
Význam molekulárně genetického vyšetření pro diagnostiku a genetické poradenství v rodinách s hyperamonémií a deficitem ornithin-karbamoyltransferázy
Importance of Molecular Genetic Analysis for Diagnosis and Genetic Counseling in Families with Hyperammonemia and Ornithine carbamoyltransferase Deficiency
Ornithine carbamolytransferase deficiency is a severe X-linked disorder presenting with acute hyperammonemic attacks.
The aim is to present clinical data and results of mutation analysis in 34 patients with ornithine carbamoyltransferase deficiency.
Patients and methods:
Nine males with severe neonatal form of the disease died in first days of life, 14 patients with late onset form manifested in the 2nd–3rd year of life or in pre-school age; one four-year-old boy and one adult male are asymptomatic. Nine heterozygotes developed symptoms of the disease, one of them in the newborn period, the remaining patients in the 2nd–3rd year of life or at the pre-school age. The diagnosis based on evaluation of analyte concentration in blood plasma and urine was confirmed by methods of mutation analysis.
Results:
The results of analyte profiling (ammonia and glutamine/glutamate concentrations in blood serum and orotic acid in urine) did not predict the phenotype and only partially correlated with the actual health state of the patient. Gross deletions and other obviously null mutations were invariably associated with the neonatal form of the disease. Some missense mutations caused neonatal form, while others were associated with late onset of the disease. Mutation analysis in patient’s families showed that affected boys usually inherit OTC mutations from their heterozygous clinically unaffected mothers, while de novo mutations occur more frequently in manifestingheterozygotes.
Conclusions:
OTC deficiency is a severe disease in which precise and fast diagnosis plays an important role. Molecular genetic diagnosis is essential for confirmation of the diagnosis, genetic counseling and possible prenatal diagnosis in affected families.
Key words:
urea cycle, OTC deficiency, mutation analysis, genotype-phenotype correlation, de novo mutation
Autori:
L. Dvořáková 1; M. Hřebíček 1; H. Vlášková 1; K. Szentiványi 2; J. Zeman 2
Pôsobisko autorov:
Ústav dědičných metabolických poruch, 1. lékařská fakulta UK a VFN, Praha
přednosta doc. MUDr. V. Kožich, CSc.
1; Klinika dětského a dorostového lékařství, 1. lékařská fakulta UK a VFN, Praha
přednosta prof. MUDr. J. Zeman, DrSc.
2
Vyšlo v časopise:
Čes-slov Pediat 2010; 65 (10): 575-579.
Kategória:
Původní práce
Súhrn
Porucha funkce ornithin-karbamoyltransferázy (OTC) je závažné onemocnění s X-vázanou dědičností, které se klinicky projevuje akutními dekompenzacemi v důsledku hyperamonémie.
Cílem studie je představit soubor klinických dat a molekulárně genetických analýz u 34 pacientů s deficitem OTC.
Soubor a metody:
Devět chlapců s těžkou neonatální formou onemocnění zemřelo již v prvních dnech života, u 14 z 16 chlapců s pozdní manifestací se onemocnění nejčastěji projevilo v batolecím nebo předškolním věku; jeden čtyřletý chlapec a jeden dospělý muž jsou dosud asymptomatičtí. Onemocnění se klinicky manifestovalo i u 9 heterozygotních dívek, u jedné v novorozeneckém věku a u ostatních v batolecím a předškolním věku. Diagnostika založená na vyšetření amoniaku a aminokyselin v krevním séru a kyseliny orotové v moči byla potvrzena metodami mutační analýzy.
Výsledky metabolických vyšetření vypovídaly jen částečně o fenotypu onemocnění a nekorelovaly vždy s aktuálním zdravotním stavem nemocného dítěte. Velké delece a další závažné mutace byly vždy spojeny s neonatální formou onemocnění. Naproti tomu některé mutace vedoucí k záměně aminokyselinového zbytku vedly k neonatální formě onemocnění, jiné jen k deficitu OTC s mírnějším fenotypem. Mutační analýza v rodinách pacientů ukázala, že klinicky postižení chlapci většinou zdědili mutaci v genu OTC od svých heterozygotních ale klinicky zdravých matek, zatímco u klinicky manifestních dívek se výrazně častěji objevila mutace de novo.
Závěr:
Včasná diagnostika deficitu OTC na metabolické úrovni hraje zásadní roli pro prognózu pacienta. Pro potvrzení diagnózy, genetické poradenství a případnou prenatální diagnostiku v postižených rodinách je nezbytná diagnostika na molekulární úrovni.
Klíčová slova:
cyklus močoviny, deficit ornithin-karbamoyltransferázy, mutační analýza, korelace genotyp-fenotyp, mutace de novo
Úvod
Ornithin-karbamoyltransferáza (OTC, E.C.2.1. 3.3, MIM *300461) je klíčovým enzymem cyklu močoviny, jehož úlohou je odstraňovat toxický amoniak vznikající při katabolismu bílkovin [1]. Enzym působí v mitochondriální matrix a je kódován genem na krátkém rameni chromozomu X (Xp21.1) [2]. V důsledku nepříznivého zešikmení inaktivace chromozomu X (podle Lyonové) mohou kromě chlapců onemocnět i heterozygotní dívky (přenašečky) [3, 4]. Předpokládaná incidence deficitu OTC je cca 1:60 000 až 1:80 000 [5].
Klinicky se u poruchy OTC rozeznávají dva základní fenotypy: neonatální typ onemocnění při kompletním deficitu OTC a onemocnění s pozdějším nástupem příznaků při částečně zachované aktivitě mutantního enzymu (parciální deficit OTC). První projevy neonatální formy onemocnění obvykle začínají po krátkém bezpříznakovém intervalu druhý až třetí den života, ale tento interval může být i kratší (12–24 hodin po narození). Charakteristická je postupná progrese příznaků: postižený novorozenec špatně pije, objevuje se hypotonie, letargie a/nebo dráždivost a tachypnoe, křeče, bezvědomí a edém mozku. Stav může rychle progredovat do obrazu sepse s krvácením do mozku nebo plic. Deficit OTC s pozdním začátkem („late onset“) se však může projevit i kdykoliv později v průběhu života akutními rychle progredujícími atakami poruch vědomí nejčastěji v průběhu interkurentních respiračních nebo gastrointestinálních infektů [1].
Hlavní toxickou látkou je amoniak a míra hyperamonémie rozhoduje o závažnosti, eventuálně reverzibilitě poškození organismu pacienta. Hladina amoniaku může stoupat velice rychle, během několika hodin až desetinásobně. V krvi bývá vysoká koncentrace glutaminu a kyseliny glutamové a nízká hladina argininu a citrulinu. Enzymologicky lze diagnózu deficitu OTC potvrdit v jaterní biopsii, ale přednost má diagnostika na molekulárně genetické úrovni, která je, stejně jako u dalších gonosomálně recesivních onemocnění, nejspolehlivější metodou pro určení heterozygocie u asymptomatických přenašeček [6, 7]. Mutační analýza je i jedinou metodou pro prenatální diagnostiku.
V tomto sdělení předkládáme souhrn klinických dat a výsledků molekulárně genetických analýz u 34 pacientů, u kterých jsme na našem pracovišti prokázali deficit OTC.
Soubor pacientů a metodika
V souboru 34 pacientů s deficitem OTC pochází 32 pacientů z 24 českých a slovenských rodin, jeden pacient z Maďarska a jeden z Japonska. Všichni se narodili v termínu a bezprostřední poporodní adaptace byla bez komplikací. Diagnostika deficitu OTC na biochemické úrovni byla založena na vyhodnocení profilu analytů v krevní plazmě a moči.
Mutační analýza byla provedena z DNA izolované z leukocytů periferní krve metodou přímého sekvenování PCR produktů všech kódujících a známých regulačních oblastí genu pro OTC a oblastí ovlivňujících sestřih [8]. Velké delece byly analyzovány metodou MLPA (multiplex ligation probe amplification assay) a s použitím DNA čipu (Affymetrix Human SNP 6.0 array). Postmortálně jsme u tří pacientů pro analýzu transkriptu použili RNA izolovanou z jaterní biopsie.
Etika
Molekulární analýzy byly prováděny na základě informovaného souhlasu pacientů a v případě dětí jejich zákonných zástupců. Studie byla schválena Etickou komisí Všeobecné fakultní nemocnice v Praze.
Léčba
V dlouhodobé terapii se uplatňuje nízkobílkovinná dieta, kterou lze doplnit směsí esenciálních L-aminokyselin. Ke zvýšení eliminace nebílkovinného dusíku se používá benzoát sodný a/nebo phenylbutyrát sodný. U pacientů s poruchou OTC je nutná i suplementace L-argininem. V období akutních infektů je nutno ještě přísněji omezit příjem bílkovin. Pokud nelze podávat sladké tekutiny perorálně nebo objeví-li se u dítěte změna chování, apatie či zvracení, nebo má-li dítě již počínající hyperamonémii, je třeba okamžitě zahájit intravenózní podávání koncentrované glukózy s inzulinem k omezení katabolismu a současně intravenózně podávat benzoát sodný a arginin-chlorid, protože při vzestupu koncentrace amoniaku hrozí edém mozku. Stoupá-li i nadále hladina amoniaku, je nutno zahájit některou z eliminačních metod – hemodialýzu nebo hemodiafiltraci.
U každého pacienta je třeba individuálně zvážit indikaci transplantace jater. V EU v současné době probíhá u kriticky nemocných novorozenců s deficitem OTC i klinická studie s transplantací fetálních jaterních buněk.
Výsledky a diskuse
Klinická a laboratorní data
Těžká neonatální forma poruchy OTC se projevila u 9 novorozenců, u 14 chlapců s částečným deficitem OTC se onemocnění projevilo až v průběhu dětství. V jedné rodině s částečným deficitem byli na základě genotypování identifikováni i dva asymptomatičtí hemizygoti. Manifestních heterozygotek bylo 9, jedna z nich je sestra pacienta s těžkou formou onemocnění, druhá je sestrou pacienta s částečným deficitem OTC. U ostatních 7 pacientek šlo o izolovaný výskyt onemocnění v rodinách.
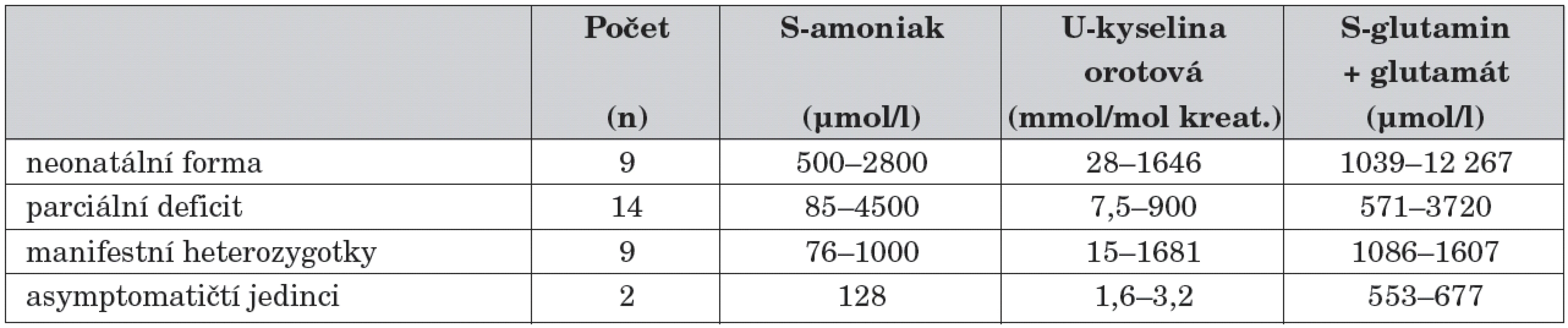
U všech probandů byla diagnóza vyslovena na základě klinických příznaků a výsledků metabolických vyšetření (tab. 1). Z tabulky vyplývá, že laboratorní nález není vždy jednoznačný. Koncentrace analytů nevypovídají o fenotypu onemocnění (např. nejvyšší hodnota amoniaku 4500 μmol/l byla zjištěna u pacienta s částečným deficitem OTC) a jen částečně korelují s aktuálním zdravotním stavem pacienta.
Analýza DNA, korelace genotyp-fenotyp
Deficit OTC byl u všech 34 pacientů potvrzen na molekulární úrovni. Celkem jsme identifikovali 22 různých mutací, jejichž charakteristika je uvedena v obrázku 1. Mutace, které vedou k úplnému deficitu enzymu (delece, předčasné terminační kodony), byly ve všech případech spojeny s těžkou neonatální formou onemocnění. U mutací měnících aminokyselinový zbytek (missense mutace) je obtížné tíži příznaků predikovat. V našem souboru bylo zjištěno osm různých missense mutací u osmnácti hemizygotů, z nichž čtyři měli neonatální formu onemocnění, dalších dvanáct částečný deficit a dva byli asymptomatičtí. Pět z osmi missense mutací bylo dříve popsáno v literatuře v asociaci se stejným fenotypem jako mají naši pacienti [9, 10].

Z literatury je známo, že neonatální fenotyp je z hlediska klinických projevů víceméně uniformní, zatímco fenotyp částečného deficitu je rozmanitý a pohybuje se od těžkých atak nastupujících v raném dětství až po asymptomatický průběh do pozdní dospělosti nebo stáří [9, 10]. Rozdílné fenotypy mohou být přitom spojeny s identickou mutací, dokonce v téže rodině, tzn. že mutace se projevuje na shodném genetickém pozadí. Tento jev jsme pozorovali v jedné rodině, ve které proband zemřel ve 14 letech, hyperamonemické kóma bylo pravděpodobně vyvoláno infekčním onemocněním s febrilním průběhem. Na druhé straně jsme v této rodině identifikovali hemizygota, který byl bez příznaků ještě v 62 letech života.
Mutace děděné a de novo mutace u chlapců a dívek
V genu OTC bylo popsáno téměř 400 různých mutací, jejichž přehled je uveden v databázi Leiden Open Variation Database (http://chromium. liacs.nl/LOVD2/). Je zajímavé, že zatímco klinicky postižení chlapci většinou zdědí mutaci v genu OTC od svých heterozygotních ale klinicky zdravých matek, u většiny klinicky manifestních dívek je obvykle nalezena mutace nová (de novo).
Při použití segregační analýzy v 35 rodinách nebyl nalezen žádný důkaz pro de novo mutaci u chlapců, zatímco pravděpodobnost, že heterozygotní dívka nese novou mutaci, byla stanovena na 57 % [11]. Podíl de novo mutací byl studován i v souboru 28 chlapců a 15 dívek [12] a v souboru 13 dívek [13]. V tabulce 2 je uvedeno srovnání s výsledky naší studie. Tuchman a kol. [12] ve své studii uvádí, že u chlapců s poruchou OTC se přenašečství prokáže u 90 % jejich matek, zatímco u matek manifestních dcer jen ve 20 %. Výsledky naší studie tento trend sice potvrzují, ale nalezené rozdíly v závislosti na pohlaví nemocných dětí již nejsou tak markantní (tab. 2).

Při hodnocení mutací z hlediska jejich dědičnosti je třeba mít na mysli, že gen OTC je na chromozomu X. Přestože není příliš pravděpodobné, že by symptomatická heterozygotka zdědila mutaci od asymptomatického otce, zcela se tato možnost vyloučit nedá.
Při nálezu de novo mutace je potřeba zohlednit i to, že u pacienta nelze odlišit skutečnou de novo mutaci od případného gonadálního mozaicismu u jednoho z rodičů. Pro potvrzení gonadálního mozaicismu u otce lze provést analýzu mutací ve spermiích, avšak průkaz gonadálního mozaicismu ve vaječnících matky není realizovatelný. Z hlediska rizika postižení dalšího potomka v rodině je proto i v případě nálezu de novo mutace indikována prenatální diagnostika.
Závěr
Deficit OTC představuje klinicky závažné onemocnění, často s nepříznivou prognózou. Včasná diagnostika, která je podmínkou pro adekvátní terapii, je založena na klinickém podezření a výsledcích metabolických analýz. Pro potřeby genetického poradenství v postižených rodinách a pro potřebu eventuální prenatální diagnostiky je nutná i diagnostika na molekulární úrovni.
Práce byla podpořena projektem IGA MZ ČR NR/9364-3.
Došlo: 2. 6. 2010
Přijato: 30. 6. 2010
RNDr. Lenka Dvořáková, CSc.
Ústav dědičných metabolických poruch
Ke Karlovu 2
128 08 Praha 2
e-mail: lenka.dvorakova@lf1.cuni.cz
Zdroje
1. Brusilow SW, Horwich AL. Urea cycle enzymes. In Scriver CR, Beaudet AL, Sly WS, Valle D (eds.). The Online Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill, Inc., 2004. Chap. 85. www.ommbid.com.
2. Horwich AL, Fenton WA, Williams KR, et al. Structure and expression of a complementary DNA for the nuclear coded precursor of human mitochondrial ornithine transcarbamylase. Science 1984; 224: 1068–1074.
3. Lyon MF. X-chromosome inactivation and human genetic disease. Acta Paediatr. Suppl. 2002; 91(439): 107–112.
4. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005; 434: 400–404.
5. Keskinen P, Siitonen A, Salo M. Hereditary urea cycle diseases in Finland. Acta Paediatr. 2008; 97(10): 1412–1419.
6. Pařízková E, Rozsíval P, Freiberger T, et al. X-vázaná hypogamaglobulinémie (Brutonova nemoc) – tři kazuistiky a molekulárně genetické studie jejich rodin. Čes.-slov. Pediat. 2004; 59: 119–122.
7. Dvořáková L, Hřebíček M, Jahnová H, et al. X-vázaná adrenoleukodystrofie u jednadvaceti českých pacientů. Čes.-slov. Pediat. 2006; 61: 129–136.
8. Luksan O, Jirsa M, Eberova J, et al. Disruption of OTC promoter-enhancer interaction in a patient with symptoms of ornithine carbamoyltransferase deficiency. Hum. Mutat. 2010; 31: E1294–1303.
9. Tuchman M, Jaleel N, Morizono H, et al. Mutations and polymorphisms in the human ornithine transcarbamylase gene. Hum. Mutat. 2002; 19: 93–107.
10. Yamaguchi S, Brailey LL, Morizono H, et al. Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Hum. Mutat. 2006; 27: 626–632.
11. Bonaïti-Pellié C, Pelet A, Ogier H, et al. A probable sex difference in mutation rates in ornithine transcarbamylase deficiency. Hum. Genet. 1990; 84(2): 163–166.
12. Tuchman M, Matsuda I, Munnich A, et al. Proportions of spontaneous mutations in males and females with ornithine transcarbamylase deficiency. Am. J. Med. Genet. 1995; 55(1): 67–70.
13. Oppliger Leibundgut E, Liechti-Gallati S, Colombo JP, et al. Ornithine transcarbamylase deficiency: ten new mutations and high proportion of de novo mutations in heterozygous females. Hum. Mutat. 1997; 9: 409–411.
Štítky
Neonatológia Pediatria Praktické lekárstvo pre deti a dorastČlánok vyšiel v časopise
Česko-slovenská pediatrie

2010 Číslo 10
- Realita liečby bolesti v paliatívnej starostlivosti v Nemecku
- Využití hodnoticích skóre a objektivních nástrojů při léčbě astmatu
- Nech brouka žít… Ať žije astma!
Najčítanejšie v tomto čísle
- Problematika zkrácené podjazykové uzdičky
- Prenatální účinky alkoholu
- Profylaxia včasnej streptokokovej sepsy novorodencov
- Fetomaternálne krvácanie ako príčina závažnej anémie novorodenca