
Klinické hodnocení nových léčiv u vzácných diagnóz v onkologii – současná situace v Evropě a u nás
Clinical evaluation of new drugs against orphan diseases in oncology – the current situation in Europe and in our country
Cancer represents one of the main causes of death among diseases across the age spectrum. Tumors in children, however, represent less than 1% of the total number of cancers in the population and in terms of the definition of orphan diseases in Europe are all children's cancers considered as orphan diseases. This is the reason why the research and development of new agents against cancer in childhood stands outside of the main interest. Every year around 30,000 new cases of cancer in children and adolescents are diagnosed in the European Unioun (EU) and approximately 80% of them achieve long-term remission using mainly conventional methods of treatment. However, almost 6,000 children and adolescents die every year of malignant tumors therefore cancer remains a major cause of morbidity and mortality. Consequently, there is a demand for new and safe drugs for children suffering from cancer which would lead to improved survival and to risk reduction of late adverse effects of cancer treatment. In the past 10 years in the EU, more in the EU-15 than in our country, 20 performed oncology trials in phase I involving adults account for only one trial in pediatric patients. The issue of new drugs clinical testing in rare cancers is very complex, complicated and for current unsatisfactory situation might be responsible various aspects. These aspects contain the legislative field, the problem of determining the correct dose of testing drug as a single agent or in combination therapy, the use of testing drug in advanced disease or already in de novo diagnosed patients, as well as equity (equal) access to new drugs being tested, the goal set for each molecule/drug in clinical trials, the conflict of interest balanced with sufficient professionalism and last but not least, the need for new methodologies and statistical approaches. The aim of this article is to describe the issue complexity of incorporation of new, modern drug for cancer patients with orphan diseases, including children.
Key words:
clinical evaluation – new drugs – orphan diseases in oncology
Authors:
Jaroslav Štěrba 1; Sylva Štěrbová 2; Daniela Kodytková 1; Dalibor Valík 3,4; Regina Demlová 3,4
Authors‘ workplace:
Klinika dětské onkologie LF MU a FN Brno, pracoviště Dětská nemocnice, přednosta prof. MUDr. Jaroslav Štěrba, Ph. D.
1; Katedra Ústavního práva PrF UP Olomouc, vedoucí katedry doc. JUDr. Jiří Jirásek, CSc.
2; Masarykův onkologický ústav Brno, ředitel prof. MUDr, Jiří Vorlíček, CSc.
3; Farmakologický ústav LF MU Brno, přednostka MUDr. Regina Demlová, Ph. D.
4
Published in:
Vnitř Lék 2014; 60(Suppl 2): 80-85
Category:
70th Birthday - prof. MUDr. Jiří Vorlíček, CSc.
Overview
Nádorová onemocnění představují i nadále jednu z hlavních příčin úmrtí mezi nemocemi napříč věkovým spektrem. Nádory u dětí však představují méně než 1 % z celkového počtu nádorů v populaci a z hlediska definice vzácných onemocnění v Evropě jsou všechny dětské zhoubné nádory vzácná onemocnění. Z tohoto důvodu stojí spíše mimo hlavní proud výzkumu a vývoje nových protinádorových léčiv. V EU je každý rok diagnostikováno kolem 30 000 nových nádorových onemocnění u dětí a mladistvých a přibližně 80 % z nich dosahuje dlouhodobých remisí dominantně konvenčními metodami léčby. Zároveň však téměř 6 000 dětí a adolescentů každý rok na zhoubné nádory umírá, a proto nádory zůstávají i nadále velmi významnou příčinou morbidity i mortality. Z tohoto důvodu trvá poptávka po nových a bezpečných léčivých přípravcích pro děti s nádory, které by vedly ke zlepšení přežití dětí se zhoubnými nádory a zároveň ke snížení rizika pozdních následků onkologické péče. V posledních 10 letech však na 20 realizovaných klinických studií fáze I v onkologii dospělých připadá pouze 1 pediatrická studie, a to spíše ve státech EU 15, než u nás. Problematika klinického testování nových léčiv u vzácných nádorových onemocnění je velmi komplexní, komplikovaná a příčiny současného nepříliš uspokojivého stavu lze spatřovat v několika oblastech. Jde zejména o oblasti legislativně právní, problém stanovení správné dávky, testování léku v monoterapii či kombinaci, zda hodnotit nová léčiva na pacientech až s pokročilým onemocněním nebo již u de novo diagnostikovaných pacientů, dále ekvita (rovnost) přístupu k novým testovaným lékům, stanovení priorit jednotlivých molekul/léků pro klinické hodnocení, vyvažování konfliktu zájmů na jedné straně a dostatečné odbornosti na straně druhé a v neposlední řadě pak potřeba nových metodologií a statistických přístupů. Cílem tohoto článku je popsat komplexnost problematiky inkorporace nových, moderních léčiv pro onkologicky nemocné pacienty se vzácnými diagnózami, včetně dětí.
Klíčová slova:
klinické hodnocení – nová léčiva – vzácné diagnózy v onkologii
Úvod
Nádorová onemocnění představují i nadále jednu z hlavních příčin úmrtí mezi nemocemi napříč věkovým spektrem. Nádory u dětí však představují méně než 1 % z celkového počtu nádorů v populaci. V Evropě je za vzácné onemocnění považována taková nemoc, jejíž prevalence je nižší než 1 : 2 000. Z hlediska této definice jsou tak všechny dětské zhoubné nádory vzácná onemocnění a z tohoto důvodu stojí spíše mimo hlavní proud výzkumu a vývoje nových protinádorových léčiv, a tedy i mimo hlavní zájem majitelů a akcionářů farmaceutických společností. Nicméně relativní vzácnost některých onemocnění by neměla znamenat, že pacienti s těmito vzácnými nemocemi by měli být léčeni léky podřadnější kvality, či dokonce léky, o jejichž bezpečnosti toho víme velmi málo.
V EU je každý rok diagnostikováno kolem 30 000 nových nádorových onemocnění u dětí a mladistvých. Přibližně 80 % z nich dosahuje dlouhodobých remisí dominantně konvenčními metodami léčby, tedy konvenční, či vysokodávkovanou chemoterapií, radioterapií a chirurgickými metodami. Zároveň však téměř 6 000 dětí a adolescentů každý rok na zhoubné nádory umírá. V EU tak dnes žije půl milionu bývalých dětských onkologických pacientů, z nichž třetina má klinicky významné pozdní následky léčby, které vyžadují prakticky trvalou zdravotní či sociální péči (postižení srdce, plic, amputace, endokrinopatie vyžadující léčbu, závažné poruchy kognitivních funkcí apod), třetina má klinicky nevýznamné zdravotní projevy (např. zhoršení sluchu pro některé frekvence apod) a pouze třetina bývalých dětských onkologických pacientů je po léčbě jejich nádoru zcela v pořádku.
Nádory tak zůstávají i nadále velmi významnou příčinou morbidity i mortality v EU, a proto trvá poptávka po nových a bezpečných léčivých přípravcích pro děti s nádory, které by vedly jednak ke zlepšení přežití dětí se zhoubnými nádory a zároveň také ke snížení rizika pozdních následků onkologické péče, kterým musí čelit jak pacienti, tak i zdravotní systémy. Realitou posledních 10 let však zůstává, že na 20 realizovaných klinických studií fáze I v onkologii dospělých připadá pouze 1 pediatrická studie, a to spíše ve státech EU 15, než u nás (obr. a graf).
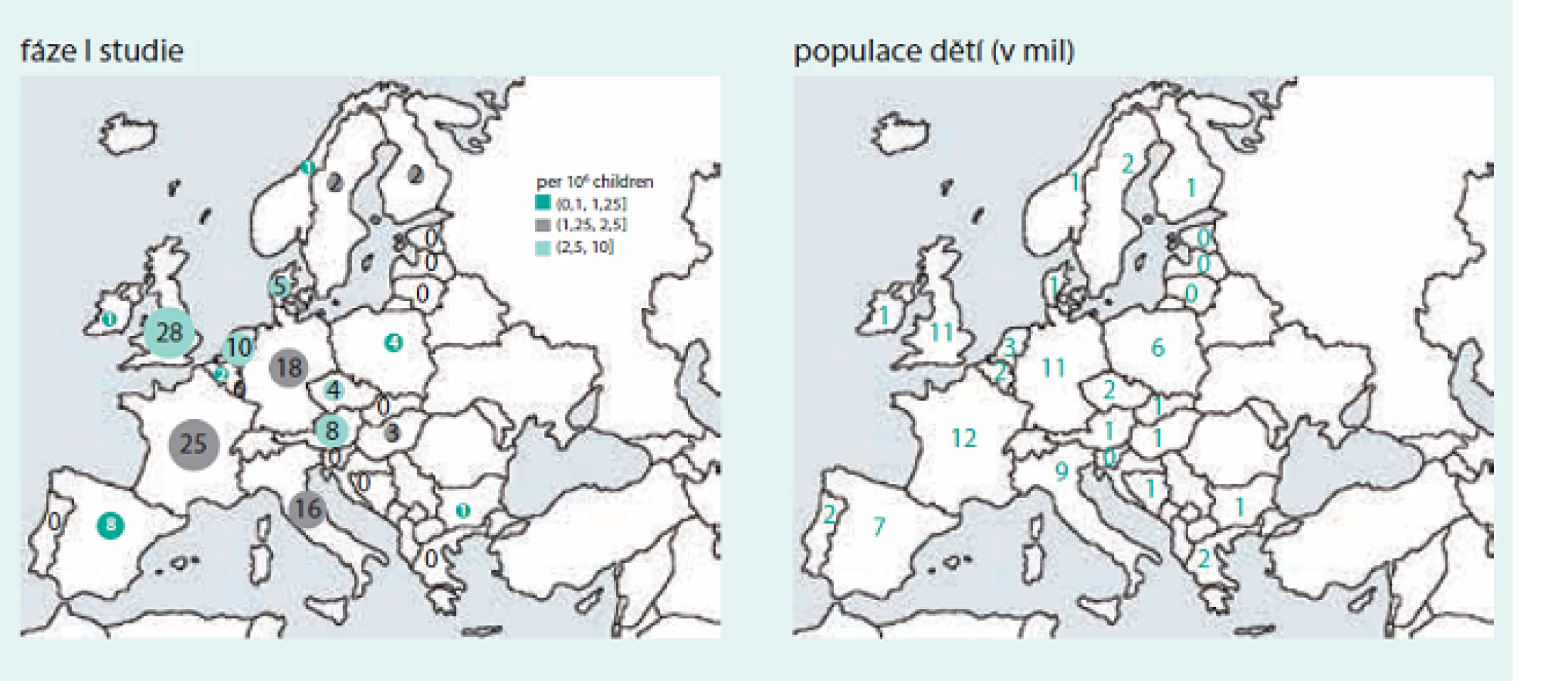

Problematika klinického testování nových léčiv u vzácných nádorových onemocnění je velmi komplexní, komplikovaná, a příčiny současného nepříliš uspokojivého stavu lze spatřovat v následujících oblastech.
- Oblast legislativně právní – registrace a systém úhrad nových léků v onkologii je založen na principu histopatologické klasifikace nemocí (tzv. condition) a nebere v potaz vlastní mechanizmus působení léku.
- Problém stanovení správné dávky pro pokračující studie fáze II a III vycházející ze studií fáze I. Tradičně jsou stanovovány maximálně tolerované dávky, jejichž dosažení však pro farmakodynamický účinek biologik není většinou potřebné a naopak vzhledem k možné toxicitě pacientovi spíše škodí [1].
- Dalším problémem je zda testovat lék v monoterapii, což je legislativně a metodicky čistší, ale méně to reflektuje reálný život, nebo v kombinacích nových léků, či tzv. add on strategiích, podle nichž se nový lék podává k již existující standardní terapii, zpravidla řadu let existující a v určité situaci dobře popsaná schémata polychemoterapie (tzv. backbone).
- Dalším problémem je, komu nové léky nabízet. Tedy zda je vhodné a správné hodnocení nových léčiv na dětech s pokročilým onemocněním po mnoha relapsech, nebo u de novo diagnostikovaných pacientů.
- Problematickou je i ekvita přístupu k novým testovaným lékům v různých částech Evropy i uvnitř jednotlivých zemí.
- Samostatnou a velmi problematickou oblastí je otázka stanovení priorit jednotlivých molekul/léků pro klinické hodnocení, v nichž se mohou velmi lišit zájmy jednotlivých farmaceutických společností i různých kooperativních pracovních skupin napříč jednotlivými zeměmi i kontinenty.
- Adekvátní vyvažování konfliktu zájmů – kdo v určité oblasti dlouhodobě a systematicky pracuje, má při dnešní absenci adekvátního financování akademického klinického výzkumu dříve nebo později konflikt zájmů. Pak ale získávají v regulačních orgánech významnější slovo farmaceuti, inženýři, ev. bývalí lékaři, kteří již ztratili kontakt s realitou současné klinické praxe a skutečných klinických potřeb, anebo ji nikdy neznali. Takoví mohou jen obtížně přispět k řešení existujících klinických problémů, které musí onkolog řešit denně na ambulancích z několika vzájemně protichůdných pohledů (názor regulační může být zcela opačný oproti názoru rodiče nebo pacienta, tj. pohled regulační – pro danou léčbu není dostatek standardních podkladů – tzv. no data vs pohled často zoufalých rodičů či pacientů „léčit teď tím, co je, nebo pro co jsou alespoň nějaká nadějná data“).
- Pro úspěšné klinické testování na malých skupinách pacientů je zde bytostná potřeba nových metodologií a statistických přístupů pro časné fáze klinického zkoušení u dětí, např. vícestupňové studie s více rameny (tzv. multistage multiarm studie) nebo tzv. adaptivní design studií.
Oblast legislativně právní
Vývoj nových léků v dnešní době by měl reflektovat především poznání v biologii nádoru a mechanizmus působení jednotlivých léků. Bohužel, současná velmi přísná legislativa, která se týká protinádorových molekul (ale pomíjí submolekulární částice, včetně fotonů či protonů), vychází při jejich klinickém zkoušení a případné následné registraci z principu pouze histopatologické klasifikace nemocí (tzv. condition). Reflektuje tak relativně dobře situaci dospělých pacientů, ale minimálně odpovídá mechanizmu působení jednotlivých tzv. cílených léků či biologik a velmi komplikuje provádění studií u dětí. Evropský regulační orgán European Medicines Agency tak mezi dubnem roku 2008 a dubnem roku 2012 musel v souladu s platnou legislativou potvrdit zastavení vývoje léků pro eventuální použití v pediatrii, původně určený pro léčbu 197 diagnóz v oblasti onkologie dospělýchi přesto, že řada molekul slibovala prospěch pro pacienty i při pediatrickém použití [2]. Jedna z těchto molekul, crizotinib, byla v USA adekvátně studována pro použití v pediatrii a již v roce 2011 schválena ke klinickému použití u dětí s neuroblastomem, či anaplastickým velkobuněčným lymfomem. V EU toto schválení zatím chybí a lék je v řadě situací používán v režimu off label.
V principu mohou při testování nových protinádorových léků u dětí nastat 3 různé situace:
- Jedná se o stejnou nemoc i stejnou cílovou strukturu, jako u onemocnění dospělého věku. Příkladem je bcr-abl u chronické myeloidní leukemie. Toto je relativně jednoduchá situace, nicméně i zde jsou důležitá pediatrická specifika. Dlouhodobé podávání imatinibu vede k poruchám růstu u dětí, data o dlouhodobých efektech obdobných molekul, jako dasatinibu, na rostoucí organizmus nejsou ještě k dispozici a dat o nilotinibu u dětí je ještě méně. Realita klinické praxe je taková, že pediatrické studie jsou významně opožděny za studiemi u dospělých i v této relativně snadné situaci, v níž máme stejnou nemoc a snažíme se cílit na stejný cíl. Dalším příkladem může být ALK či CD30 u anaplastických velkobuněčných lymfomů.
- Jindy se jedná se o odlišné onemocnění, často i s různou histogenezí, ale máme možnost působit v nádorové buňce na stejný cíl jako u zcela jiného nádorového onemocnění dospělého věku. Např. ALK u neuroblastomu, IGF1R u neuroblastomu, nefroblastomu, Ewingova sarkomu, B-RAF u gliomů nízkého stupně malignity, histiocytóz, ev. VEGF u řady pediatrických nádorů, či SHH inhibitory u meduloblastomu.
- Další situací je existence specifických pediatrických cílů, jejichž protipóly v dospělé onkologii zatím neznáme. Sem patří např. N-Myc u neuroblastomu, PAX3-FKHR, PAX7-FKHR u rabdomyosarkomu, ev. translokace EWS genu u Ewingova sarkomu.
A stále je zde požadavek, že potřebujeme léky nejen účinnější, ale také bezpečnější, a to i z dlouhodobého hlediska.
Problém stanovení správné dávky
Závažnou a velmi problematickou oblastí klinického testování léků pro děti je stanovení správné dávky protinádorového léku pro dětské pacienty. Současná legislativa motivuje farmaceutické společnosti k provádění pediatrických studií, ale již méně k jejich úspěšnému ukončování, či uvádění léků do reálné pediatrické praxe.
Design klinických studií je i v současnosti založen na tradičním paradigmatu studií fáze I-III. Primárním cílem studií fáze I zůstává v naprosté většině případů stále nalezení tzv. maximálně tolerované dávky (MTD), jež zejména u cílených léčiv postrádá své farmakologické opodstatnění. Tento přístup byl smysluplný při vývoji cytostatik: vztah dávky a účinku a potažmo i toxicity byl lineární, u cílených léčiv nové generace je však zásadní nalézt optimální biologickou dávku, která je schopna aktivovat/inhibovat daný cíl – target, nikoliv dávku způsobující neopodstatněnou toxicitu. Můžeme spekulovat, že „lpění“ na MTD u cílených léčiv může být ekonomicky zajímavější pro firmu, rozhodně však ne pro pacienty.
Klasickým příkladem je imatinib, jehož MTD nebyla nikdy stanovena, a možná proto je natolik úspěšný v klinické praxi. V této souvislosti je naprosto na místě se ptát, je-li toxicita skutečně to, k čemu nám rodiče dávají informovaný souhlas při realizaci klinických studií časných fází u svých dětí. Realitou dále zůstává, že dávky pro zahájení časných fází klinického zkoušení se odvozují od dávky pro dospělé v určitém procentu vzhledem k váze, či tělesnému povrchu. Tento způsob stanovení jen velmi málo reflektuje často zásadně odlišnou farmakokinetiku u malých dětí, ve srovnání s dospělými. Dávkování řady úspěšných biologik se dnes realizuje v podstatě na principu metronomické léčby, včetně již zmiňovaného imatinibu [3].
Přes všechny tyto výhrady je zde objektivní potřeba, aby podstatně více protinádorových léků vstupovalo do časných fází klinického zkoušení. Ideálně se tak děje formou spolupráce s kooperativními mezinárodními pracovními skupinami dětské onkologie, které často ve svých protokolech takovýto prostor pro testování nových léků nechávají, např. BFM, či COG (www.bfm-international.org, www.childrensoncologygroup.org).
Klinická onkologie je však také oborem, který má nejvyšší podíl selhání nových léků v následných randomizovaných studiích fáze III.
Příčiny tohoto stavu mohou být následující:
- Design klinických studií, ev. zvolená kombinace léků bývá nevhodná, nebo příliš toxická, vedená také snahou o časné zařazení nových léků pro děti s nově diagnostikovaným onemocněním po předchozím úspěchu v léčbě recidiv. Např. v současné době byla zastavena pro nadměrnou toxicitu randomizovaná studie, která testovala možnost časného zařazení clofarabinu do léčby de novo diagnostikovaných dětí s vysoce rizikovou akutní lymfoblastickou leukemií (http://www.cancer.gov/clinicaltrials/search/view?cdrid=706370&version=HealthProfessional). U většiny pediatrických studií také nemáme ještě k dispozici adekvátní biomarkery.
- Problematické je také zahajování klinických studií v situaci, v níž není moc jasné, v jaké situaci, či jaké populaci bude nový lék nabízen v klinické praxi, tedy jaká má být definitivní pozice nového preparátu. Stává se to zejména tehdy, je-li celý proces řízen spíše snahou jednotlivých firem uplatnit to, co mají k dispozici (v tzv. pipeline), než to, co je skutečně klinicky zapotřebí. Příkladem zde může být izotretinoin, který zcela selhal v léčbě relabujících neuroblastomů, ale stejný lék statisticky signifikantně zlepšil přežití dětí s vysoce rizikovými neuroblastomy, pokud byl zařazen jako udržovací terapie dětí s vysoce rizikovými neuroblastomy po dosažení klinické remise předchozí intenzivní chemoterapií [4]. Dalším příkladem léku, jehož testování není vedeno reálnou klinickou potřebou, ale jen jeho dostupností, je ponatinib [5].
- Velkou pozornost vyžaduje ale i celá oblast preklinického testování, ze které v klinických studiích nutně musíme vycházet. Zde je nutno podtrhnout příliš velkou závislost na buněčných liniích, které nereflektují nádorovou heterogenitu, včetně nádorové vaskularizace a mikroprostředí nádoru včetně komplikovaných vztahů mezi imunitní odezvou hostitele a nádorem. Problematická je také reprodukovatelnost jednotlivých preklinických nálezů mezi různými laboratořemi, což situaci dále komplikuje. Pro některé preparáty, jak je zmiňováno již výše, nejsou navíc k dispozici žádné prediktivní biomarkery.
Jednou z inovativních strategií, jak zvýšit pravděpodobnost, že nová léčba prokáže svůj přínos v realizovaných klinických studiích, jsou tzv. basket trial. Ty jsou designovány na principu přiřazení léčebné strategie na základě předem stanovené konkrétní mutace receptoru nebo signální dráhy konkrétního pacienta s konkrétním nádorem, nikoliv tedy podle histopatologie a lokalizace nádoru. Umožňují tak iniciovat jednu klinickou studii u různých diagnóz, a v případě testování většího počtu různých aberací jednotlivých cílů tak využít několika kohort s různými hodnocenými léčivy. Tento postup se jeví nejen vědecky smysluplnější s možným profitem účinnosti pro reálného pacienta, ale šetří i čas, náklady i administrativní zátěž při provádění klinických studií.
Lék v monoterapii vs kombinace
Velmi komplikovanou oblastí je, jak stanovovat doporučenou dávku léku pro následující studie fáze II a III v souvislosti s další souběžně podávanou léčbou. Tedy zda definovat dávku léku pro monoterapii, jako samostatný lék, či při definování dávky počítat s podáváním léku v kombinaci. Zde nastupuje celé další spektrum problémů – firmě k finančnímu prospěchu může někdy stačit i negativní pediatrická studie, tedy i studie nového léku v monoterapii pro vícečetné relapsy, což není situace, v níž by se dal očekávat největší přínos nového léku v reálné klinické praxi.
Jestliže přijmeme strategii add on, tedy kombinaci nového léku s něčím dalším, pak tedy s čím? Stávající standard of care? Tedy kombinace s nějakou známou, existující chemoterapií, založené na maximálně tolerovaných dávkách? Řada léčebných režimů v dětské onkologii je již dnes na hraně toho, co jsou děti schopny klinicky tolerovat a toxická úmrtí, způsobená akutními komplikacemi léčby, nejsou výjimkou a stále se pohybují v řádu několika procent. A k tomu bychom měli přidat další toxicitu nového preparátu? Již dříve zmiňovaný imatinib a komplikovaná historie jeho začleňování do léčby Ph+ ALL a problémy s jeho registrací regulačními orgány je dobrým příkladem komplikovaného vývoje v této oblasti [6], a není proto divu, že se objevují úvahy o kombinaci cílené léčby novými preparáty, právě s metronomickou terapií [1].
Chceme-li kombinovat různé moderní preparáty, pak vstupují do hry i komplikované vztahy mezi jednotlivými farmaceutickými firmami, které jsou ještě ochotny podpořit vývoj svých vlastních preparátů, ale již méně ochotně vývoj preparátů v kombinaci při nutné spolupráci s konkurenční společností. Celosvětově je nedostatečná podpora klinických studií iniciovaných samotnými výzkumníky ze strany nejrůznějších grantových agentur a administrativní a regulační zátěž je pak zpravidla nad možnosti akademických institucí.
Občas nastává situace, v níž má několik různých farmaceutických firem k dispozici několik různých léků cílících na stejnou strukturu u velmi malých skupin pacientů, jako např. pediatrický gastrointestinální stromální nádor, maligní melanom s mutací BRAF V600E, avšak tyto malé podskupiny pediatrických pacientů nejsou adekvátně velkým trhem pro farmaceutické firmy.
Komu nové léky nabízet?
Tedy na jaké populaci pacientů je vhodné testovat nové léky? U dětí s relabujícími, či rezistentními nádory? Není to poněkud jiná situace, než kde nakonec nové efektivnější a bezpečnější léky chceme? Řada desítky let úspěšně používaných cytostatik by dnes v klinickém testování velmi pravděpodobně neuspěla. Výše jsme již zmínili situaci s retinoidy u dětí s neuroblastomy.
Jaká je situace u nově diagnostikovaných pacientů? Jsme tady ochotni riskovat zhoršení výsledků tam, kde máme 90% EFS 5 let od diagnózy? Např. náhrada antracyklinů u některých NHL za monoklonální protilátky s cílem snížit rizika pozdních následků?
Dalším nezodpovězeným problémem je, zda máme testovat nové, cílené léky pouze u subpopulací s určitým předdefinovaným cílem. Pacienti s definovanou přítomnou cílovou strukturou odpoví až v 50 %, pacienti bez této cílové struktury pouze v asi 4 %, ale tento způsob testování může ukázat i nepředpokládané mechanizmy působení léku.
Ekvita přístupu k novým testovaným lékům
Problém ekvity, tedy jakéhosi ideálního stavu nastolení rovných příležitostí k dosažení zdravotního potenciálu jedince, je nejlépe dokumentován asi absencí nějakého Phase I/II konsorcia ve střední a východní Evropě. V USA je takových skupin dokonce 5, naproti tomu v Evropě existuje pouze jedna, která však pokrývá jen omezenou část teritoria a pacientů. Při zachování ekvity v přístupu k léčbě může nastat i situace, že by pacienti z Brna či Hodonína jezdili na experimentální léčbu do Frankurtu či Paříže. Tato představa však není příliš realistická.
Vyvažování konfliktu zájmů
Situace v této oblasti je velmi citlivá a připomíná kyvadlo hodin, které čas od času se dostává do určitých extrémních poloh.
Problematika designu klinických studií a jejich statistického hodnocení
Významnou otázkou je, jak lépe studovat stále se zmenšující skupiny pacientů v rámci klinických studií s novými léky. Určitou možnost nabízejí vícestupňové studie s více rameny – tzv. Multi-Arm Multi-Stage design, což bylo poprvé použito u pacientek s ovariálními karcinomy a u pacientů s karcinomem prostaty – v každé studii bylo použito 5 různých ramen a několik na sebe logicky navazujících fází [7].
Tyto formy studií mohou nabídnout i určitou flexibilitu: experimentální ramena, která ukazují nedostatečnou efektivitu, jsou ukončována předčasně a mohou být nahrazena novými preparáty (tzv. pick up winner a drop up looser strategie).
Je však zapotřebí velmi opatrně posuzovat léčebné stratifikace, protože s postupujícím poznáváním nádorové biologie se dnes ze vzácných nádorů stávají skupiny různých, ještě vzácnějších onemocnění. Příkladem může být současný zánik kategorie supratentoriální PNET a její rozpadnutí do několika biologicky velmi odlišných podskupin.
Na druhou stranu, rozvoj klinických studií založených na lepším poznání a pochopení nádorové biologie by mohl vést také k jiným definicím studovaných populací, s možností určitého grupování histologicky odlišných nemocí, ale sdílejících zásadní společné biologické charakteristiky.
Příkladem inovativně připravované studie může být nová pediatrická studie pro anaplastické velkobuněčné lymfomy testující relativní efektivitu crizotinibu a vinblastinu v kombinaci se standardní chemoterapií.
Nicméně, zde je zapotřebí od statistiků ještě mnoho metodologické práce, s cílem pomoci lépe navrhovat klinické studie a interpretovat jejich výsledky tak, aby klinici, pacienti, farmaceutické firmy i regulační orgány byly schopny přijímat nejlepší možná rozhodnutí v situacích, které nejsou tradičními způsoby hodnocení klinických studií zcela jednoznačné.
Od farmaceutických firem potřebují klinici racionálně časný přístup k novým lékům, klinici i rodiny pacientů se vzácnými diagnózami potřebují vzbudit a udržet zájem farmaceutických firem o rozvoj účinnějších a bezpečnějších léků pro pacienty se vzácnými skupinami nádorových onemocnění, a také podporu při financování akademických klinických studií. Klinici, pacienti i jejich rodiny dále potřebují spolupráci mezi jednotlivými farmaceutickými firmami s cílem testovat současně více přípravků více firem, jak jsme již komentovali v posledním odstavci.
Od kliniků starajících se o pacienty se vzácnými nádory je oprávněně možno požadovat omezování používání léků schválených pro dospělé v režimu off label a raději děti zařazovat do klinických studií. Pokud jsou ovšem tyto studie k disposici v reálném čase a prostoru tak, aby na ně pacienti vůbec dosáhli.
Významnými hráči jsou zde především pacientské a rodičovské organizace, které by měly tlačit na realizaci prospektivních klinických studií jako nejlepší cestě ke zlepšování výsledků péče. Tyto studie však musí být prospektivní, s dlouhodobým sledováním účinnosti, ale i toxicity léčby, protože vedlejší účinky, které jsou patrné u dospělých, ještě nemusí být prediktivní pro toxicitu, kterou vidíme u vyvíjejících se organizmů, zejména při dlouhodobém sledování.
Souhrn
Problematika inkorporace nových, moderních preparátů pro onkologicky nemocné děti je velmi složitá, komplikovaná. Vyžaduje navíc dlouhodobou, systematickou spolupráci klinických lékařů, mezinárodních kooperativních onkologických skupin, farmaceutických společností, rodičovských, pacientských organizací, i regulačních orgánů (národních i nadnárodních), což s sebou zcela pochopitelně nese řadu úskalí a komplikací. Avšak v zájmu pacientů se vzácnými typy nádorových onemocnění, včetně onkologicky nemocných dětí na snahy o časné nabízení adekvátně testovaných léčiv i těmto malým skupinám nemocných nemůžeme a nesmíme rezignovat.
Podpořeno RECAMO CZ.1.05/2.1.00/03.0101. Lékařská fakulta Masarykovy University, projekt LM 2011017. Projekt velké infrastruktury CZECRIN LM2013034.
prof. MUDr. Jaroslav Štěrba, Ph.D.
jsterb@fnbrno.cz
Klinika dětské onkologie LF MU a FN Brno, pracoviště Dětská nemocnice
www.fnbrno.cz
Doručeno do redakce 19. 9. 2014
Přijato po recenzi 30. 9. 2014
Sources
1. André N, Carré M, Pasquier E. Metronomics: towards personalized chemotherapy? Nat Rev Clin Oncol 2014; 11(17): 413–431.
2. Vassal G, Geoerger B, Morland B. Is the European Pediatric Medicine Regulation Working for Children and Adolescents with Cancer? Clin Cancer Res 2013; 19(6): 1315.
3. André N, Pasquier E, Kamen B. Can targeted therapy be successful without metronomic scheduling? Curr Top Med Chem 2012;12(15): 1639–1642.
4. Masetti R, Biagi C, Zama D et al. Retinoids in pediatric onco-hematology: the model of acute promyelocytic leukemia and neuroblastoma. Adv Ther 2012; 29(9): 747–762.
5. Mathisen MS, Kantarjian HM, Cortes J et al. Practical issues surrounding the explosion of tyrosine kinase inhibitors for the management of chronic myeloid leukemia. Blood Rev 2014; 28(5): 179–187.
6. Schultz KR, Carroll A, Heerema NA et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children‘s Oncology Group study AALL0031. Leukemia 2014; 28(7): 1467–1471.
7. Parmar MK, Barthel FM, Sydes M et al. Speeding up the evaluation of new agents in cancer. J Natl Cancer Inst 2008; 100(17): 1204–1214.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2014 Issue Suppl 2
Most read in this issue
- Diferenciální diagnostika eozinofilie
- Paliativní medicína a dobrá smrt
- Dispenzarizace nemocných po léčbě karcinomu prsu, kolorekta a prostaty
- Ph-negativní myeloproliferativní onemocnění s trombocytemií ve světle účinku léčby přípravkem Thromboreductin® v datech registru ke konci roku 2013