
Familiárna stredomorská horúčka – prvé skúsenosti na Slovensku
Familial Mediterranean Fever – First Experiences in Slovakia
Familial Mediterranean fever (FMF) is the most prevalent genetically determined autoinflammatory disease. FMF significantly decreases the quality of life and limits life expectancy due to the development of amyloidosis in affected individuals. Prevalence of FMF is highest in the south-eastern Mediterraneans. In other parts of the world, its occurance is often restricted to high-risk ethnic groups. In Central Europe, experience with FMF is scarse to none, as in the case of Slovakia, where no cases have been reported, so far. Herein we report the first five patients (3 adults and 2 children, 4 native Slovaks) in whom the diagnosis of FMF could be confirmed in Slovakia. Our experience demonstrates that FMF does occur in low-risk populations in Central Europe. Due to low prevalence and lack of experience, FMF diagnosis may be significantly delayed (4.5–30 years) and undiagnosed cases are to be expected in our population.
Key words:
familial mediterranean fever – amyloidosis – colchicin – Slovakia
Authors:
Tomáš Dallos 1; Lucia Lukáčiková Gálová 2; Eva Macejková 3; Jozef Sedlačko 4; Nataša Toplak 5; Maruša Debeljak 6; Hasmik Sargsyan 7; Denisa Ilenčíková 1; László Kovács 1
Authors‘ workplace:
II. detská klinika Lekárskej fakulty UK a DFNsP, Bratislava, Slovenská republika, prednosta prof. MUDr. László Kovács, DrSc., MPH
1; Kardiologická klinika FN Nitra, Slovenská republika, prednosta MUDr. Pavol Poliačik, PhD.
2; Reumatologická ambulancia Bella s. r. o., Banská Bystrica, Slovenská republika, vedúci pracoviska MUDr. Eva Macejková
3; III. interná klinika Lekárskej fakulty UK a UN Bratislava, Slovenská republika, prednosta prof. MUDr. Viliam Bada, CSc.
4; Oddelenie alergológie, reumatológie a klinickej imunológie Detskej univerzitnej nemocnice Ljubljana, Slovinsko, prednosta prof. Tadej Avčin, MD, PhD.
5; Centrum lekárskej genetiky Detskej univerzitnej nemocnice Ljubljana, Slovinsko, vedúci pracoviska Mirjana Župančič, mag. med. bioch.
6; Detské centrum pre FMF Štátnej lekárskej univerzity Jerevan, Arménsko, vedúci pracoviska Arman A. Babloyan, MD
7
Published in:
Vnitř Lék 2014; 60(1): 80-85
Category:
Case Report
Overview
Familiárna stredomorská horúčka (FMF) je najčastejšie geneticky podmienené autoinflamačné ochorenie. Významne zhoršuje kvalitu a limituje dĺžku života rozvojom systémovej amyloidózy. Najvyššia prevalencia je v juhovýchodnom Stredomorí, v iných častiach sveta je výskyt väčšinou viazaný na etnický pôvod z týchto oblastí. V strednej Európe sú skúsenosti s FMF obmedzené, na Slovensku doposiaľ chýbali úplne. Predstavujeme kazuistiky prvých 5 pacientov (3 dospelí, 2 deti, z toho 4 slovenského etnika), u ktorých bola FMF potvrdená na Slovensku. Naša skúsenosť potvrdzuje, že FMF sa v strednej Európe vyskytuje aj u nerizikových etník. Stanovenie diagnózy FMF je však v dôsledku nízkej prevalencie a nedostatku skúseností s týmto ochorením v našich podmienkach často zdĺhavé (4,5–30 rokov). Dá preto sa predpokladať, že v našej populácii je viac nediagnostikovaných jedincov.
Kľúčové slová:
familiárna stredomorská horúčka – amyloidóza – kolchicín – Slovensko
Úvod
Familiárna stredomorská horúčka (Familial Mediterranean Fever – FMF, OMIM 249100) je najdlhšie známe, najčastejšie sa vyskytujúce a prvé autoinflamačné ochorenie, u ktorého bola odhalená jeho genetická podstata [1,2]. Prevalencia FMF je vysoká (1 : 200–1 : 500) najmä v juhovýchodnom Stredomorí a na Blízkom východe. V dôsledku migrácie arménskeho, židovského, arabského a tureckého etnika sa u príslušníkov týchto etník vyskytuje aj v USA, Francúzsku (Arabi) a Nemecku (Turci) a sporadické prípady boli popísané aj v odľahlých krajinách ako je Japonsko [3]. V strednej Európe je evidovaných do 60 pacientov (z toho 11 geneticky potvrdených) najmä v balkánskych krajinách [4]. Prípady na území Slovenska neboli doposiaľ zdokumentované.
V tejto práci prezentujeme kazuistiky prvých pacientov, u ktorých bola FMF diagnostikovaná na Slovensku, pričom len jeden patril k rizikovému etniku. Chceme poukázať na výskyt FMF u príslušníkov slovenskej národnosti a na osudy pacientov, ktoré boli poznačené aj nedostatkom skúseností lekárov s týmto ochorením.
Kazuistiky
Štyria pacienti boli slovenskej národnosti a pochádzali z južnej časti stredného Slovenska. V žiadnej z rodín sa nevyskytla konsangvinita. Dve pacientky (2. a 3.) boli v príbuzenskom vzťahu, u ďalších 2 bola rodinná anamnéza negatívna. Jeden pacient bol cudzinec, ktorý patril k rizikovému etniku a v jeho rodine sa pravdepodobne vyskytli už 3 prípady FMF. Klinické údaje všetkých pacientov uvádzame v tabuľke.
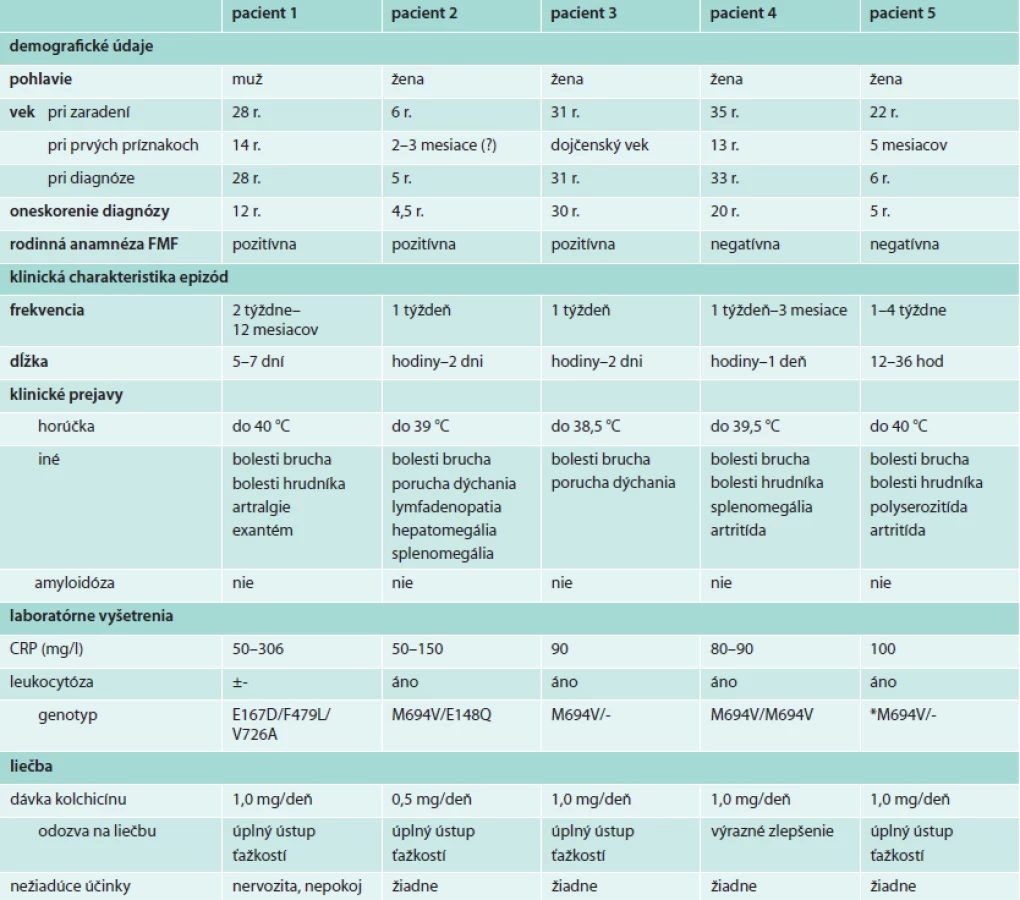
Kazuistika 1
26-ročný muž egyptskej národnosti bol hospitalizovaný pre týždeň trvajúce horúčky do 40 °C a bolesti brucha. Klinicky bola prítomná difúzna palpačná citlivosť brucha s peritoneálnym dráždením. Krvný obraz bol fyziologický s hraničnou leukocytózou, bola prítomná hyperfibrinogenémia a vysoká zápalová aktivita (CRP 306 mg/l). EKG, RTG hrudníka, sonografia a CT brucha boli bez patológie. Vzhľadom na vysoké zápalové parametre bola začatá empirická antibiotická liečba s následným ústupom klinických ťažkostí a poklesom zápalových parametrov v priebehu 48 hod.
Počas posledných 3 rokov, kedy žil na Slovensku, bol opakovane vyšetrený pre podobné ťažkosti a liečený antibiotikami s dobrým efektom. Zhodné ťažkosti rôznej intenzity však udával od 14. roku života; v nepravidelných intervaloch (2 tyždne až 1 rok) sa u neho objavovali epizódy 5–7 dní trvajúcich horúčok do 40 °C sprevádzaných výraznými tupými migrujúcimi bolesťami brucha a hrudníka, artralgiami ramenných, bedrových, kolenných a členkových kĺbov. Počas epizód mal sklon k obstipácii a eruktáciám, ale niekedy pozoroval aj riedke stolice s prímesou hlienov. Ako dieťa mával bolesti zápästí, 3-krát aj artritídu kolena s výpotkom. Udával, že po dlhšej chôdzi ho bolievajú lýtka.
Podobné klinické príznaky si pamätal u viacerých členov rodiny: otca a matky, ktorí sa neliečili, a u sestry, u ktorej bola prednedávnom začatá liečba kolchicínom pre podozrenie na FMF. Na základe klinických príznakov a pozitívnej rodinnej anamnézy sme vyslovili podozrenie na FMF. Vzhľadom na etnický pôvod pacienta sme začali terapeutický pokus kolchicínom a indikovali genetickú analýzu, ktorá potvrdila, že pacient je zložený heterozygot s 3 známymi heterozygotnými mutáciami (E167D/F479L/V726A) v géne MEFV. Amyloidózu nemal. Pod liečbou kolchicínom je bez ťažkostí a epizódy FMF sa neopakovali, ale liečbu pre subjektívne psychické ťažkosti po niekoľkých mesiacoch vysadil. Vzhľadom na problematickú spoluprácu sme mu odporúčali ďalšiu dispenzarizáciu na špecializovanom pracovisku v Egypte.
Kazuistika 2
3-ročné dievča sa predstavilo s chronickými bolesťami brucha a recidivujúcimi epizódami horúčok. Bola prvým dieťaťom zdravých nepríbuzných rodičov z fyziologickej gravidity s nenápadnou perinatálnou anamnézou. Ako 3-mesačná mala niekoľko epizód sťaženého dýchania s premodraním, ale bez poruchy vedomia, ktoré boli na základe polysomnografického vyšetrenia hodnotené ako syndróm alveolárnej hypoventilácie pri centrálne podmienených apnoických pauzách. Neurológ konštatoval generalizovanú hypotóniou bez topickej symtomatológie, odchýlok v EEG a štrukturálnych abnormít v sonografickom a MRI znázornení mozgu. Bola vylúčená vrodená srdcová chyba a gastroezofágový reflux.
Od nástupu do kolektívu bola opakovane hospitalizovaná pre horúčnaté stavy s vysokou zápalovou aktivitou nejasnej etiológie. Horúčky do 39 °C sa vyskytovali v nepravidelných intervaloch približne 1-krát za týždeň, trvali niekoľko hodín až 2 dni s následným spontánnym ústupom. Nemávala klinické prejavy akútnej infekcie, artralgie či artritídu ani kožné zmeny, ale pri horúčkach sa zvýrazňovali bolesti brucha niekedy sprevádzané hnačkou. Poruchy dýchania už neviedli k desaturáciám, ale matka popisovala sťažené dýchanie dieťaťa počas febrilít. Opakovane bola konštatovaná mierna neprogredujúca krčná, axilárna a ingvinálna lymfadenopatia. Medzi epizódami bolo dieťa bez ťažkostí a až na pretrvávanie pobolievania brucha, bolo väčšinou čulé, dobre naladené a usmiate, primerane sa psychomotoricky vyvíjalo až neskôr sa začal mierne spomaľovať rast (výška -1,5 SD a -1,7 SD v 2., resp. 5. roku života).
Zistili sme ľahkú mikrocytovú anémiu a hraničnú sideropéniu. Počas febrilít bola prítomná mierna leukocytóza s neutrofíliou bez výskytu patologických fenotypov leukocytov. Neboli zistené biochemické známky orgánového postihnutia. Zápalové parametre boli zvýšené ((FW 30/40–50/80, CRP 50–150 mg/l) a počas celého sledovania ani medzi epizódami febrilít nikdy nepoklesli na normálne hodnoty. Kultivačnými a sérologickými vyšetreniami sa nepodarilo potvrdiť infekciu. Vylúčili sme závažný primárny bunkový aj humorálny imunodeficit, cyklickú neutropéniu, deficit komplementu a cystickú fibrózu. Pre nižšie počty CD8+ (10–16 %) a CD3+ (48–55 %) subpopulácií T-lymfocytov v minulosti užívala imunomodulačnú liečbu s čiastočným efektom.
Sonograficky sa zobrazila retroperitoneálna lymfadenopatia a mierna hepatosplenomegália, avšak v čreve sa nepodarilo ani pomocou značkovaných leukocytov (Leukoscint/SPECT-CT) preukázať zápalové zmeny v zmysle nešpecifického zápalového ochorenia čreva. Endoskopické vyšetrenie sme preto neindikovali. Vzhľadom na pretrvávajúce bolesti brucha, hypotrofiu, hraničný rast a sklon k sideropénii ako aj slabú pozitivitu protilátok proti gliadínu podstúpila biopsiu tenkého čreva s hraničným histologickým nálezom pre celiakiu, ktorý viedol k indikácii bezlepkovej diéty bez efektu na klinický stav dieťaťa.
Nemala artritídu, koža bola bez exantému a prejavov vaskulitídy, oftalmologické vyšetrenie vylúčilo uveitídu a vaskulitídu na očnom pozadí. Chýbali klinické a laboratórne prejavy systémového ochorenia spojiva, autoprotilátky boli negatívne. Dobrý klinický stav ostro kontrastoval s trvalo zvýšenými zápalovými parametrami. Pre epizodický charakter febrilít so známkami aktivácie nešpecifickej imunity sme predpokladali syndróm periodických horúčok. V rodine sa podobné ťažkosti od útleho veku vyskytli u sestry matky dieťaťa (pacientka v kazuistike 3). Keďže klinické príznaky zapadali do širšieho spektra manifestácie FMF, indikovali sme analýzu génu MEFV, ktorá potvrdila nosičstvo dvoch známych mutácií (zložený heterozygot E148Q/M694V). Po začatí liečby kolchicínom (0,5 mg/deň), ktorú toleruje bez výraznejších ťažkostí, ustúpili epizódy febrilít ako aj bolesti brucha, dieťa začalo prospievať a zápalové parametre prvýkrát klesli na fyziologické hodnoty.
Kazuistika 3
31-ročná žena (teta pacientky v kazuistike 2) od dojčenského veku trpela bolesťami brucha a horúčkami. Podobne ako jej neter mala bližšie nešpecifikovanú poruchu dýchania v dojčenskom veku. Jej detstvo bolo poznačené početnými vyšetreniami, aj pre podozrenie na náhlu brušnú príhodu a hospitalizáciami, ktoré neodhalili príčinu jej ťažkosti. V dospelosti sa sťažovala na nepravidelne sa vyskytujúce krátke epizódy difúznych bolestí brucha sprevádzané pocitom sťaženého dýchania, myalgiami, artralgiami a zvýšenou telesnou teplotou. Počas epizód bývali zvýšené zápalové parametre (napr. CRP 90 mg/l) s následným spontánnym poklesom bez antibiotickej liečby. Napriek pretrvávajúcim ťažkostiam prestala z frustrácie navštevovať lekárov.
Pacientku sme vyšetrili po potvrdení diagnózy FMF u jej netere. Genetické vyšetrenie aj u nej potvrdilo nosičstvo heterozygotnej mutácie M694V. Vzhľadom na prítomnosť klinických príznakov zlučiteľných s diagnózou FMF sme za}čali liečbu kolchicínom s dobrým efektom. Napriek vyššiemu veku nebola u nej prítomná mirkoalbuminúria ako prejav možnej amyloidózy. Detailnejšia anamnéza odhalila, že brat jej matky mal dlhodobo ťažkosti s obličkami, na ktoré exitoval.
Kazuistika 4
Dievča zo 4. fyziologickej gravidity a nepozoruhodným perinatálnym obdobím bolo prvýkrát hospitalizované v 6. mesiaci života pre folikulárnu angínu a obojstranný zápal stredného ucha. Ďalšie angíny mala v 4.–6. roku života, prečo ako 6-ročná podstúpila adenotonzilektómiu. V 13. roku života sa u nej vyskytli opakovane epizódy horúčok sprevádzané bolesťami dolných končatín a začervenaním pravého členka, neskôr aj septický stav so zvýšenými zápalovými parametrami bez dôkazu etiologického agens a lokalizácie infekcie. Odvtedy sa niekoľkokrát do mesiaca opakovali ataky horúčok s bolesťami brucha a epizodickými artritídami kolien, členkov a ramien, neskôr sa pridali aj bolesti na hrudníku. Medzi 21. a 32. rokom života podstúpila početné a opakované odborné vyšetrenia vrátane neurologického, kardiologického, pneumologického, imunologického, urologického, hematologického, ortopedického, infektologických a reumatologických. Bola vylúčená infekčná etiológia a endokarditída. Urológ našiel angiolipóm ľavej obličky. Imunológ konštatoval známky dysregulácie špecifickej imunity. Zistila sa splenomegália. Hoci autoprotilátky boli negatívne, bolo vyslovené podozrenie na nediferencované systémové ochorenie spojiva. I keď MRI vyšetrenie zobrazilo subakromiálnu burzitídu vpravo, reumatické ochorenie sa nepotvrdilo. CT hrudníka neodhalilo príčinu bolestí na hrudníku ani v oblasti chrbtice. Ortopéd však predpokladal vertebrogénny algický syndróm pri skolióze, ktorý zdanlivo podporoval aj MRI nález multietážových protrúzií intervertebrálnych diskov cervikálnej a lumbálnej chrbtice a Schmorlových uzlov v oblasti distálnej Th chrbtice.
Nasledovalo obdobie so zvýšenou frekvenciou aták febrilít s myalgiami, artralgiami, bolesťami brucha a hrudníka. Gravidita v 29. roku života bola poznačená recidivujúcimi febrilitami a skončila spontánnym abortom v 3. mesiaci. Pacientka s prosbou o pomoc oslovila svoju kamarátku z detstva lekárku internej medicíny, ktorá preštudovala zdravotnú dokumentáciu a vzhľadom na súbor klinických príznakov a trvalo zvýšenú zápalovú aktivitu (CRP 85 mg/l) vyslovila podozrenie na FMF. Genetická analýza génu MEFV potvrdila homozygotnú mutáciu M694V. Napriek rizikovému genotypu a dlho neliečenému ochoreniu neboli prítomné známky postihnutia obličiek amyloidózou. Pod liečbou kolchicínom došlo k výraznému zlepšenou klinického stavu: jednodňové ataky febrilít s artralgiami sa vyskytujú už len raz za 2–3 mesiace, pacientka zvládla graviditu a bez komplikácií porodila zdravé dieťa. Genetické vyšetrenie príbuzných potvrdilo heterozygotnú mutáciu M694V u otca, matky aj mladšieho brata, ktorí sú ale bez klinických ťažkostí, a teda nevyžadujú liečbu.
Kazuistika 5
Eutrofické dieťa z 2. fyziologickej gravidity vo veku 4 mesiacov prekonalo akútnu otitídu s komplikovaným priebehom, ktorá si vyžiadala antrummastoidektómiu. V priamej následnosti sa začali objavovať v krátkych 7-dňových intervaloch epizódy horúčok trvajúce 12–36 hod. Podstúpila komplexné vyšetrenia na viacerých klinických pracoviskách, ktoré preukázali len ľahkú hypogamaglobulinémiu v triedach IgA a IgG, prítomnosť antigénu HLA-B27 a zvýšenú zápalovú aktivitu. Na základe týchto vyšetrení sa predpokladalo systémové ochorenie spojiva a pacientka užívala nesteroidové antiflogistiká a glukokortikoidy bez efektu na výskyt horúčnatých stavov. Tieto sa opakovali približne v mesačných intervaloch, dieťa bolo vždy febrilné, schvátené, malo bolesti brucha a miestami vykazovalo známky polyserozitídy a artritídy veľkých kĺbov. Medzi epizódami bola bez ťažkostí.
Vo veku 5,5 rokov absolvovala ďalšie komplexné vyšetrenia vrátane nefrologického, kardiologického, hematologického aj s punkciou kostnej drene, imunologického a zobrazovacie vyšetrenia, ktoré neviedli k objasneniu diagnózy. Vzhľadom na pokračujúce ťažkosti rajónny reumatológ zvážil možnosť familiárnej stredomorskej horúčky a začal terapeutický pokus kochicínom. Liečba bola okamžite účinná – epizódy febrilných stavov sa pod liečbou už nevyskytovali. Dodatočná genetická analýza na zahraničnom pracovisku vo veku 10 rokov potvrdila prítomnosť heterozygotnej mutácie M694V. V 14. roku života bol u pacientky diagnostikovaný Hodgkinov lymfóm a po chemoterapii a rádioterapii je až do veku 22 rokov v remisii ochorenia. Naďalej užíva kolchicín, je bez febrilných epizód a nedošlo u nej k rozvoju amyloidózy.
Diskusia
V poslednej dekáde sme svedkami objasnenia podstaty viacerých porúch univerzálnych mechanizmov prirodzenej imunity, ktoré vyúsťujú do jej spontánnej a nekontrolovanej aktivácie prejavujúcej sa v podobe autoinflamačných ochorení. Dostupná a vysoko účinná liečba (blokáda IL1, ev. TNFα) z nich robí medicínsky veľmi atraktívne nozologické jednotky. Ich zriedkavý výskyt a s tým súvisiaci nedostatok skúseností sťažujú ich diagnostiku a vytvárajú situáciu, v ktorej je dostupná liečba, ale málokedy sa rozpozná pacient, ktorý ju vyžaduje.
FMF je prototypom autoinflamačného ochorenia, ktoré je známe viac ako 100 rokov. Aj naše skúsenosti potvrdzujú, že jeho nízka prevalencia v etnicky nedisponovanej populácii sa dodnes podpisuje na zložitom osude postihnutých pacientov. Našich 5 kazuistík demonštruje len niekoľko možných, individuálne veľmi odlišných, a predsa charakteristických priebehov FMF. Pacient egyptskej národnosti s recidivujúcimi epizódami horúčok, bolesťami brucha a zvýšenými zápalovými parametrami predstavuje pri pozitívnej rodinnej anamnéze štandardnú klinickú situáciu, ktorá v endemickej oblasti musí viesť k vylúčeniu FMF. Striedavý priebeh, krátke alebo naopak netypicky dlhé trvanie a spontánny ústup ťažkostí spôsobujú, podobne ako v prezentovanom prípade, významné oneskorenie diagnózy aj v týchto krajinách. Na FMF je však potrebné myslieť aj u príslušníkov rizikových etník žijúcich u nás. Takýto prístup viedol k stanoveniu správnej diagnózy aj u 2 známych pacientov s FMF arménskeho [5], resp. sýrskeho pôvodu (osobná komunikácia s prof. MUDr. P. Doležalovou, CSc.) v Českej republike. Klinické diagnostické kritériá validované na rizikových populáciách predstavujú účinný skríningový nástroj a pri príslušnom etniku umožňujú aj diagnostické použitie kolchicínu [6–8].
V geografických oblastiach s nízkou prevalenciou môže klinický obraz FMF v dôsledku genetických a environmentálnych faktorov podliehať variáciám. U pôvodného obyvateľstva, ale aj u príslušníkov rizikových etník tu býva vyšší výskyt nejednoznačných a atypických priebehov a manifestácia v neskoršom veku (15,6 ± 2,5 rokov), turecké deti žijúce v Nemecku majú napriek zhodnému veku miernejší priebeh ochorenia a japonskí pacienti často vyžadujú nižšiu dávku kolchicínu na dosiahnutie remisie [9]. Tieto údaje poukazujú na to, že kritériá vyvinuté pre endemické etniká, môžu byť len obmedzene použiteľné v nerizikových populáciách (špecificita 45,0 % vs 98,8 %). Vo všeobecnosti však platí, že všetky kritériá vykazujú aj v týchto populáciách vysokú senzitivitu (86,5–100 %) a v nerizikovom etniku sú spojené skôr s falošnou pozitivitou. Navyše u detí, u ktorých môžu byť recidivujúce horúčky jediným prejavom ochorenia [10], použitie kritérií s vysokou senzitivitou a nízkou špecificitou v tejto vekovej kategórii naráža na problém častého výskytu podobných stavov odlišnej etiológie. To odzrkadľuje aj nižšia špecificita kritérií Tel Hashomer (kritéria stanovená v nemocnici Tel ha-Šomer v Izraeli) u detí v porovnaní s dospelými (54,6 % vs 98,8 %) v samotnej židovskej populácii. Ale aj kritériá pre deti vyvinuté v Turecku, ktoré vykazovali vyhovujúce parametre v tejto populácii, sa v západoeurópskej populácii správali podobne ako kritériá pre dospelých (senzitivita 100 %, špecificita 50 %) [11]. Naši 4 slovenskí pacienti splnili príslušné kritériá a ak by patrili k rizikovému etniku, bol by u nich indikovaný terapeutický pokus kolchicínom. Avšak pre nízku špecificitu všetkých doposiaľ validovaných kritérií sa takýto postup v nerizikových populáciách v súčasnosti neodporúča. Z našich pacientov bol použitý len u 6-ročnej pacientky v čase, keď nebola dostupná analýza génu MEFV, ktorá sa doplnila dodatočne a popri dobrej klinickej odpovedi na liečbu potvrdila diagnózu FMF.
Všetky diagnostické kritériá sú založené výlučne na prítomnosti klinických príznakov, neobsahujú žiadne laboratórne parametre a nerátajú ani s možnosťou potvrdenia diagnózy molekulárne-genetickým vyšetrením. To zdôrazňuje dôležitosť klinických príznakov, ktoré je potrebné objektivizovať v akútnej fáze ochorenia a zohľadňuje skutočnosť, že genetická analýza je len obmedzene prínosná. Na druhej strane je situácia mimo endemických oblastí o to zložitejšia, že tu je vyššie zastúpenie menej častých mutácií, ktoré môžu viesť k miernejšiemu priebehu alebo atypickým manifestáciám FMF, ktoré sa len vzdialene ponášajú na FMF. Boli popísaní pacienti s rekurentnými myalgiami, rekurentným bilaterálnym eryzipeloidným exantémom, palindromickým reumatizmom a livedoidnou mikroangiopatiou, u ktorých sa potvrdila FMF. Genetická analýza v týchto populáciách môže vyžadovať sekvenáciu celého génu MEFV s cieľom odhaliť zriedkavejšie mutácie. U slovenských pacientov sme potvrdili výlučne dobre známe a časté mutácie. Taktiež tu môže byť častejší výskyt mutácií pre iné autoinflamačné ochorenia. Niektorí autori predpokladajú, že FMF môže byť výsledkom kombinácie heterozygotnej mutácie v géne MEFV a mutácie resp. polymorfizmu v ďalších génoch (digénová dedičnosť) [12].
Rodinná anamnéza je dôležitým diagnostickým kritériom ako v prípade 1. pacienta z predisponovaného etnika. V našich podmienkach bola prínosná len pre 3. pacientku – tetu dieťaťa s FMF. V tejto rodine musíme predpokladať, že aj rodičia dieťaťa sú heterozygoti pre mutáciu M694V (matka) resp. E148Q (otec), ale vzhľadom na chýbanie klinických ťažkostí a malú pravdepodobnosť amyloidózy u nich nie je indikovaná genetická analýza. V rodine, kde takáto analýza bola vykonaná u všetkých príbuzných (4. pacientka), boli odhalení 3 asymptomatickí heterozygoti (fenotyp 3), ktorí podľa platných odporúčaní nevyžadujú ani sledovanie. 4-ročný syn 3. pacientky (bratranec 2. pacientky) môže byť nosičom mutácie M694V a v prípade rozvoja klinických príznakov kompatibilných s FMF, bude u neho doplnená genetická analýza.
Podľa niektorých autorov je vzhľadom na nízky záchyt mutácií genetické testovanie na FMF len obmedzene použiteľné u pacientov s rekurentnými horúčkami v západoeurópskych populáciách [13]. Napriek tomu je potrebné konštatovať, že v regiónoch, kde úplne chýbajú skúsenosti s FMF, genetická analýza predstavuje dnes už dostupný a často jediný spoľahlivý spôsob potvrdenia diagnózy FMF aj u etnicky nedisponovaného pacienta. Prikláňame sa preto k názoru [14], že charakteristický obraz krátkych, nepredvídateľných, spontánne ustupujúcich epizód febrilít s prejavmi serozitídy a laboratórnym obrazom bakteriálnej infekcie by aj v nerizikových populáciách mal viesť k indikácii takéhoto vyšetrenia a to aj pri vyššej pravdepodobnosti negatívneho výsledku. Nevýhodou je, že FMF je možné potvrdiť, ale nikdy nie vylúčiť na základe genetického vyšetrenia, keďže boli popísaní pacienti splňujúci diagnostické kritériá, u ktorých napriek dobrej odozve na liečbu kochicínom, nebola odhalená mutácia v géne MEFV. Z praktického hľadiska je nutné doplniť, že genetickú analýzu je tč. možné uskutočniť len na zahraničných pracoviskách, ktoré túto službu poskytujú v rámci budovania medzinárodných registrov pacientov s autoinflamačnými ochoreniami [15].
FMF predstavuje v našich podmienkach raritné ochorenie a vyskytuje sa väčšinou u príslušníkov rizikových etník [6]. Naše skúsenosti sú však dôkazom, že jeho „mediteránny pôvod“ by nás nemal odradiť od zvažovania takejto diagnózy aj u slovenského pacienta, a to aj pri spoľahlivo preukázanom slovenskom pôvode a absencii konsangvinity u rodičov. Naopak, výskyt v slovenskej populácii je z historického hľadiska dokonca zdôvodniteľný – 150 rokov tureckej okupácie a veľká židovská komunita celkom iste zanechali stopy aj v našom genóme. I keď sa jedná len o malú vzorku pacientov, výlučná prítomnosť mutácií častých aj v tureckej populácii u našich pacientov podporuje správnosť tejto hypotézy. Jedným z možných vysvetlení nízkeho výskytu je aj skutočnosť, že mierny klinický priebeh nemusí spôsobovať pacientovi klinické ťažkosti takej intenzity, ktoré by ho nútili k hľadaniu diagnózy. Naopak, môžu spôsobiť frustráciou podmienené odmietanie lekárskej starostlivosti (3. pacientka). Nedá sa preto vylúčiť, že v našej populácii je viac doposiaľ nediagnostikovaných pacientov. Význam stanovenia správnej diagnózy pritom presahuje len prínos v zmysle zvýšenia kvality života a spočíva predovšetkým v možnosti zavedenia účinnej prevencie amyloidózy a prevencii zbytočných vyšetrení a chirurgických výkonov. Liečba kolchicínom sa aj u našich pacientov ukázala ako účinná a bezpečná vo všetkých vekových kategóriách.
Záver
FMF je v našich podmienkach zriedkavé, ale nie vylúčené ochorenie, a to aj u nášho domáceho etnika. Hoci sa manifestuje vždy pred 20. rokom života, je potrebné o tomto v našich podmienkach pravdepodobne „poddiagnostikovanom“ ochorení uvažovať aj v dospelom veku. Nepredvídateľné, krátke a spontánne ustupujúce epizódy horúčok sprevádzané bolesťami brucha či na hrudníku a vysokými zápalovými parametrami, ale aj ich nevysvetlené pretrvávanie medzi epizódami, by mali aj pri negatívnej rodinnej anamnéze viesť k zváženiu tejto diagnózy. Analýza génu MEFV môže byť veľmi nápomocná, je dostupná a v konečnom dôsledku umožní začatie celoživotnej liečby kolchicínom, ktorá má rozhodujúci význam pre kvalitu života aj prognózu pacienta.
Analýza génu MEFV bola podporená grantmi VEGA 1/0342/08 slovenskej Vedeckej grantovej agentúry a L3–4150 a P3–0343 Slovinskej grantovej agentúry. Za trpezlivosť a spoluprácu ďakujeme našim pacientom a ich rodičom.
MUDr. Tomáš Dallos, PhD.
dallos@dfnsp.sk
II. detská klinika LF UK a populácii, DFNsP, Bratislava, Slovensko
www.detskaklinika.sk
Doručeno do redakce: 7. 5. 2013
Přijato po recenzi: 2. 9. 2013
Sources
1. French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997; 17(1): 25–31.
2. The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997; 90(4): 797–807.
3. Tomiyama N, Higashiuesato Y, Oda T et al. MEFV mutation analysis of familial Mediterranean fever in Japan. Clin Exp Rheumatol 2008; 26(1): 13–17.
4. Toplak N, Doležalová P, Constantin T et al. Periodic fever syndromes in Eastern and Central European countries: results of a pediatric multinational survey. Pediatr Rheumatol Online J 2010; 8 : 29. Dostupné z DOI: <http://doi: 10.1186/1546–0096–8-29>.
5. Doležel Z, Macků M, Schuller M et al. Mohli jsme být v diagnsotice rychlejší? Pediatr pro Praxi 2009; 10(4): 260–265.
6. Livneh A, Langevitz P, Zemer D et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 1997; 40(10): 1879–1885.
7. Yalçinkaya F, Ozen S, Ozçakar ZB et al. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology (Oxford) 2009; 48(4): 395–398.
8. Kondi A, Hentgen V, Piram M et al. Validation of the new paediatric criteria for the diagnosis of familial Mediterranean fever: data from a mixed population of 100 children from the French reference centre for auto-inflammatory disorders. Rheumatology (Oxford) 2010; 49(11): 2200–2203.
9. Caglayan AO, Demiryilmaz F, Ozyazgan I et al. MEFV gene compound heterozygous mutations in familial Mediterranean fever phenotype: a retrospective clinical and molecular study. Nephrol Dial Transplant 2010; 25(8): 2520–2523.
10. Padeh S, Livneh A, Pras E et al. Familial Mediterranean fever in children presenting with attacks of fever alone. Rheumatol 2010; 37(4): 865–869.
11. Touitou I. The spectrum of Familial Mediterranean Fever (FMF) mutations. Eur J Hum Genet 2001; 9(7): 473–483.
12. Marek-Yagel D, Berkun Y, Padeh S et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum 2009; 60(6): 1862–1866.
13. Tchernitchko D, Moutereau S, Legendre M et al. MEFV analysis is of particularly weak diagnostic value for recurrent fevers in Western European Caucasian patients. Arthritis Rheum 2005; 52(11): 3603–3605.
14. Touitou I. Diagnostic value of MEFV gene analysis in familial Mediterranean fever must still be assessed in non-classically affected populations. Arthritis Rheum 2004; 50(4): 1354–1355. [Comment on: Cazeneuve C, Hovannesyan Z, Geneviève D et al. Familial Mediterranean fever among patients from Karabakh and the diagnostic value of MEFV gene analysis in all classically affected populations. Arthritis Rheum 2003; 48(8): 2324–2331.]
15. Toplak N, Frenkel J, Ozen S et al. An international registry on autoinflammatory diseases: the Eurofever experience. Ann Rheum Dis 2012; 71(7): 1177–1182.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2014 Issue 1
Most read in this issue
- Vliv konzumace alkoholu na srdeční elektrofyziologii
- Familiární středozemní horečka v České republice
- Familiárna stredomorská horúčka – klinický obraz, diagnóza a liečba
- Mezinárodní doporučení pro léčbu těžké sepse a septického šoku 2012 – komentovaný výběr