
Selektivní modulátory progesteronového receptoru a jejich terapeutické využití
Selective progesterone receptor modulators and their therapeutical use
Currently developed selective progesterone receptor modulators (SPRMs) are steroid derived compounds with a bulky radical substitution at carbon 11. They interact with progesterone receptor and exert antagonistic or/and agonistic effects. Mifepristone was approved for pregnancy termination and ulipristal acetate as emergency contraception and pharmacological therapy of uterine fibroids. SPRMs inhibit endometrial proliferation and myoma growth, this suggests a therapeutical effect in cases of endometriosis and other estrogen-dependent diseases.
Keywords:
progesterone – progesterone receptor –selective progesterone receptor modulators – agonists and antagonists of progesterone – pregnancy termination – emergency contraception
Autoři:
D. Driák 1
![]() ; B. Sehnal 1; I. Švandová 2
; B. Sehnal 1; I. Švandová 2
Působiště autorů:
Gynekologicko-porodnická klinika 1. LF UK a Nemocnice Na Bulovce, Praha, přednosta prof. MUDr. M. Halaška, DrSc.
1; Oddělení neurobiologie, Katedra fyziologie, Přírodovědecká fakulta UK, Praha
2
Vyšlo v časopise:
Ceska Gynekol 2013; 78(2): 175-181
Souhrn
V současnosti vyvinuté selektivní modulátory progesteronového receptoru (SPRM) jsou steroidní sloučeniny s velkým substituentem na uhlíku 11. Interagují s progesteronovým receptorem a vykazují antagonistické nebo/a agonistické účinky. Mifepriston byl schválen k ukončení gravidity a ulipristalacetát jako pohotovostní kontracepce a farmakologická léčba děložních myomů. SPRM inhibují proliferaci endometria a růst myomů a předpokládá se léčebný efekt v případech endometriózy a jiných estrogen-dependentních chorob.
Klíčová slova:
progesteron – progesteronový receptor – selektivní modulátory progesteronového receptoru – agonisté a antagonisté progesteronu – ukončení těhotenství – pohotovostní kontracepce
ÚVOD
Progesteron je C21 steroid, který je secernován corpus luteum, placentou a v malých kvantech rostoucím foliklem. Vykazuje fyziologické funkce na urogenitální, kardiovaskulární, nervový a jiné systémy, nejdůležitějšími cílovými orgány jsou děloha, prsy a mozek. Stěžejní funkce progesteronu je příprava dělohy na těhotenství (gestaci) a jeho udržení. Progesteron hraje klíčovou roli v maturaci a diferenciaci mléčné žlázy, v endometriu a myometriu antagonizuje účinky estrogenu. Mechanismus účinku je zprostředkován vazbou na cytoplazmatický progesteronový receptor (PR), transportem komplexu hormon-receptor do jádra a indukcí transkripce cílových genů [4]. Cytoplazmatický PR existuje ve 2 hlavních izoformách A a B. Izoforma B je delší o 165 aminokyselin. PR A zajišťuje kontrolu estrogeny indukované proliferace endometria a inhibuje izoformu B.PR B zprostředkuje proliferaci a diferenciaci epitelu mléčné žlázy [2].
V nepřítomnosti progesteronu se PR vyskytuje v heteromerickém komplexu s různými proteiny, včetně proteinu teplotního šoku 90 (hsp90). Vazba hsp90 na ligand vázající doménu PR je nezbytná pro správnou konformaci PR a jeho udržení ve vysokoafinitním stavu pro progesteron. Po vazbě steroidu se proteiny teplotního šoku uvolní, receptor dimerizuje a váže se na hormon-responzibilní element promotorové oblasti příslušného genu v jádře. Byly rovněž prokázány negenomické (rychlé) účinky progesteronu. Ty se mohou realizovat cestou PR lokalizovaných na plazmatické membráně buňky nebo vazbou progesteronu do alternativního, negenomického vazebného místa na cytoplazmatickém PR [10].
Důležitost progesteronu v ženské reprodukci vedl k vývoji syntetických ligandů s agonistickými a antagonistickými účinky. Antiprogestiny, dnes označované jako selektivní modulátory progesteronového receptoru (selective progesterone receptor modulators, SPRM) jsou steroidní sloučeniny s objemným substituentem na C11 – viz obr. 1. Navázáním menších radikálů na C11 se zvýší gestagenní účinek, přidáním složitějších substituentů vzniknou sloučeniny s vysokou afinitou k PR, ale bez schopnosti jeho aktivace. Historicky první byl mifepriston; dnes je již známa řada molekul, které se vážou na PR a působí na něj antagonisticky, agonisticky a nejčastěji smíšeně: ulipristalacetát, lonaprisan, telapristonacetát, lilopriston, onapriston, asoprisnil a další [2]. Do budoucna se jeví perspektivně v roli pohotovostní antikoncepce a dlouhodobé antikoncepce bez estrogenů, kromě toho k regulaci nepravidelného krvácení při progestinových metodách a jako léčba endometriózy a myomů [8]. Pro použití v gynekologii byly schváleny zatím pouze dva SPRM, mifepriston jako historicky první a „čistý“ antagonista progesteronu k ukončení těhotenství a ulipristalacetát se smíšenou aktivitou na PR k emergentní kontracepci a farmakologické léčbě děložních myomů [2]. Studie s asoprisnilem byly v roce 2005 ve fázi III vzhledem ke změnám endometria naopak přerušeny.
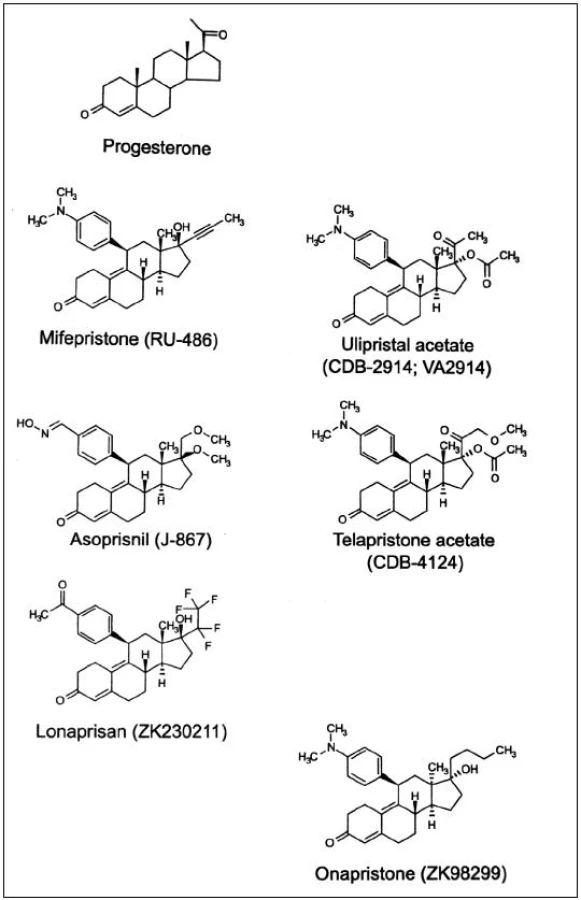
HISTORIE
V roce 1970 francouzská firma Roussel-Uclaf pod vedením doktora Édouarda Sakize a profesora Étienne-Émile Baulieu zahájila výzkum nových steroidních molekul s hormonálními a antihormonálními účinky. Při vývoji antagonistů glukokortikoidního receptoru v dubnu 1980 chemik Georges Teutsch syntetizoval RU-38486. Kódové označení RU-38486 jako 38 486. sloučenina vyrobená firmou Roussel-Uclaf od roku 1949 bylo brzy změněno na RU-486. Byl tak objeven první antiprogesteron mifepriston, 19-norsteroid substituovaný na C11β s velmi silnou antiprogesteronovou aktivitou. V následujícím roce na 7. mezinárodním farmakologickém kongresu v Tokiu D. Philibert, R. Deraedt a G. Teutsch představili světu RU-38486 jako antiglukokortikoidní sloučeninu s antiprogestinovým účinkem. V roce 1982 vědci oznámili objev francouzské akademii věd a ve stejném roce byla látka poprvé použita v klinické praxi k ukončení těhotenství, a to profesorem Walterem Herrmannem v Ženevě. V roce 1987 Baulieu navrhl termín kontragestace pro antigestační působení RU-486 a přípravek se stal kontragestivem.
„Potratová pilulka“ pod firemním názvem Mifegyne byla povolena ve Francii od září 1988, v říjnu 1988 však kvůli protestům antiinterrupčních skupin ve Francii, Německu a USA firma její distribuci přerušila. Za dva dny francouzský ministr zdravotnictví Claude Evin nařídil firmě distribuci obnovit a označil RU-486 za „morální vlastnictví žen“. V roce 1988 byla „interrupční pilulka“ schválena v Číně, v roce 1991 ve Velké Británii, v roce 1992 ve Švédsku.
Protipotratová hnutí podporovaná Bushovou administrativou bránila zavedení v USA a RU-486bylo povoleno až v roce 2000 za vlády Billa Clintona. V roce 2009 byl mifepriston schválen přes tvrdý odpor Vatikánu i v Itálii. Dnes se mifepriston po-užívá ve více než 29 zemích světa, v Evropě jím bylo provedeno více než 1,5 milionu potratů.
Po mifepristonu následoval vývoj dalších steroidních progesteronových ligandů, zatímco nesteroidní ligandy byly objeveny teprve nedávno [2].
ÚČINKY
Stejně jako progesteron SPRM interagují s cytoplazmatickým PR. V závislosti na své unikátní struktuře je účinek SPRM smíšený, agonisticko-antagonistický. Některé části molekuly mohou navodit nebo stabilizovat inaktivní konformaci receptoru, jiné zase zvyšovat afinitu PR k progesteronu. Závisí i na relativní tkáňové koncentraci koaktivátorů a korepresorů [2].
Všechny selektivní modulátory PR potenciálně blokují ovulaci a vykazují podobný efekt na růst leiomyomů a změny endometria. V endometriálních změnách byly popsány jemné odlišnosti, což může odpovídat vlivu agonistického/antagonistického působení. Navrhovaná nová klasifikace ligandů PR je založena na transkripční aktivitě in vitro a demonstruje unikátní účinek každého SPRM [2].
Kontracepční mechanismus SPRM pravděpodobně tkví v kompetitivní blokádě účinků progesteronu, působí luteolyticky a antigestačně. Předpokládá se, že působí centrálně na hypofýzu (blokáda vzestupu LH, a tím ovulace) i periferně na úrovni endometria (desynchronizace, porucha maturace) a snad i na tubární funkce. Jejich aplikace není spojena s deplecí estradiolu [8].
MIFEPRISTON
Afinita mifepristonu k PR je více než 2,5krát silnější než afinita progesteronu a ke glukokortikoidnímu receptoru 3krát větší, než vykazuje dexametazon. Nemá účinky estrogenní, androgenní, mineralokortikoidní, glukokortikoidní, antiestrogenní ani antimineralokortikoidní. Za nepřítomnosti progesteronu může mít mifepriston progestační efekt na endometrium a prs. Je slabým antiandrogenem. Po podání jednorázové dávky 20–600 mg p.o. je mifepriston rychle a kompletně absorbován z gastrointestinálního traktu, prochází presystémovým metabolismem a jeho biologická dostupnost je 70 %. Maximální koncentrace 1,98 ng/ml je dosaženo za 1,3 hodiny. Mifepriston je z 98 % vázán na plazmatické proteiny, na albumin, a především na α1-kyselý glykoprotein. Farmakokinetika je nelineární, snad díky nasycování vazby na α1-kyselý glykoprotein. Vylučování je zpočátku pomalé s poločasem mezi 12–72 hodinami, posléze rychlejší, s poločasem 18 hodin, konečný poločas činí méně než 90 hodin. Metabolismus probíhá v játrech primárně cestou demetylace a hydroxylace 17-propynyl řetězce na 3 hlavní metabolity, z nichž jeden je schopný vazby na PR podobně jako rodičovská molekula. Vzhledem k tomu, že je mifepriston metabolizován na cytochromu P450 (CYP3A4), induktory enzymů (induktor vede v buňce ke tvorbě většího množství enzymu přímou vazbou na regulační sekvence jeho genu), jako fenytoin, fenobarbital, karbamazepin aj., mohou koncentraci mifepristonu snižovat. Naopak inhibující sloučeniny, jako je ketokonazol, itrakonazol či grapefruitová šťáva, mohou sérové koncentrace zvyšovat. Vylučování probíhá v 83 % stolicí, v 9 % močí. Z lékových interakcí je nejdůležitější snížení účinku glukokortikoidů.
Současné schválené indikace mifepristonu zahrnují:
- ukončení těhotenství až do 63. dne amenorey (v kombinaci s prostaglandiny),
- indukci abortu ve 13.–24. týdnu (v kombinaci s prostaglandiny),
- přípravu děložního hrdla před mechanickou dilatací při chirugické interrupci v I. trimestru – 200 mg mifepristonu p.o. 36–48 hodin před výkonem,
- vypuzení mrtvého plodu – 600 mg mifepristonu denně po dobu 2 dní vede k porodu během 72 hodin ve více než 60 %.
Kromě toho se mifepriston testuje jako dlouhodobá antikoncepce bez estrogenů, léčba myomů, endometriózy a dalších gynekologických, ale vzhledem k antiglukokortikoidnímu účinku i negynekologických chorob, jako je m. Cushing, m. Alzheimer, psychózy (tab. 1).
![Terapeutické možnosti selektivních modulátorů progesteronového receptoru [2, 7, 8]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/8bb297ced154f46f70618110e3da7384.png)
Kontraindikace pro použití mifepristonu představuje přecitlivělost na kteroukoli složku přípravku, nepotvrzené intrauterinní těhotenství nebo jeho velikost přesahující limit, ektopická gravidita, přítomnost IUD, onemocnění nadledvin, dlouhodobá kortikoterapie, poruchy krevní srážlivosti, porfýrie, těžké a nedostatečně léčitelné astma, adrenální selhání. V případech kombinací s prostaglandiny je kontraindikací i přecitlivělost na prostaglandiny a jejich kontraindikace. Dále vzhledem k nedostatku dat mifepriston vyžaduje zvláštní opatrnost u pacientek se selháním ledvin, jater, malnutricí, poruchami krevní srážlivosti, anémií, kardiovaskulárními chorobami, antikoagulační a kortikoidní léčbou a u kojících žen.
Použití mifepristonu jako kontraceptiva je možné v různých fázích menstruačního cyklu. Při podání ve folikulární fázi v závislosti na dávce inhibuje folikulární vývoj nebo inhibuje ovulaci. Dávka do 50 mg mifepristonu vede k opoždění maturace folikulu, která pokračuje po odeznění vlivu mifepristonu, a k ovulaci dochází později. Podání 5 mg mifepristonu ještě před selekcí dominantního folikulu nemá na folikulární vývoj žádný vliv. Stejná dávka aplikovaná při průměru dominantního foliklu 12–14 mm zpomaluje jeho růst na 12–48 hodin. Vyšší dávky (3 mg/kg) vyvolají kolaps dominantního folikulu a oddalují ovulaci až do rekrutace nového folikulu [5]. Podání 10 mg mifepristonu při folikulárním průměru větším než 15 mm do doby 2 dny před ovulací vede k opoždění nebo inhibici peaku LH [11]. Při aplikaci mifepristonu těsně před ovulací je již na blokádu ovulace pozdě a k ruptuře folikulu může dojít, avšak vysoké dávky mifepristonu alterují tubární a uterinní funkce. Studie na zvířatech ukázaly akcelerovaný transport vajíčka tubou po léčbě mifepristonem. Dávka 200 mg mifepristonu v době, kdy již vzestup LH odstartoval, vyvolá změny ve vývoji endometria v luteální fázi, nerozvíjí se normální sekreční změny, vakuolizace je minimální nebo žádná, počet mitóz zvýšený. Podání 200 mg mifepristonu v časné luteální fázi znamená změny v endometriální glandulární apoptóze, sekreční aktivitě, expresi steroidních, a především progesteronových receptorů, složení a množství uterinní tekutiny. Aplikace 25 a více mg mifepristonu ve střední a pozdní luteální fázi vede k odlučování endometria a vaginálnímu krvácení během několika dní, asi v 50 % dochází k luteolýze [5].
Při použití mifepristonu jako časného abortiva se rozvíjí v endometriální decidui edém, nekróza, alterace kapilár, klesá koncentrace hCG, chorion se odlučuje od děložní stěny. Nastává luteolýza s poklesem koncentrací estradiolu i progesteronu.
Mifepriston se testoval v klinických studiích v dávkách 10, 25, 100 nebo 600 mg p.o. jako postkoitální antikoncepce. Byl účinný při použití až do 120 hodin po nechráněném styku, vedlejší účinky, jako nepravidelné krvácení, nauzea, zvracení, bolesti hlavy a napětí v prsou, byly méně časté než u jiných hormonálních postupů. Oddálení začátku menstruace se jeví jako hlavní nežádoucí účinek, je přímo úměrné dávce mifepristonu a znejisťuje ženu obávající se otěhotnění [8]. Dávky 10–50 mg jsou efektivní a dobře tolerované [12].
K dlouhodobé antikoncepci se testují nižší dávky mifepristonu – 0,5 až 2 mg denně způsobuje opoždění ovulace a alteraci endometria, ale nemá vliv na vývoj folikulů. Dávka 2–10 mg blokuje vývoj folikulů, prahová denní dávka mifepristonu k zablokování ovulace je 2 mg. Dávka 50 mg jednou týdně se jeví rovněž dostatečně efektivní. Přestože cysticky dilatované žlázky imponují na ultrazvuku jako ztluštělé endometrium, atypie nebyly nalezeny a ve většině studií zjišťovaná amenorea by mohla být výhodou.
Mechanismus účinku mifepristonu je komplexní a působí na několika úrovních. Zablokováním působení progesteronu dochází k odloučení embrya, poklesu hCG a sekundárně k luteolýze. Zvyšuje se aktivita myometria, kontrakce se objevují za 24–36 hodin po aplikaci mifepristonu, za 24 hodin se začíná zvyšovat senzitivita k prostaglandinům s maximem (až pětinásobně) za 36–48 hodin. Na děložním čípku se projevuje zrací a otevírací efekt.
Patnáctileté zkušenosti s kombinací mifepriston a následně za 24–48 hodin misoprostol p.o. jsou tak dobré, že některé evropské země (Švýcarsko, Švédsko, Francie) zlegalizovaly medikamentózně indukované potraty v domácím prostředí. Ze všech aplikačních schémat se nejvíce ustálily dva standardní režimy: 600 mg (3 tbl.) mifepristonu + 400 μg (2 tbl.) misoprostolu do 49. dne amenorey nebo 200 mg mifepristonu + 800 μg misoprostolu do 63. dne amenorey [1]. Srovnatelné výsledky mají i jiné režimy: 200 mg mifepristonu p.o. + 800 μg misoprostolu aplikovaného bukálně do 63 dnů amenorey [15] nebo 200 mg mifepristonu p.o. + 400 μg misoprostolu bukálně do 63 dnů amenorey [14].
Pacientka je předem důkladně poučena o principu metody a možných vedlejších účincích mifepristonu (nauzea, zvracení, únava, vzácněji reakce jako vyrážka a otok v obličeji) a téměř pravidelných doprovodných příznaků potratu (krvácení, děložní stahy). Krvácení začíná zpravidla 1–2 dny po požití mifepristonu a trvá průměrně 12, ale i více dní. Aplikace prostaglandinu nese s sebou riziko dalších vedlejších účinků (nauzea, zvracení, průjem, horečka, závrať) a příznaků probíhajícího abortu. Po aplikaci léků následuje nezbytné, ideálně ambulantní sledování pacientky po dobu 14-21 dnů od aplikace mifepristonu, kdy se gynekologickým vyšetřením a ultrazvukem a/nebo hodnocením koncentrace β-hCG ověřuje, že došlo k úplnému vypuzení plodu a zástavě krvácení. V případě přetrvávajícího krvácení, bolestí a nekompletního vypuzení nebo pokračování těhotenství se doporučuje provést chirurgickou evakuaci děložní dutiny. Přibližně u 3–4 % pacientek dochází k potratu ještě před podáním prostaglandinu, většina žen potratí během 3 hodin po podání prostaglandinu, část pak během následujících několika dní. Až v 1,4 % případů hrozí silné krvácení vyžadující akutní kyretáž. Po celou dobu procedury by pacientka měla být v dosahu a kontaktu se zdravotnickým zařízením, které farmakologickou interrupci provádí, dodržovat klidový režim a instrukce lékaře.
S přibývajícími týdny počet nekompletních vypuzení progresivně stoupá až na 23 % [9].
ULIPRISTALACETÁT
Zatímco použití mifepristonu k emergentní kontracepci je z různých důvodů limitováno a bylo schváleno v Rusku a Číně, až překvapivě rychle byl v celé Evropské unii dne 15. května 2009 registrován přípravek ellaOne (ella v USA) obsahující 30 mg ulipristalacetátu. Ulipristalacetát je perorálně účinný SPRM, který byl vyvinut speciálně pro emergentní kontracepci a schválen k užití až do 120 hodin po nechráněném pohlavním styku. Kromě vysoké afinity k PR, kde vykazuje antagonistické a částečně agonistické účinky, má minimální afinitu k androgennímu a žádnou afinitu k estrogenním a mineralokortikoidním receptorům. Váže se v určité míře na glukokortikoidní receptor, antiglukokortikoidní účinky u člověka však nebyly pozorovány, antagonistický efekt je ve srovnání s mifepristonem podstatně redukovaný [12].
Ulipristalacetát se velmi rychle a téměř kompletně resorbuje z trávicího ústrojí, biologická dostupnost se blíží 100 %, dosahuje průměrné maximální koncentrace 176 ng/ml přibližně 1 hodinu po p.o. podání. Zvýšené žaludeční pH a vysoký obsah tuků ve stravě snižuje absorpci, ale nebylo prokázáno, že by měnilo efektivitu [12]. Prahová dávka inhibující ovulaci je 5 mg [8]. Vazba na bílkoviny včetně albuminu, α1- kyselého glykoproteinu a HDL činí 96,7-99,5 % a biologický poločas 32 hodin. Ulipristalacetát je extenzivně metabolizován v játrech zejména cytochromem P450 (CYP3A4), z metabolitů je pouze mono-demetylovaný derivát farmakologicky účinný. Kvůli možným interakcím se nedoporučuje současné podávání s induktory mikrosomálních enzymů, účinné inhibitory CYP3A4 mohou naopak zvyšovat expozici ulipristalacetátem [12]. Nedoporučuje se kombinovat s jinými nouzovými přípravky, jako modulátor progesteronového receptoru může snižovat jejich účinnost i účinnost kombinovaných i čistě progestinových přípravků.
Mechanismus účinku tkví nejspíše v inhibici nebo zpoždění ovulace, což je způsobeno zpožděním nástupu peaku LH nebo jeho oddálením, pokud již vzestup koncentrace LH započal [12]. Studie sledující koncentrace LH a ultrazvukovou folikulometrii 5 dní po podání levonorgestrelu, placeba a ulipristalacetátu demonstrovala výhodu oproti levonorgestrelu. Při velikosti folikulu 18 a více mm levonorgestrel již ovulaci nezablokuje (levonorgestrel 12 %, placebo 13 %), zatímco ulipristalacetát i přes peak LH v 60 % oddálí ovulaci o 5 a více dní [3, 6]. Na kontracepčním účinku se mohou se podílet i změny endometria. Při použití do 24 hodin došlo k otěhotnění v 0,9 % (pro srovnání u levonorgestrelu ve 2,5 %), při aplikaci do 3 dnů činilo těhotenské číslo 1,8 % (u levonorgestrelu 2,6 %). Přípravek ellaOne je signifikantně účinnější v zabránění nechtěného těhotenství než levonorgestrel, rozdíl je patrnější v intervalu do 24 hodin od nechráněného styku, což je kritická doba pro podání emergentní kontracepce [6, 13].
Držitelem registrace ellaOne je francouzská firma Laboratoire HRA Pharma, ale v ČR jej propaguje zavedená firma Richter-Gedeon. Klinická data se stále ještě shromažďují, SÚKL v následujících 5 letech eviduje všechna otěhotnění, bezpečnost byla stanovena zatím pouze pro ženy od 18 let. Nejčastější vedlejší účinky jsou bolest hlavy, dysmenorea a nauzea [6]. Asi u 20 % pacientek dochází k nepravidelné menstruaci. Pro časté závrati, event. ospalost a rozmazané vidění se po použití nedoporučuje řídit motorová vozidla nebo obsluhovat stroje. Před podáním má být vyloučeno těhotenství a při laktaci se nedoporučuje nejméně 36 hodin kojit, neboť lipofilní ulipristal se může vylučovat do mléka a riziko pro kojence není vyloučeno.
V únoru roku 2012 byl registrován přípravek Esmya tbl. obsahující 5 mg ulipristal acetátu od firmy PregLem. Byl schválen k léčbě děložních myomů v dávce jednou denně p.o. kontinuálně na dobu 3 měsíců. V ČR Esmyu prezentuje zavedená farmaceutická firma Richter Gedeon.
ZÁVĚR
Selektivní modulátory progesteronového receptoru se smíšenými agonistickými a antagonistickými účinky jsou relativně novou skupinou sloučenin, které zasahují významným způsobem do ženské reprodukce. Mifepriston a ulipristalacetát mají schválené gynekologické indikace. Některé ze selektivních modulátorů progesteronového receptoru představují perspektivní látky, jejichž terapeutické použití může v budoucnosti přesáhnout antikoncepční a abortivní působení a uplatnit se i při dalších gynekologických i negynekologických chorobách.
MUDr. Daniel Driák, Ph.D.
Gynekologicko-porodnická klinika 1. LF UK
Nemocnice Na Bulovce
Budínova 2
180 81 Praha 8
e-mail: driak@seznam.cz
Zdroje
1. Aubény, E. Medical abortion at home. Eur J Contracept Reprod Health Care, 2008, 13, suppl 2, p. 189–190.
2. Bouchard, P., Chabbert-Buffet, N., Fauser, BCJM. Selective progesterone receptor modulators in reproductive medicine: pharmacology, clinical efficacy and safety. Fertil Steril, 2011, 96, 5, p. 1175–1189.
3. Brache, V., Cochon, L., Jesam, C., et al. Immediate pre-ovulatory administration of 30 mg ulipristal acetate significantly delays follicular rupture. Hum Reprod, 2010, 25, 9, p. 2256–2263.
4. Ganong, WF. Přehled lékařské fyziologie. 20. ed. Praha: Galén, 2005, 890 s.
5. Gemzell-Danielsson, K., Marions, L. Mechanisms of action of mifepristone and levonorgestrel when used for emergency contraception. Hum Reprod Update, 2004, 10, 4, p. 341–348.
6. Glasier, AF., Cameron, ST., Fine, PM., et al. Ulipristal acetate versus levonorgestrel for emergency contraception: a randomised non-inferiority trial and meta-analysis. Lancet, 2010, 375, 9714, p. 555–562.
7. Chabbert-Buffet, N. Selective progesterone receptor modulators. Eur J Contracept Reprod Health Care, 2010, 15, suppl 1, p. 7–8.
8. Chabbert-Buffet, N., Ouzounian, S., Kairis, AP., Bouchard, P. Contraceptive applications of progesterone receptor modulators. Eur J Contracept Reprod Health Care, 2008, 13, 3, p. 222–230.
9. Kreutner, AK. Infekční komplikace po interrupci. Komentář: Charvát, M. Gynek po prom, 2001, 1, 6, s. 72–75.
10. Mani, SK., Mermelstein, PG., Tetel MJ., Anesetti G. Convergence of multiple mechanisms of steroid hormone action. Horm Metab Res, 2012, 44, 8, p. 569–576.
11. Marions, L., Hultenby, K., Lindell, I., et al. Emergency contraception with mifepristone and levonorgestrel: mechanism of action. Obstet Gynecol, 2002, 100, 1, p. 65–71.
12. McKeage, K., Croxtall, JD. Ulipristal acetate: a review of its use in emergency contraception. Drugs, 2011, 71, 7, p. 935–945.
13. Scott, LJ., McKeage, K., Croxtall, JD. Ulipristal acetate: a guide to its use in emergency contraception. Drugs Ther Perspect, 2012, 28, 2, p. 6–9.
14. Tsereteli, T., Chong, E., Tsertsvadze, G., et al. A comparison of two dosages of buccal Misoprostol following Mifepristone for early medical abortion. Eur J Contracept Reprod Health Care, 2008, 13, suppl 2, p. 43–44.
15. Winikoff, B., Creinin, M., Crowden, W., et al. Expanding options for in-the-mouth Misoprostol administration following Mifepristone in medical abortion. Eur J Contracept Reprod Health Care, 2008, 13, suppl 2, p. 44.
Štítky
Detská gynekológia Gynekológia a pôrodníctvo Reprodukčná medicínaČlánok vyšiel v časopise
Česká gynekologie

2013 Číslo 2
- Ne každé mimoděložní těhotenství musí končit salpingektomií
- Mýty a fakta ohledně doporučení v těhotenství
- I „pouhé“ doporučení znamená velkou pomoc. Nasměrujte své pacienty pod křídla Dobrých andělů
- Gynekologické potíže pomáhá účinně zvládat benzydamin
- Jak podpořit využití železa organismem bez nežádoucích účinků
Najčítanejšie v tomto čísle
- Transfusion-related acute lung injury (TRALI) – přehledový článek
- Opakované potrácení – přehledový článek
- Význam stanovení proteinu p16 v managementu prekanceróz děložního hrdla
- Hyperlipidémie v těhotenství