
Trombofilní stavy
Thrombophilia
Venous thromboembolism is still major medicinal problem. In the last couple of years, the significant progress concerning the etiopathogesis of this disease has been made, which could enable us to explain the cause of many cases of venous thrombosis, mainly with the familiar occurrence. The knowledge of criteria of the indication for thrombophilia work–up and especially pragmatic clinical interpretation of results are very important. These results together with perfect knowledge of family and personal history of venous thrombosis can help us to assess thrombophilic potential of every individual and enable us to choose the correct strategy of thromboprophylaxis.
Key words:
venous thromboembolism – thrombophilia – prophylaxis of venous thrombosis
Authors:
P. Ďulíček
Authors‘ workplace:
Oddělení klinické hematologie II. interní kliniky Lékařské fakulty UK a FN, Hradec Králové, přednosta prof. MUDr. Jaroslav Malý, CSc.
Published in:
Vnitř Lék 2005; 91(7 a 8): 819-825
Category:
128th Internal Medicine Day - 21rd Vanysek's Day Brno 2005
Overview
Venózní tromboembolizmus je stále významným zdravotnickým problémem. V posledních letech došlo k zásadnímu rozvoji poznatků v etiopatogenezi tohoto onemocnění, které umožnily objasnit příčiny vzniku řady žilních trombóz, zejména familiárně se vyskytujících. Důležitá je znalost indikačních kritérií na vyšetření vrozeného trombofilního stavu a zejména racionální klinická interpretace získaných výsledků. To nám společně s dokonalou znalostí rodinné a osobní anamnézy žilní trombózy umožní co nejlépe zhodnotit trombofilní potencionál každého jedince a zvolit správnou strategii prevence tohoto onemocnění.
Klíčová slova:
venózní tromboembolizmus – trombofilní stavy – prevence žilní trombózy
Venózní tromboembolizmus (VTE) představuje v rozvinutých zemích závažný zdravotnický problém. Po ischemické chorobě srdeční a cévní mozkové příhodě je na 3. místě ve výskytu mezi kardiovaskulárními chorobami [1]. VTE často postihuje i jedince v produktivním věku a je tedy i problémem sociálně ekonomickým. V poznání etiopatogeneze VTE došlo v posledních 10 letech k významným a zásadním objevům, které umožnily objasnit příčinu vzniku trombózy v celé řadě případů, zejména familiárně se vyskytujících. Kongenitálním trombofilním stavům je proto v poslední době věnována velká pozornost.
Incidence venózního tromboembolizmu
Stanovením incidence VTE se zabývalo v posledních letech několik studií. Anderson se svými spolupracovníky [2] stanovil incidenci 107 prokázaných VTE na 100 000 jedinců. Také Nordström v prospektivní studii v Malmö [3] stanovil incidenci 1,6 na 1000 jedinců. Ve Spojených státech amerických [4] je incidence VTE též udávána 1/1 000. Ve všech zmíněných studiích převládá výskyt hluboké žilní trombózy nad plicní embolií.
Incidence VTE závisí na věku, s přibývajícím věkem incidence stoupá [2,3]. Před 40. rokem věku je udávána 1/10 000 jedinců, po 75. roce pak 1/100.
Etiopatogeneze venózního tromboembolizmu
Již před více než 100 lety Virchow popsal 3 základní faktory, jejichž ovlivnění vede k riziku žilní trombózy [5] – žilní stáza, cévní stěna, změny v krevních elementech.
Jakékoliv faktory, jak vrozené, tak získané, které postihují 1 či více z těchto 3 faktorů, vychylují rovnováhu hemostatických mechanizmů směrem k trombóze. Pokud dojde k „překročení prahu“ obranyschopnosti organizmu, dojde ke vzniku žilní trombózy. Všechny 3 faktory jsou tedy ovlivněny interakcí mezi geny a zevním prostředím [6]. Generace trombinu s tvorbou fibrinu a s jeho degradací jsou dynamickým procesem, jehož aktuální interakce rozhoduje o překročení výše uvedeného prahu a tedy k vytvoření trombu. V posledních 20 letech byl v poznání patogeneze trombózy učiněn významný pokrok zejména v pochopení multifaktoriální příčiny vzniku žilní trombózy, kdy má interakce mezi vrozenými a získanými faktory za následek vznik trombu v daném čase a na daném místě.
Rizikové faktory #žilního tromboembolizmu
Rizikové faktory VTE lze dělit na vrozené a získané. Někteří autoři je dělí na faktory dispoziční a expoziční. K dispozičním faktorům patří věk, rodinná a osobní anamnéza, varixy a žilní insuficience, obezita, srdeční nedostatečnost, zhoubné bujení, vrozené a získané trombofilní stavy. K expozičním faktorům patří především operace, gravidita a šestinedělí, užívání léků (u žen zejména hormonální antikoncepce a hormonální substituční terapie), úraz, imobilizace, dehydratace. Některé dispoziční faktory lze ovlivňovat přímo (obezitu), některé vůbec (vrozené trombofilní stavy). Významnost jednotlivých rizikových faktorů byla zjišťována v řadě studií. Mezi nejuznávanější patří svým designem a velikostí studie SIRIUS, provedená Samamou [7]. Více se používá rozdělení rizikových faktorů VTE na vrozené a získané, tak jak je uvedeno v tab. 1 (upraveno dle Rosendaala) [8].
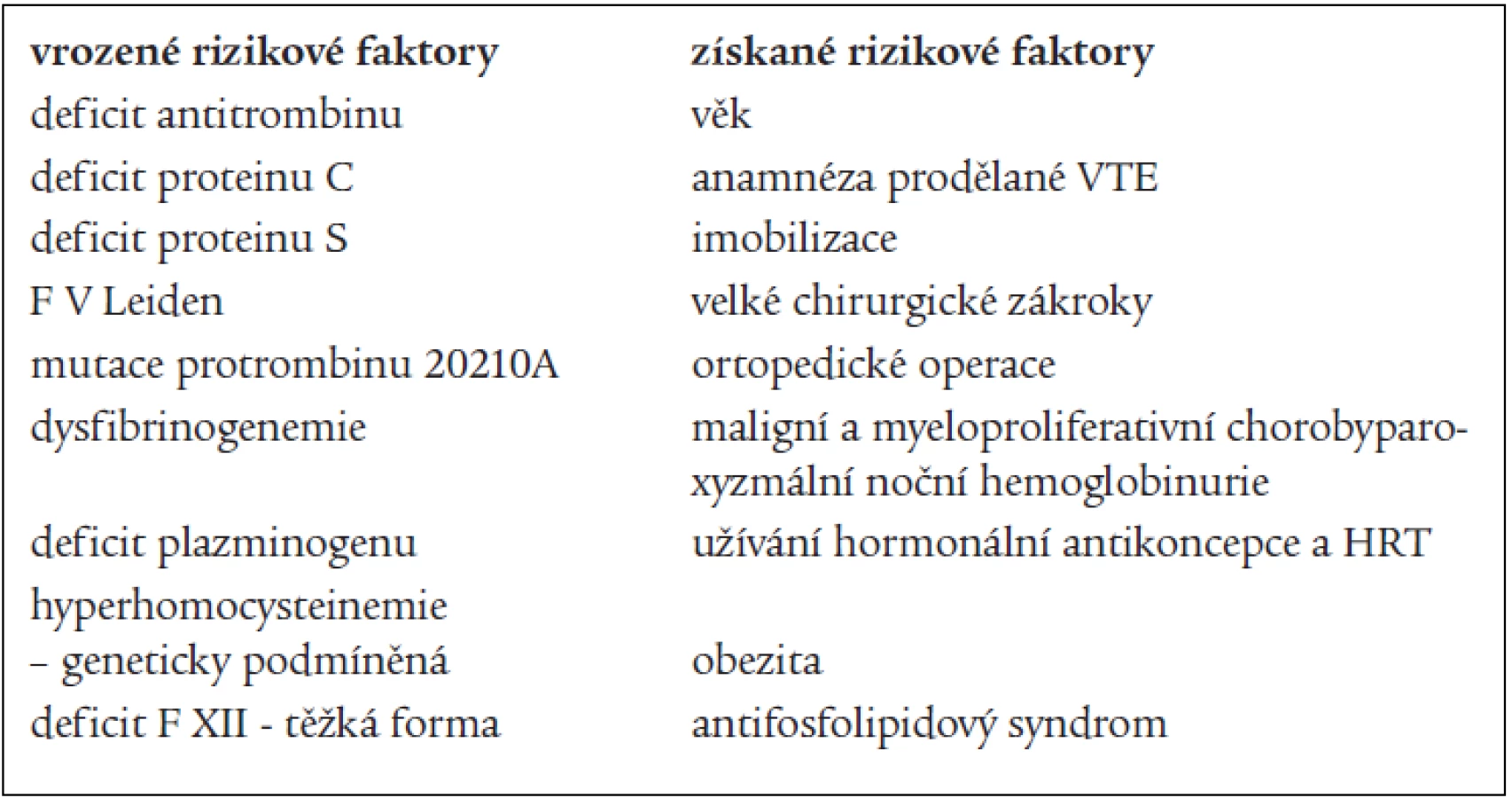
Některé rizikové faktory mají příčinu jak vrozenou, tak získanou, ev. neznámou. Sem lze zařadit např. hyperhomocysteinemii [9], vysokou aktivitu F VIII [10], přítomnost rezistence na aktivovaný protein C (APC-R) bez mutace F V Leiden [8]. Mezi trombofilní stavy patří i vysoká aktivita F IX [11], F XI [12], vyšší hladina fibrinogenu a pravděpodobně i vyšší aktivita TAFI (trombinem aktivovaný inhibitor fibrinolýzy) [13]. Z hlediska volby správné sekundární profylaxe VTE, tedy určení délky užívání antikoagulační terapie, je důležité zhodnocení rizikového faktoru z hlediska trvání – jedná-li se o přechodný stav (přiložení sádry, imobilizace, užívání hormonální antikoncepce) nebo o trvalý rizikový faktor (vrozený trombofilní stav).
O faktu, že v populaci existuje řada jedinců s vrozeným sklonem ke krvácení, se ví již stovky let, ale o tom, že jsou v populaci i jedinci s vrozeným sklonem k VTE, se dlouho nevědělo. Zájem o vrozené trombofilní stavy stoupá po roce 1965, kdy Egeberg [14] popsal vztah kongenitálního deficitu antitrombinu III (AT) k rozvoji VTE. Tento zájem je dále akcelerován v 80. letech minulého století, kdy Griffin et al [15] popsali kongenitální deficit proteinu C a Schwarz [16] s Compem [17,18] deficit proteinu S.
Zásadním zlomem v zájmu o trombofilní stavy byl v roce 1993 Dahlbäckův objev rezistence na aktivovaný protein C (APC-R) [19]. V roce 1994 Bertina z Leidenu zjistil, že za tuto rezistenci na aktivovaný protein C je zodpovědná mutace F V, označovaná dle místa objevu jako mutace leidenská [20].
Poslední popsanou mutací, která predisponuje jedince k VTE, je mutace protrombinová – F II20210A, popsaná v roce 1996 Poortem [21]. Ostatní vrozené trombofilní stavy jsou vzácnější. Jednotlivé trombofilní stavy se liší navzájem zejména frekvencí výskytu v populaci a svojí trombofilností, tedy mírou pohotovosti k rozvoji VTE. Některé z nich vedou k tzv. „získání funkce“, kdy dochází k větší generaci trombinu. Do této skupiny patří např. APC–R s mutací F V Leiden a mutace F II20210A. U některých naopak dochází ke „ztrátě funkce“. Sem patří deficity přirozených inhibitorů koagulace. První skupina – F V Leiden a mutace F II20210A patří mezi stavy velmi frekventní, méně trombofilní a jsou způsobeny jednou bodovou mutací, zatímco trombofilní stavy v druhé skupině jsou poměrně vzácné, více trombofilní a jsou způsobeny různými genetickými defekty.
Jak lze trombofilní stav tedy definovat? Přesto, že dosud neexistuje jediná mezinárodně uznávaná definice [22], lze definovat trombofilní stav jako stav, který má tendenci ke vzniku VTE a který může být geneticky podmíněn, může být získaný, či obojí. Na této definici se shodla před 9 lety skupina expertů WHO [23]. V Severní Americe jsou pojmem trombofilie označováni jedinci s výskytem spontánní VTE, jedinci s rekurentní VTE, jedinci s VTE v mladém věku či jedinci s VTE, jejíž tíže neodpovídá vyvolávajícímu stimulu. Stanovení definice trombofilního stavu je významné pro stanovení indikačních kritérií pro vyšetření jedince na vrozený trombofilní stav a na antifosfolipidový syndrom (APS). To má jak medicínský, tak ekonomický význam. Na našem pracovišti jsou na kongenitální trombofilní stav vyšetřeni všichni jedinci splňující následující kritéria:
- VTE pod 50 let věku – spontánní VTE i VTE v rizikové situaci
- jedinci s rekurentními VTE příhodami
- jedinci s VTE v neobvyklé lokalizaci: v portální, slezinné, renální, dolní duté žíle apod
- jedinci s rekurentními záněty povrchových žil bez přítomnosti varixů
- jedinci s pozitivní rodinnou anamnézou výskytu VTE před 50. rokem věku. V každém případě se jedná o objektivně prokázanou trombotickou příhodu (zobrazovací metodou).
Nyní se krátce zmíníme o jednotlivých vrozených trombofilních stavech.
Antitrombin (AT) – dříve označován jako antitrombin III (AT III) je nejsilnějším inhibitorem trombinu a je též inhibitorem ostatních serinových proteáz – F IXa, F Xa, F XIa, F XIIa a též komplexu TF/F VIIa. Heparin reakci mezi antitrombinem a serinovými proteázami značně urychluje. Antitrombin je syntetizován v játrech, má biologický poločas 48 hodin. Vrozený deficit antitrombinu má prevalenci v populaci 1 : 2000 až 1 : 5000, a patří tedy mezi stavy s poměrně malou frekvencí výskytu, ale naopak mezi stavy značně trombofilní. Ve studiích, které stanovovaly prevalenci jednotlivých trombofilních stavů ve skupině neselektovaných jedinců s první VTE příhodou v životě, byla frekvence výskytu deficitu antitrombinu 0,5–1,0 % [24,25]. Relativní riziko VTE je u jedinců s kongenitálním deficitem antitrombinu 25–50krát větší než u jedince bez trombofilního stavu [8].
Protein C – je K1 vitamin dependentní inhibitor, který je syntetizován v játrech a má krátký biologickým poločas (5–7 hodin). Protein C je aktivován komplexem trombinu s trombomodulinem na povrchu endotelu. Aktivovaný protein C (APC) pak vede k vysoce selektivní proteolýze F Va a F VIIIa a je jedním z hlavních inhibitorů krevního srážení. Prevalence výskytu proteinu C se vyskytuje v populaci v 0,2–0,3 % [26,27]. Frekvence výskytu deficitu proteinu C ve skupině neselektovaných jedinců s první VTE se udává kolem 3 % [24,25,28]. Relativní riziko VTE je u jedinců s kongenitálním deficitem proteinu C 10–15krát větší [8].
Protein S – je další K1 vitamin dependentní protein, který slouží jako kofaktor aktivovaného proteinu C při proteolýze F Va a F VIIIa. Je syntetizován v játrech (podstatně méně v endotelu) a má biologický poločas 48 hodin. V plazmě se vyskytuje ve 2 formách – ve volné formě, která je funkčně aktivní (40 %) a ve formě vázané na C4bBP (složka komplementu) v 60 %. Tato část nemá kofaktorovou aktivitu.
Prevalence výskytu deficitu proteinu S v populaci není přesně známa, což je ovlivněno i výběrem metody stanovení. Nejvíce je doporučováno stanovení prevalence výskytu pomocí měření antigenu volného proteinu S [29]. V „Leiden Thrombophilia Study“ [28] byla nízká hladina antigenu volného proteinu S nalezena ve 3 % u nemocných s VTE a v 2,1 %, resp. v 1,3 % v kontrolní skupině. Z tohoto faktu vyplývá, že deficit proteinu S je jen mírným rizikovým faktorem pro VTE (zvyšující riziko VTE asi 2krát). Tento názor stran rizikovosti VTE je zpochybňován názory, které udávají prevalenci výskytu deficitu proteinu S v populaci nižší než 1–2 % [22].
Deficity přirozených inhibitorů koagulace v homozygotní formě představují velmi těžké, závažné protrombotické stavy, které se v případě deficitu proteinu C a S manifestují již po narození – jako neonatální purpura fulminans [30], později jako recidivující VTE a mohou vést i k warfarinem indukované kožní nekróze [31]. Homozygotní forma deficitu AT l. typu je neslučitelná se životem. Deficity antitrombinu, proteinu C a S se diagnostikují jen v 5–10 % všech nemocných s VTE [25,32]. Do roku 1993 tak velká část familiárních trombofilních stavů zůstávala nevysvětlena.
APC rezistence a mutace F V Leiden
V roce 1993 objevil Dahlbäck se svými spolupracovníky [19] u 3 nepříbuzenecky vázaných jedinců s žilní trombózou dosud nepopsaný defekt v antikoagulačním systému proteinu C, který vedl k špatné odpovědi plazmy na aktivovaný protein C – k APC rezistenci. Aktivovaný protein C (APC) je klíčový enzym v systému proteinu C. Mezi komponenty tohoto systému patří: trombomodulin, protein C, protein S, endoteliální receptor pro protein C (EPCR), F V a skupina inhibitorů APC. Systém proteinu C je jedním z přirozených antikoagulačních mechanizmů, který slouží k zabraňování nárůstu trombu a okluzi cév. Aktivovaný protein C pak vede k proteolytické inaktivaci dvou aktivovaných koagulačních faktorů, vázaných na fosfolipidy, a to F Va a F VIIIa. V roce 1994 Bertina [20] prokázal, že APC-R je způsobena bodovou mutací genu F V (G→A v nukleotidu 1691) v exonu 10, což vede k záměně aminokyseliny argininu v pozici 506 za glutamin. Faktor V s touto mutací je označován F VQ506 nebo dle místa objevu jako F V Leiden. Dědičnost této mutace je autozomálně dominantní [33].
Rezistence na aktivovaný protein C je více než v 90 % případů způsobena mutací F V Leiden. V ostatních případech se jedná buď o jiné vrozené příčiny – F V Cambridge (Arg 306 →Thr) popsaný poprvé Williamsonem [34] či homozygotní formou HR2 haplotypu v genu F V [35]. Prevalence F V Cambridge je v populaci velmi malá, velmi malá je i prevalence mezi nemocnými s trombózou.
Častěji se jedná o tzv. „získanou APC-R“, která se nejčastěji vyskytuje v souvislosti s těhotenstvím [36], při užívání hormonální antikoncepce [37], u nádorových onemocnění [38], akutních zánětů [39], cirkulující protilátky typu lupus antikoagulans (LA) [40,41] či u nemocných s akutní trombotickou příhodou [42]. Zatímco jedinci s F V Cambridge nemají větší riziko VTE a ani HR2, haplotyp se u jedinců s podezřením na trombofilní stav nevyšetřuje, tato tzv. „získaná APC-R“ je rizikovým faktorem pro VTE, jak to prokázali např. Visser et al v Leidenské trombofilní studii [43]. V této studii je prokázáno, že existuje závislost mezi stupněm APC rezistence a rizikem VTE, kdy jedinci s APC-R v dolní čtvrtině pozitivity mají 4krát větší riziko VTE než jedinci v horní čtvrtině pozitivity APC-R. V této studii je frekvence výskytu APC-R 36 % mezi jedinci s VTE, po korekci s F V Leiden pozitivními zůstává stále 24 % jedinců. Znamená to, že tato získaná APC-R (bez F V Leiden) se nachází u každého 10. jedince přicházejícího s VTE v neselektovaném souboru jedinců. Rizikovost získané APC-R pro VTE byla potvrzena v rozsáhlé populační studii, která zahrnovala více než 15 000 jedinců. Zde jedinci s APC-R bez F V Leiden měli 2krát větší riziko VTE než jedinci bez APC-R [44].
Byla též popsána mutace F V (Arg 306 Gly) Hong-Kong, která však nevede k APC-R [45].
Příčiny APC-R bez mutace F V Leiden přehledně shrnuje tab. 2.

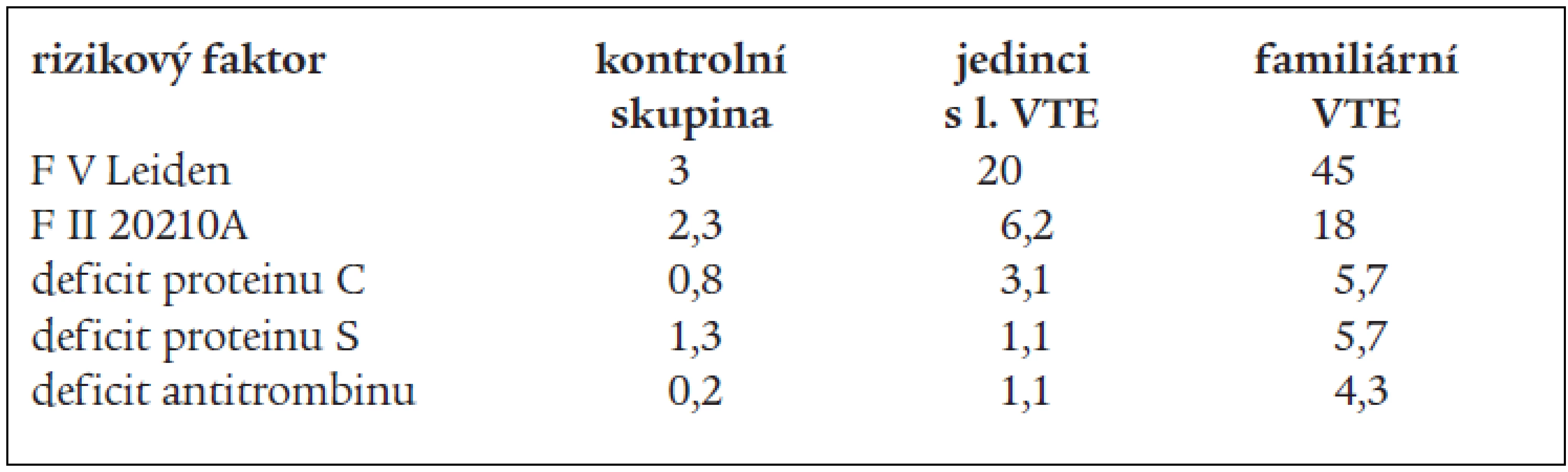
Protrombinová mutace F 20210A popsaná v roce 1996 Poortem et al se vyskytuje v europoidní populaci kolem 2 % v závislosti na geografické distribuci (vyšší prevalence na jihu Evropy než na severu) [46] a jedinci s touto mutací mají 2–3krát větší relativní riziko VTE [21]. Tato mutace byla nalezena v 18 % mezi jedinci s VTE mezi selektovanými nemocnými s familárním výskytem VTE a v 6,2 % u jedinců (v neselektovaném výběru) s první VTE příhodou [21].
V tab. 3 (dle Bertiny) [47] jsou shrnuty údaje o prevalenci trombofilních stavů (včetně F V Leiden, které jsou odvozeny z leidenské trombofilní studie).
Mezi získané trombofilní stavy patří i antifosfolipidový syndrom. Tento syndrom popsali v roce 1986 Hughes a Harris [48]. Je definován kritérii klinickými a laboratorními. Mezi klinická kritéria patří:
- výskyt arteriální či žilní trombózy (či trombózy v malých cévách), postihující kterýkoliv orgán;
- gynekologické komplikace: rekurentní aborty – 3 a více po sobě jdoucích abortů v 1. trimestru gravidity do 10. týdne gravidity, dále jeden a více potratů v 10. a po 10. týdnu gravidity, s normální morfologií plodu a nakonec 1 a více předčasných porodů do či v 34. týdnu gravidity, opět s morfologicky normálním plodem;
- trombocytopenie, ale toto kritérium se nověji z klinických kritérií vylučuje.
Mezi laboratorní kritéria patří průkaz přítomnosti protilátky typu lupus antikoagulans nebo průkaz přítomnosti antikardiolipinových protilátek či protilátek proti β2-glykoproteinu I, a to ve třídě IgG či IgM ve středních a vysokých titrech [49,50].
Antifosfolipidové protilátky obecně představují široké spektrum autoprotilátek, které mají rozdílnou afinitu a specificitu k cílovému antigenu [51]. Jsou namířeny proti různým typům fosfolipidů, fosfolipidům vázajících proteiny, nebo obou. Pro splnění diagnózy APS je nutno splnit alespoň jedno klinické a jedno laboratorní kritérium, které musí být potvrzeno opakovanou pozitivitou s časovým odstupem 6 týdnů až 3 měsíců. Jedinci s protilátkou LA mají větší riziko VTE. Mezi nemocnými s VTE byla tato protilátka popsána v 5–15 % [24,52,53].
Vysoká hladina F VIII
Vysoká hladina (aktivita) F VIII je rizikovým faktorem pro VTE. Toto riziko stoupá se zvyšující se hladinou F VIII. Jedinci s aktivitou F VIII nad 150 % mají 6krát větší riziko VTE než jedinci s aktivitou F VIII pod 100 % [10]. Aktivita F VIII větší než 150 % se však vyskytuje v normální populaci v 11 %. Vysoká aktivita byla připisována probíhajícímu zánětu (např. u posttrombotického syndromu), ale další práce ukázaly, že i po vyloučení tohoto vlivu (komparací F VIII s hodnotou CRP) [54] zůstává vysoká aktivita F VIII rizikovým faktorem pro VTE. Vysvětlení pro tuto vysokou hladinu není zcela jasné a vzhledem k familiárnímu výskytu se nabízí genetická predispozice [55], i když žádná genová mutace nebyla dosud prokázána [56,57].
Obecně je znám fakt, že jedinci s krevní skupinou 0 mají nižší hladinu F VIII než jedinci s krevní skupinami A,B, či AB.
Hyperhomocysteinemie
Mírně zvýšená hladina homocysteinu je spojena s vyšším rizikem venózního tromboembolizmu [9,58,59,60]. V Nizozemí byla nalezena hladina homocysteinu vyšší než 18,5 μmol/l u 5 % zdravých jedinců a je spojena s 2krát větším rizikem VTE [9]. Stejné riziko bylo nalezeno ve studii v Itálii, zde však byla tato hladina homocysteinu nalezena u 10 % zdravých jedinců [59]. Příčiny vyšší hladiny homocysteinu mohou být jak vrozené, tak získané [61]. Z vrozených příčin jsou to genové mutace, které vedou k defektům enzymů, které se uplatňují vmetabolizmu homocysteinu – např. cystathion β-synthasa (CS). Ze získaných příčin vede k vyšší hladině homocysteinu nízký přísun vitaminu B6, B12, kyseliny listové [62,63,64]. Nejčastější genetickou mutací, která může vést k vyšší hladině homocysteinu, je termolabilní varianta enzymu methylen tetrahydrofolát reduktázy – MTHFR C677T [65,66]. Vlastní mutace MTHFR C677T je velmi prevalentní v normální populaci a názory na rizikovost této mutace se vyvíjely. Samotná mutace MTHFR C677T byla ve starších pracích považována za rizikový faktor pro VTE [67], pozdější práce však tuto rizikovost vyvrátily [68,69]. Fakt, že vlastní mutace MTHFR C677T bez zvýšené hladiny homocysteinu není rizikovým faktorem pro VTE, a to jak v heterozygotní, tak v homozygotní formě jasně prokázala práce Browna [70]. Tato mutace tedy nepatří tedy do základního spektra vyšetření u nemocných s podezřením na trombofilní stav [22].
Při hodnocení výsledků trombofilního stavu je nutno vždy zvážit následující informace:
- termín odběru vzorků v časovém vztahu k výskytu VTE
- současnou medikaci (včetně ev. přetrvávání vlivu hormonální antikoncepce)
- metodiku vyšetření
V případě funkčních vyšetření nutno počítat, že k definitivnímu stanovení diagnózy vrozeného trombofilního stavu bude nutno vyšetření zopakovat.
Shrnutí
Volba délky antikoagulační terapie po prodělaném venózním tromboembolizmu vychází z následujících faktů: počet VTE, okolnosti vzniku (spontánní či v rizikové situaci), tíže příhody, lokalizace trombózy a eventuálně na přítomnosti trombofilního stavu (dle nálezu). V případě pozitivního nálezu pak zveme na vyšetření všechny první pokrevní příbuzné.
Závěr
Venózní tromboembolizmus je stále závažné, nicméně správnou profylaxí eliminovatelné onemocnění. Tohoto cíle lze dosáhnout jedině při co nejpřesnějším ohodnocení míry rizika venózního tromboembolizmu u každého jedince.
doc. MUDr. Petr Dulíček, Ph.D.
www.fnhk.cz
e-mail: brambpav@fnhk.cz
Doručeno do redakce: 11. 3. 2005
Přijato k otištění: 11. 3. 2005
Sources
1. Goldhaber SZ. Epidemiology of pulmonary embolism and deep vein thrombosis. In: Tuddenham EGD, Bloom AL, Forbes CD et al. Hemostasis and Thrombosis. Edinburg: Churchill Livingstone 1994 : 1327–1333.
2. Anderson FA, Wheeler HB, Goldberg RJ et al. A population-based perspective of the hospital incidence and case–fatality rates of deep vein thrombosis and pulmonary embolism. Arch Intern Med 1991; 151 : 933–938.
3. Nordström M, Lindblad B, Berqvist D et al. A prospective study of the incidence od deep vein thrombosis with a defined urban population. J Intern Med 1992; 232 : 155–160.
4. Silverstein MD, Heit JA, Mohr DN et al. Trends in the incidence of deep vein thrombosis and pulmonary embolism. Arch Intern Med 1998; 158 : 585–593.
5. Virchow R Thrombose und Embolie. Vol Gesammelte Abhandlungen zur wissenschaftlichen Medizin. Berlin: Verlag Maxirsch 1865 : 219.
6. Bertina RM Molecular risk factors for thrombosis. Thromb Haemost 1999; 82 : 601–609.
7. Samama MM. An epidemiologic study of risk factors for deep vein thrombosis in medical outpatients – the Sirius Study. Arch Intern Med 2000; 160 : 3415–3420.
8. Rosendaal FR. Risk factors for venous thrombotic disease. Thromb Haemost 1999; 82 : 610–619.
9. den Heyer M, Koster T, Bloom HJ et al. Hyperhomocysteinemia as a risk factor for deep–vein thrombosis. N Engl J Med 1996; 334 : 759–762.
10. Koster T, Blann AD, BriËt E et al. Role of clotting factor VIII in effects of von Willebrand factor on occurrence of deep-vein thrombosis. Lancet 1995; 345 : 152–155.
11. van Hylckama Vlieg A, van der Linden IK, Bertina RM et al. High levels of factor IX increase the risk of venous thrombosis. Blood 2000; 95 : 3678–3682.
12. Meijers JCM, Tekelenburg WLH, Bouma BN et al. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med 2000; 342 : 696–701.
13. van Tilburg NH, Rosendaal FR, Bertina RM. Thrombin activatable fibrinolysis inhibitor and the risk for deep vein thrombosis. Blood 2000; 95 : 2855–2859.
14. Egeberg O Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh 1965; 13 : 516–530.
15. Griffin JH, Evatt B, Zimmerman TS et al. Deficinecy of protein C in congenital thrombotic disease. J Clin Invest 1981; 68 : 1370–1373.
16. Schwarz HP, Fisher M, Hapmeier P et al. Plasma protein S deficiency in familial thrombotic disease. Blood 1994; 64 : 1297–1300.
17. Comp PC, Nixon RR, Cooper MR et al. Familial protein S deficiency is associated with recurrent thrombosis. J Clin Invest 1984; 74 : 2082.
18. Comp PC, Esmon CT. Recurrent venous thromboembolism in patients with a partial deficiency of protein S. N Engl J Med 1984; 311 : 1525–1528.
19. Dahlbäck B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C; prediction of a cofactor
to activated protein C. Proc Natl Scad Sci USA 1993; 90 : 1004–1008.
20. Bertina RM, Koeleman BPC, Koster T et al. Mutation in blood coagulation factor V associtated with resistance to activated protein C. Nature 1994; 369 : 64–67.
21. Poort SR, Rosendaal FR, Reitsma PH et al. A common genetic variation in the 3’-untraslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an in venous thrombosis and an increase in venous thrombosis. Blood 1996; 88 : 3698–3703.
22. Walker ID, Greaves M, Preston FE. Guideline. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114 : 512–528.
23. Lane DA, Mannucci PM, Bauer KA et al. Inherited thrombophilia. Part I. Thromb Haemost 1996; 76 : 651–652.
24. Mateo J, Oliver A, Borrell M et al. Laboratory evaluation and clinical characterisation of 2132 consecutive unselected patients with venous thromboembolism – results of the Spanish multicentre study
on thrombophilia (EMET – Study). Thromb Haemost 1997; 77 : 444–451.
25. Heijboer H, Brandjes DPM, Büller HR et al. Deficiencies of coagulation inhibiting and fibrinolytic proteins in outpatients with deep vein thrombosis. N Engl J Med 1990; 332 : 1512–1516.
26. Miletich JP, Sherman L, Broze GI. Absence of tbrombosis in subjects with heterozygous protein C deficiency. N Engl J Med 1987; 317 : 991–996.
27. Tait RC, Walker ID, Reitsma PH et al. Prevalence of protein C deficiency in the healthy population. Thromb Haemost 1995; 73 : 87–93.
28. Koster T, Rosendaal FR, BriËt E et al. Protein C deficiency in a controlled series of unselected outpatients. An infrequent but clear risk factor for venous thrombosis, Leiden Thrombophilia Study. Blood 1995; 85 : 2756–2761.
29. Faioni EM, Valsecchi C, Palla A et al. Free protein S deficiency is a risk factor for venous thrombosis. Thromb Haemost 1997; 78 : 1343–1346.
30. Seligsohn U, Berger A, Abend M et al. Homozygous protein C deficiency manifested by massive venous thrombosis i the newborn. N Engl J Med l984; 310 : 559–562.
31. McGehee WG, Klotz TA, Epstein DJ et al. Coumarin necrosis associated with hereditary protein C deficiency. Ann Intern Med 1984; 101 : 59–60.
32. Malm J, Laurell M, Nilsson IM et al. Thromboembolic disease – critical evaluation of laboratory investigation. Thromb Haemost 1992; 740 : 9–15.
33. Svensson Pj, Dahlbäck B Resistance to activated protein C as a basis for thrombosis. N Engl J Med 1994; 330 : 517–522.
34. Williamson D, Brown K, Luddigton et al. Factor V Cambridge: a new mutation (Arg 306-Thr) associated with resistance to activated protein C. Blood 1998; 91 : 1140–1144.
35. Bernardi F, Faioni EM, Castoldi E et al. A factor V genetic component differing from factor F R506Q contributes to activated protein C phenotype. Blood 1997; 90 : 1552–1557.
36. Meinardi JR, Henkens CMA, Heringa MP et al. Acquired APC resistance related to oral contraceptives and pregnancy and its possible implications for clinical practise. Blood Coag Fibrinol 1997; 8 : 152–154.
37. Olivieri O, Friso S, Manzato F et al. Resistance to activated protein C in healthy women taking oral contraceptives. Br J Haematol 1995; 91 : 465–470.
38. Green D, Maliekel K, Sushko E et al. Activated protein C in cancer patients. Thromb Haemost 1997; Suppl: 316.
39. Mathonnet F, de Mazancourt P, Denninger MM et al. Role of factor VIII on activated protein C resistance ratio in inflammatory diseases. Br J Haematol 1996; 95 : 423–425.
40. Dahlbäck B. Resistance to activated protein C caused by the R506Q mutation in the gene for factor V is a common risk factor for venous thrombosis. J Intern Med 1997; 242(Suppl 740): 1–8.
41. Nojima J, Kuratsune H, Suehisa E et al. Acquired activated protein C resistance is associated with the co-existence of anti-prothrombin antibodies and lupus anticoagulant activity in patients with systematic lupus erythematosus. Br J Haematol 2002; 118 : 57–83.
42. Svensson PJ, Zöller B, Dahlbäck B. Evaluation of original and modified APC-resistance tests in unselected outpatients with clinically suspected trombosis and in healthy controls. Thromb Haemost 1997; 77 : 332.
43. de Visser MCH, Rosendaal FR, Bertina RM. A reduced sensitivity for activated protein C in the absence of factor V Leiden increases the risk of venous thrombosis. Blood 1999; 93 : 1271–1276.
44. Rodeghiero F, Tosseto A. Activated protein C resistance and factor V Leiden mutation are independent risk factors for venous thromboembolism. Ann Intern Med 1999; 130 : 643–650.
45. Chan WP, Lee CK, Kwong YL et al. A novel mutation of Arg 306 of factor V gene in Hong Kong Chinese. Blood 1998; 91 : 1135.
46. Rosendaal FR, Doggen CJM, Zivelin A et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost 1998; 79 : 706–708.
47. Bertina RM. Genetic approach to thrombophilia. Thromb Haemost 2001; 86 : 92–103.
48. Hughes GRV, Harris EN, Gharavi AE. The anticardiolipin syndrome. J Rheumatol 1986; 13 : 486–489.
49. Galli M, Comfurius P, Barbui T et al. Anticoagulant activity of β2-glycoprotein I is potentiated by a distinct subgroup of anticardiolipin antibodies. Thromb Haemost 1992; 68 : 217.
50. Roubey RAS. Immunology of the antiphospholipid antibody syndrome. Arthritis Rheum 1996; 39 : 1444–1454.
51. Levine JS, Branch D, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346 : 752–763.
52. Ginsberg JS, Weles PS, Brill–Edwards P et al. Antiphospholipid antibodies and venous thromboembolism. Blood 1995; 86 : 3685–3691.
53. Simioni P, Prandoni P, Zanon E ta al. Deep venous thrombosis and lupus anticoagulant. Thromb Haemost 1996; 76 : 187–189.
54. O’Donnell J, Tuddenham EGD, Manning R et al. High prevalence of elevated F VIII levels in patients reffered for thrombophilia screening: role of increased synthesis and relationship to the acute phase reaction. Thromb Haemost 1997; 77 : 825–828.
55. Kamphuisen PW, Houwing-Duistermaat JJ, van Houwelingen JC et al. Familial clustering of factor VIII and von Willebrand factor levels. Thromb Haemost 1998; 79 : 323–327.
56. Mansveld EPG, Laftan M, McVey JH et al. Analysis of the F8 gene in individuals with high plasma factor levels and associated venous thrombosis. Thromb Haemost 1998; 80 : 561–565.
57. Kamphuisen PW, Eikenboom JCJ, Vos HL et al. Two highly variable CA repeat polymorphism in the factor VIII gene and the risk of venous thrombosis. Blood 1998; 90 : 97a.
58. Falcon CR, Cattaneo M, Panzeri D et al. High prevalence of hyperhomocysteinemia in patients with juvenile venous thrombosis. Arterioscler Thromb 1994; 14 : 1080–1083.
59. Simioni P, Prandoni P, Burlina A et al. Hyperhomocysteinemia and deepvein thrombosis: a case control study. Thromb Haemost 1996; 76 : 883–886.
60. Den Heijer M, Rosendaal FR, Blom HJ et al. Hyperhomocysteinemia and venous thrombosis: a meta analysis. Thromb Haemost 1998; 80 : 874–877.
61. D’Angelo A, Selhub J Homocysteine and thrombotic disease. Blood 1997; 90 : 1–11.
62. Kang SS, Wong PWK, Norusis M. Homocysteinemia due to folate deficiency. Metabolism 1987; 36 : 458–462.
63. Rees MM, Rodgers GM. Homocysteinemia: association of a metabolic disorder with vascular disease and thrombosis. Thrombosis Research 1993; 71 : 337–359.
64. Ubbink JB, Vermaak WJ, van der Merwe et al. Vitamin B12, vitamin B6, and folate nutritional status in men with hyperhomocysteinemia. Am J Clin Nutr 1993; 57 : 47–53.
65. Engbertsen AMT, Franken DG, Boers GHJ et al. Thermolabile 5,10–methylenetetrahydrofolate reductase as a cause of mild hyperhomocysteinemia. Am J Hum Genet 1995; 56 : 142–150.
66. Frosst P, Blom HJ, Milos R et al. A candidate genetic risk factor for vascular disease a common mutation in methylenetetrahydrofolate reductase. Nat Genet 1995; 10 : 111–113.
67. Arruda V, von-Zuben P, Chiaparini L et al. The mutation Ala 677 Val in the methylene tetrahydrofolate reductase gene: a risk factor for arterial disease and venous thrombosis. Thromb Haemost 1997; 77 : 818–821.
68. Salden A, Keeney S, Hay C et al. The C677T MTHFR variant and the risk of venous thrombosis (Letter). Br J Haematol 1997; 99 : 472.
69. Tosseto A, Missiaglia E, Frezzato M et al. The VITA project: C677T mutation in the methylen – tetrahydrofolate reductase gene and the risk of venous thromboembolism. Br J Haematol 1997; 97 : 804–806.
70. Brown K, Luddington R, Baglin T. Effect of the MTHFRC677T variant on the risk of venous thromboembolism: interaction with factor F V Leiden and prothrombin (F2G20210A) mutations. Br J Haematol 1998; 103 : 42–44.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2005 Issue 7 a 8
Most read in this issue
- Potransfuzní reakce
- Trombocytózy a trombocytemie
- Antifosfolipidový syndrom – diagnostika a léčba
- Protidestičková léčba