostikován u matky a syna [27].
![Schéma 1. Patogeneze nestabilní hemoglobinopatie. Upraveno dle [21].](https://pl-master.mdcdn.cz/media/image/2a163e3239f678a8c64cd268d24dcbec.jpg?version=1537793176)
(ii) Záměny proximálního (F8) nebo distálního (E7) histidinu za tyrosin vedou k permanentní oxidaci ferohemu (Fe2+) na ferrihem (Fe3+) a k methemoglobinemii (viz dále). Záměny kontaktních histidinů za jiné aminokyseliny než tyrosin vedou k různé míře oxidace ferohemu a k různému stupni nestability molekuly Hb. U Hb Haná (β 63 His-Asn) došlo k záměně distálního histidinu za hydrofilní amid - asparagin [28,29]. To vede k porušení stérické zábrany řetězce v hem-kontaktní oblasti a k průniku molekuly vody do hemové kapsy s následnou oxidativní denaturací Hb. Hb Haná byl zjištěn v moravské rodině u 2 generací rodu s rozdílnými klinickými projevy u postižených jedinců.
(iii) Některé Hb varianty jsou vysoce nestabilní a není je možné identifikovat ani pomocí HPLC ani elektroforézou, ba ani testy stability, protože se rozpadnou během vyšetření. Jak bylo uvedeno v části věnované dominantním β-talasemiím, takovéto Hb varianty (hypernestabilní globiny) mohou vést v důsledku nerovnováhy α a β-řetězců k talasemickému fenotypu. U Hb Hradec Králové je pozice 115 β-globinového řetězce normálně obsazena alaninovým zbytkem, který se společně s dalšími aminokyselinami G a H helixů podílí na kontaktu mezi α - a β-řetězci. Substituce těchto zbytků většími, hydrofilními nebo pozitivně nabitými zbytky způsobuje konformační změny v α-β-kontaktech, a vede tak k poruše stability Hb.
Diferenciální diagnostika
U nestabilních hemoglobinopatií často nacházíme zvýšenou hladinu methemoglobinu. Musíme je proto odlišit od kongenitálních methemoglobinemií (viz dále). Nestabilní Hb varianty mohou mít změněnou afinitu ke kyslíku, a je proto nutné je odlišit (v případech, kdy nezpůsobují anémii) i od variant vedoucích k familiárním erytrocytózám s mutacemi destabilizujícími deoxykonformaci Hb.
Léčba
Většina nemocných s nestabilním hemoglobinem nevyžaduje žádnou léčbu. Nemocným s chronickou hemolýzou je vhodné podávat kyselinu listovou, jejíž spotřeba při hemolýze roste. Transfuze jsou indikovány jen při těžké hemolytické krizi nebo během aplastické krize. V indikovaných případech může u nemocných s těžkou hemolýzou zlepšit anémii a klinický průběh splenektomie. Předpovídat klinický efekt splenektomie je ale obtížné. Při rozhodování může pomoci znalost efektu splenektomie u jiných nemocných se stejným typem hemoglobinopatie. U dětí do 6 let věku splenektomii neprovádíme z důvodu zvýšeného infekčního rizika. Podobně jako u jiných hemolytických anémií je i zde častá cholelitiáza, která někdy vyžaduje chirurgické řešení. Preventivně by se nemocní měli vyvarovat oxidativně působícím lékům (zvláště sulfonamidům) a infektům, které, pokud vzniknou, by měly být okamžitě intenzivně léčeny.
Methemoglobinemie
Methemoglobinemie (také familiární cyanózy) jsou způsobeny hemoglobinovými variantami, označovanými souborně jako Hb M, a dále rozlišenými podle místa výskytu jako Hb M-Saskatoon, Hb M-Milwaukee ap. Nejčastěji jsou způsobeny záměnou proximálního nebo distálního histidinu (tj. hem-kontaktní aminokyseliny, obr. 3) v α - nebo β-řetězcích za tyrozin. Met-Hb obsahuje atom železa hemové skupiny v oxidované formě, která nemůže reverzibilně vázat kyslík. Pacienti mají cyanózu a anémii. Snížení dodávky kyslíku do tkání může být kompenzováno mírnou polycytemií. Laboratorní stanovení methemoglobinu je založeno na fotometrickém měření hemolyzátu před a po redukci kyanidem draselným [21]. Nedávno jsme diagnostikovali Hb M-Saskatoon u rodiny ze Slovenské republiky (A. Krotká, J. Lazúr, Hematologické oddelenie FN L. Pasteura, Košice, dosud nepublikováno).
Tato práce vznikla za podpory grantů IGA NR/7799-3 a MSM 6198959205.
doc. RNDr. Vladimír Divoký, Ph.D.
www.fnol.cz
e-mail: divoky@tunw.upol.cz
Doručeno do redakce: 17. 5. 2005
Přijato k otištění: 17. 5. 2005
Sources
1. Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet 2001; 2 : 245-255.
2. Nienhuis AW, Stamatoyannopoulos G. Hemoglobin switching. Cell 1978; 1 : 307-315.
3. Steinberg MH, Forget BG, Higgs D et al (eds). Disorders of Hemoglobin: Genetics, Pathophysiology and Clinical Management. Cambridge: University Press 2001.
4. Antonarakis SE, Kazazian HH Jr, Orkin SH. DNA polymorphism and molecular pathology of the human globin gene clusters. Hum Genet 1985; 69 : 1-14.
5. Shear HL, Grinberg L, Gilman J et al. Transgenic mice expressing human fetal globin are protected from malaria by a novel mechanism. Blood 1998; 92 : 2520-2526.
6. Clegg JB, Weatherall DJ. Thalassemia and malaria: new insights into an old problem. Proc Assoc Am Physicians 1999; 111 : 278-282.
7. Allen SJ, O'Donnell A, Alexander ND et al. α+-thalassemia protects children against disease caused by other infections as well as malaria. Proc Natl Acad Sci USA 1997; 94 : 14736-14741.
8. Indrak K, Brabec V, Indrakova J et al. Molecular characterization of beta-thalassemia in Czechoslovakia. Hum Genet 1993; 88 : 399-404.
9. Indrák K, Divoký V, Brabec V et al. Molekulárně-genetická charakteristika α-, β - a δβ-thalassémií u 139 heterozygotů z 56 nepříbuzných rodin českého a slovenského původu. Vnitř Lék 1993; 39 : 969-978.
10. Indrák K, Divoký V, Brabec V et al. Dominantní β-talasemické alely v české a slovenské populaci [β-talasemické mutace v 112 (T-A) a 121 (G-T) a nestabilní hemoglobinová varianta Hradec Králové nebo α2β2 115 (Gl7) Ala-Asp]. Vnitř Lék 1994; 40 : 223-230.
11. Popovich BW, Rosenblatt DS, Kendall AG et al. Molecular characterization of an atypical β-thalassemia caused by a large deletion in the 5' β-globin gene region. Am J Hum Genet 1986; 39 : 797-810.
12. Divoky V, Indrak K, Mrug M et al. A novel mechanism of β-thalassemia: The insertion of L1 retrotransposable element into β-globin IVS II. Blood 1996; 88 : 148a.
13. Kimberland ML, Divoky V, Prchal J et al. Full-length human L1 insertions retain the capacity for high frequency retrotransposition in cultured cells. Hum Mol Genet 1999; 8 : 1557-1560.
14. Stamatoyannopoulos G, Woodson R, Papayannopoulou T et al. Inclusion-body β-thalassemia trait. A form of β-thalassemia producing clinical manifestations in simple heterozygotes. N Engl J Med 1974; 290 : 939-943.
15. Kazazian HH Jr, Dowling CE, Hurwitz RL et al. Dominant thalassemia-like phenotypes associated with mutations in exon 3 of the β-globin gene. Blood 1992; 79 : 3014-3018.
16. Thein SL. Is it dominantly inherited β-thalassaemia or just a β-chain variant that is highly unstable? Br J Haematol 1999; 107 : 12-21.
17. Divoky V, Svobodova M, Indrak K et al. Hb Hradec Kralove (Hb HK) or α2β2 115 (G17) Ala→Asp, a severely unstable hemoglobin variant resulting in a dominant β-thalassemia trait in a Czech family. Hemoglobin 1993; 17 : 319-328.
18. Divoky V, Gu LH, Indrak K et al. A new β-thalassaemia nonsense mutation (codon 112, T→A) not associated with a dominant type of thalassaemia in the heterozygote. Br J Haematol 1993; 83 : 523-524.
19. Cooley TB, Lee P. A series of cases of splenomegaly in children with anemia and peculiar bone changes. Trans Am Ped Soc 1925; 37 : 29.
20. Kutlar A, Huisman THJ. Detection of hemoglobinopathies. In Hommes FA (ed). Techniques in Diagnostic Human Biochemical Genetics: A Laboratory Manual. New York: Wiley-Liss 1991 : 519-560.
21. Huisman TH (ed). The hemoglobinapathies. In Methods in hematology. Vol. 15. New York: Churchill Livingstone 1986.
22. Indrák K, Brabec V, Divoký V et al. Strukturní varianty hemoglobinu nalezené v České republice. Vnitř Lék 1995; 41 : 13-20.
23. Indrak K, Brabec V, Wilson JB et al. Hb Köln or α2β2 98 (FG5) Val-Met in a Czechoslovakian. Hemoglobin 1991; 15 : 133-135.
24. Wiedermann B, Indrak K, Wilson JB et al. Hb Saint Louis or α2β2 28 (Bl0) Leu-Gln in a Czechoslovakian male. Hemoglobin 1986; 10 : 673-676.
25. Brabec V, Indrak K, Fortova H et al. Hb Nottingham or α2β2 98 (FG5) Val-Gly in a Czech child. Ann Hematol 1994; 69 : 93-95.
26. Divoký V, Hammerová T, Sakalová A et al. Nestabilní hemoglobin Santa Ana nebo α2β2 88 (F4) Leu-Pro identifikovaný u slovenské dívky. Vnitř Lék 1996; 42 : 38-41.
27. Indrák K, Divoký V, Kynčlová E et al. Hemoglobin Sydney α2β2 (E11) Val-Ala a hemoglobin Olomouc α2β286 (F2) Ala-Asp v českých rodinách. Přínos sekvenační analýzy DNA pro zpřesnění diagnostiky hemoglobinopatií. Vnitř Lék 1998; 44 : 347-349.
28. Divoky V, Pospisilova D, Luhovy M et al. HB-HANA or α2β2 63(E7) His-Asn, a new unstable haemoglobin variant with variable clinical manifestation. Br J Haematol 1996; 93(Suppl): 88.
29. Divoký V, Luhový M, Divoká M et al. Hemoglobin Haná nebo α2β2 (E7) His-Asn: nová nestabilní varianta hemoglobinu s paradoxně rozdílnou klinickou manifestací u kuřáků a nekuřáků z téže rodiny. Vnitř Lék 1997; 43 : 267-272.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2005 Issue 7 a 8
Most read in this issue
- Potransfuzní reakce
- Trombocytózy a trombocytemie
- Antifosfolipidový syndrom – diagnostika a léčba
- Protidestičková léčba