
Deficit transkripčního faktoru GATA-2: nová imunodeficience se širokým fenotypovým spektrem. První pacienti diagnostikovaní v České republice a přehled literatury
Deficit of GATA-2 transcription factor: new immunodeficiency syndrome with broad phenotype. First patients diagnosed in the Czech Republic and review of the literature
Defects in the zinc-finger transcription factor GATA-2 gene have been recently identified in variable phenotypes associated with myeloid malignancies - myelodysplastic syndrome and acute myeloid leukaemia (MDS/AML): DCML (dendritic cell, monocyte, B-lymphocyte and NK-lymphocyte) deficiency or MonoMAC syndrome (monocytopenia with Mycobaterium avium complex infections), Emberger syndrome (early onset primary lymphoedema, multiple warts, sensorineural deafness, dysmorphism); and familial MDS/AML with no additional known phenotype.
In the Czech Republic there have been 2 patients diagnosed with GATA-2 deficiency so far. The first patient was investigated at the age of 12 years for recurrent urinary tract infections, aphtous stomatitis, herpetic skin infections and bronchial asthma; leukopenia, mild macrocytic anaemia and markedly low number of B lymphocytes with normal immunoglobulin levels were found. In the following 10 years bicytopenia progressed into a hypoplastic MDS. The girl underwent successful transplantation of haematopoietic stem cells from her HLA-identical healthy sister. Retrospectively, a heterozygous mutation (c.1187 G>A, p.R396Q) in GATA-2 has been identified. The second patient is 17 year-old boy investigated for an advanced interstitial pulmonary process with cystic remodelling of pulmonary parenchyma accompanied by signs of vasculitis. Suspicion of GATA-2 genetic defect was based on profound monocytopenia and B-cell lymphopenia. By molecular genetic analysis of GATA-2 heterozygous mutation (c.1081 C>T, p.R361C) was revealed.
Our experience corresponds with the published data on the broad phenotypic variability in patients with GATA-2 mutations. Establishment of the diagnosis of GATA-2 deficiency should lead to closer follow-up of the affected individuals and earlier allogeneic haematopoietic stem cell transplantation in case of clonal progression (MDS/AML).
Key words:
primary immunodeficiency, GATA-2 deficiency, MonoMAC, dendritic cell, monocyte, B lymphocyte, and natural killer lymphocyte deficiency (DCML), Emberger syndrome; pulmonary alveolar proteinosis, haematopoietic stem cell transplantation
Autoři:
A. Janda 1; E. Mejstříková 2; U. Salzer 1; J. Grimová 3; T. Svobodová 4; M. Suková 2; M. Nováková 2; P. Hubáček 2,9; Z. Zemanová 5; J. Čermák 6; Z. Černá 7; A. Vítek 6; A. Šedivá 8; O. Hrušák 2; J. Starý 2
Působiště autorů:
Centre of Chronic Immunodeficiency (CCI), University Medical Centre and University of Freiburg, Freiburg im Breisgau, Německo
1; Klinika dětské hematologie a onkologie UK 2. LF a FN Motol, Prahapřednosta prof. MUDr. J. Starý, DrSc.
2; Onkologická klinika UK 1. LF a Fakultní Thomayerovy nemocnice, Prahapřednosta MUDr. T. Buchler, Ph. D.
3; Pediatrická klinika UK 2. LF a FN Motol, Prahapřednosta prof. MUDr. J. Lebl, CSc.
4; Ústav lékařské biochemie a laboratorní diagnostiky UK 1. LF a VFN, Prahapřednosta prof. MUDr. T. Zima, DrSc., MBA
5; Ústav hematologie a krevní transfuze, Prahaředitel prof. MUDr. M. Trněný, CSc.
6; Dětská klinika Lékařské fakulty Univerzity Karlovy, Plzeňpřednosta doc. MUDr. J. Kobr, Ph. D.
7; Ústav imunologie UK 2. LF a FN Motol, Prahapřednostka prof. MUDr. J. Bartůňková, DrSc., MBA
8; Ústav lékařské mikrobiologie UK 2. LF a FN Motol, Prahapřednosta doc. MUDr. P. Dřevínek, Ph. D.
9
Vyšlo v časopise:
Čes-slov Pediat 2013; 68 (2): 101-112.
Kategorie:
Kazuistika
Souhrn
V roce 2011 byla popsána nová imunodeficience postihující transkripční faktor GATA-2. Tento deficit byl zjištěn u několika syndromů spojených s výskytem myeloidních malignit – myelodysplastického syndromu a akutní myeloidní leukemie (MDS/AML). Konkrétně jde o tyto jednotky: DCML (Deficit dendritických buněk, monocytů, B lymfocytů a NK buněk), MonoMAC (monocytopenie, B a NK lymfopenie, plicní alveolární proteinóza a zvýšená náchylnost k infekcím vyvolaných papilomaviry, herpesviry a aviárními kmeny mykobakterií), Embergerův syndrom (primární lymfedém dolních končetin a/nebo genitálu, mnohočetné bradavice, senzorineurální hluchota, mírný dysmorfismus); a familiální MDS/AML bez dalšího známého fenotypu.
V České republice byli dosud diagnostikováni 2 pacienti s GATA-2 deficitem. U první pacientky byly klinickým projevem recidivující infekce močových cest, aftózní stomatitida, herpetické kožní infekce a bronchiální astma. V laboratorním vyšetření ve 12 letech věku dominovala leukopenie s nápadnou B lymfopenií, mírná makrocytární anemie a normální hladina sérových imunoglobulinů. V průběhu následujících 10 let bicytopenie progredovala do obrazu hypoplastického MDS. Ve věku 22 let dívka podstoupila úspěšnou transplantaci hematopoetických buněk od HLA-identické zdravé sestry. Retrospektivně u ní byla detekována heterozygotní mutace c.1187 G>A, p.R396Q v GATA-2. Druhým pacientem je 17letý chlapec vyšetřovaný pro pokročilý intersticiální plicní proces s cystickou přestavbou plicního parenchymu, se známkami vaskulitidy. Podezření na GATA-2 deficit bylo vysloveno na základě výrazné monocytopenie a B-lymfopenie. Molekulárně genetickým vyšetřením byla v GATA-2 nalezena heterozygotní mutace c.1081 C>T, p.R361C.
Zkušenost s našimi pacienty koresponduje s publikovanými údaji o velké fenotypové šíři této imunodeficience. Průkaz GATA-2 deficitu by měl vést k důsledné monitoraci postižení krvetvorby a k časné indikaci alogenní transplantace hematopoetickými buňkami v případě klonání progrese (MDS/AML).
Klíčová slova:
vrozená imunodeficience, GATA-2 deficit, MonoMAC, deficit dendritických buněk, monocytů, B lymfocytů a NK buněk (DCML), Embergerův syndrom, plicní alveolární proteinóza, transplantace hematopoetických buněk
ÚVOD
Obor primárních imunodeficiencí se dynamicky rozvíjí. Výzkumnou aktivitu v této oblasti odráží skutečnost, že v rámci aktualizované klasifikace těchto nemocí, zveřejněné v roce 2011 Mezinárodní unií imunologických společností (IUIS), bylo ke stávajícím chorobám přidáno více než 15 nových nemocí [1]. V současné době je tedy v rámci vrozených poruch imunity popsáno přes 150 nozologických jednotek.
Jednou z horkých novinek je nález deficitu transkripčního faktoru GATA-2, který je kromě projevů imunodeficitu asociován s rozvojem myelodysplastického syndromu (MDS), respektive akutní myeloidní leukemie (AML). V následujícím textu stručně shrnujeme známá fakta o této poruše a popisujeme průběh onemocnění u prvních dvou českých pacientů, u kterých byla tato nemoc zjištěna.
Rodina transkripčních faktorů GATA
Transkripční faktor je termín užívaný pro protein, který má schopnost spouštět či jinak regulovat přepis (transkripci) DNA do mRNA. Tento proces probíhá prostřednictvím vazby transkripčního faktoru na specifickou sekvenci DNA v oblasti promotoru či enhanceru daného genu. V souhře s ostatními proteiny je regulována aktivita RNA polymerázy, která zajišťuje přepis genetické informace. Ovlivnění transkripce probíhá také nepřímo změnou přístupnosti DNA k transkripci ovlivněním reakcí určujících vazbu DNA na histony (acetylace/deacetylace histonů). Studium transkripčních faktorů je komplikováno vysokou komplexností systému – jeden gen může být regulován více transkripčními faktory a naopak jeden transkripční faktor se může podílet na regulaci více genů. Vztah transkripčního faktoru k určitému genu, respektive skupině genů, je dán přítomností domény umožňující vazbu na specifickou sekvenci DNA (DNA-binding domain). Specificita vazby této domény a podobnost její terciární struktury u jednotlivých transkripčních faktorů se používá k jejich klasifikaci.
Šest GATA transkripčních faktorů patří do rodiny tzv. zinc-finger-domain proteinů. Klíčové domény u této skupiny, které jsou potřebné jak pro vazbu na DNA, tak pro interakci s ostatními proteiny, vytvářejí krátké smyčky podobné prstům stabilizované molekulou zinku (odtud anglický název). Název GATA odkazuje na sekvenci nukleotidů, kterou tato skupina transkripčních faktorů rozpoznává při vazbě na cílové geny [2].
Transkripční faktory GATA ovlivňují osud buněk v různých orgánových systémech. GATA-1, 2, 3 se zásadně uplatňují v regulaci krvetvorby, konkrétně GATA-1 je důležitý pro erytropoezu, vývoj megakaryocytů a eozinofilů; GATA-3 se podílí na řízení diferenciace T lymfocytů. GATA-2 je naopak zásadní pro regulaci homeostázy kmenových a pluripotentních buněk. V rámci více diferencovaných hematopoetických progenitorů je důležitý pro vývoj myeloidní řady. Mimo hematopoetický systém byla zjištěna exprese těchto tří transkripčních faktorů také v endotelu, placentě, varlatech, prsní žláze, prostatě, v buňkách vnitřního ucha, hypofýze a některých typech neuronů. GATA-4, 5, 6 jsou naopak aktivní v buňkách srdečního svalu a ve střevě [2, 3].
Somatická mutace v druhém exonu transkripčního faktoru GATA-1, který se nachází na X chromozomu, je patognomickou mutací u prakticky všech případů tranzientní myeloproliferace (TMD) a akutní megakaryo-cytární leukemie u pacientů s Downovým syndromem (MIM 190685). Vrozená mutace GATA-1 byla popsána jako příčina anemie spojené s neutropenií a anomáliemi destiček (XLANP, MIM 300835), X-vázanou trombocytopenií s beta-thalasemií (XLTT, MIM 314050) a X-vázanou trombocytopenií spojenou s dys-erytropoetickou anemií (XLTDA,MIM 300367). Mutace v GATA-3 byly zjištěny u syndromu hypoparatyreodismu, senzorineurální hluchoty a renální dysplazie (HDRS, MIM 146255).
GATA-2 deficit (MIM 137295)
První popis lidského onemocnění asociovaného se zárodečnou mutací v GATA-2 zveřejnila australská skupina profesora H. S. Scotta na konci roku 2010. Jednalo se o popis 18 pacientů ze 4 rodin s familiárním výskytem MDS a AML bez dalšího jasného fenotypu jak v rámci hematopoetického systému, tak mimo něj [4]. Následovala práce skupiny profesora S. Hollanda z amerického Národního zdravotního institutu (NIH, National Institute of Health) týkající se GATA-2 poškození u 20 pacientů s tzv. MonoMAC syndromem (monocytopenie, B a NK lymfopenie, plicní alveolární proteinóza a zvýšená náchylnost k infekcím vyvolaných papilomaviry, herpesviry a aviárními mykobakteriemi – odtud pochází druhá polovina názvu syndromu: Mycobacterium Avis Complex – a tendence k rozvoji myeloidní malignity) [5]. Paralelně byl defekt GATA-2 nalezen pomocí celoexomového sekvenování britskou skupinou u 4 pacientů s nově popsaným syndromem deficitu dendritických buněk, monocytů, B lymfocytů a NK buněk (DCML, z anglického Dendritic Cell Monocyte Lymphopenia) [6, 7]. Ve stejné době popsala londýnská skupina mutace v GATA-2 u 14 pa-cientů s Embergerovým syndromem (primární lymfedém dolních končetin a/nebo genitálu manifestující se v dětství, mnohočetné bradavice, senzorineurální porucha sluchu, mírný dysmorfismus a rozvoj MDS/AML) [8].
Poškození transkripčního faktoru GATA-2 je tedy společným jmenovatelem tří nozologických jednotek: syndromu familiární MDS//AML, DCML (MIM 614172, v databázi OMIM je termín MonoMAC používán jako synonymum DCML) a Embergerova syndromu (MIM 614038).
Ve shodě s aktuální klasifikací primárních imunodeficiencí IUIS [1] považujeme v tomto textu GATA-2 deficit za jednu nemoc se širokým fenotypovým spektrem.
Klinické příznaky GATA-2 deficitu
V literatuře byly dosud publikovány údaje o přibližně 60 pacientech s GATA-2 deficitem. V následujících odstavcích stručně shrnujeme publikované informace [4–12].
Nejvýraznějším společným rysem popisovaných případů byla zvýšená náchylnost k rozvoji myeloidní malignity. U více než 60 % pacientů došlo k manifestaci MDS s případnou progresí do AML, a to s mediánem věku kolem 30 let.
Většina pacientů trpěla zvýšenou náchylností k infekcím, a to nezávisle na postižení krvetvorby. Popisovány byly atypické netuberkulózní mykobakteriální infekce, především tzv. aviární mykobakteriózy i infekce ostatními mykobakterii. U jednoho pacienta se vyskytla diseminovaná BCGitida po očkování. Relativně častý byl nález chronických a obtížně léčebně ovlivnitelných infekcí lidským papilomavirem (HPV) a herpesvirem (HSV).Vzácněji se objevily: diseminovaná infekce virem varicella-zoster (VZV), chronická aktivní infekce virem Epsteina-Barrové (EBV) a infekce parvovirem B19 nebo virem parainfluenzy. Jeden pacient zemřel na H1N1 chřipku. Zajímavé je, že běžná virová onemocnění v raném dětství proběhla u pacientů bez pozoruhodností. Z dalších etiologických agens byly detekovány: Aspergillus spp., Cryptococcus neoformans, Histoplasma capsulatum a Mycoplasma spp. Bakteriální infekce byly většinou lehčího rázu, respektive byly dobře terapeuticky zvládnutelné. Výjimkami byla ojedinělá těžká infekce Serratia marcescens a meningitida vyvolaná Haemophilus influenzae.
Častěji se u pacientů vyskytovaly bradavice na různých místech těla, více v oblasti genitálií. Jeden pacient měl těžké akné. U několika pacientů došlo k rozvoji autoimunitních projevů: sterilní granulomy v kůži, erythema nodosum, panikulitida, artritida a stavy připomínající systémový lupus erythematodes, primární biliární cirhózu a roztroušenou sklerózu. U 7 pacientů s mediánem věku 42 let (rozmezí 25–60 let) se rozvinula plicní alveolární proteinóza (PAP) charakterizovaná abnormální akumulací surfaktantu v plicních sklípcích. U několika pacientů se vyskytl primární lymfedém dolních končetin a/nebo genitálu, a to jednostranný (častěji vlevo) nebo oboustranný. Popisován je také častější výskyt solidních nádorů (především carcinoma in situ), které vznikly nejspíše vlivem chronické infekce HPV. Dále se vyskytly metastatizující melanom, bazaliom, skvamózní kožní karcinom a mnohočetný EBV-pozitivní leiomyosarkom. U některých pacientek byly zaznamenány vícečetné potraty a předčasné porody. Několik pacientů s rozsáhlými delecemi zasahujícími GATA-2 i okolní geny trpělo mentální retardací. Klinické příznaky a abnormality laboratorních paramentů u GATA-2 deficitu jsou zachyceny v tabulce 1.
![Přehled charakteristických znaků vyskytujících se u pacientů s GATA-2 deficitem. Adaptováno z práce Bigley et al. [7] s pomocí údajů z dalších publikací [4–6, 8–12].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/429d3b823fb6d04ae8c5c6f74ddeb52f.png)
Abnormality laboratorních parametrů u GATA-2 deficitu
Nápadné změny v zastoupení některých buněčných subpopulací v periferní krvi vedly k zavedení termínu DCML, resp. MonoMAC. Charakteristickým imunologickým nálezem je drastické snížení cirkulujících monocytů, plazmacytoidních (pDC, CD123+) i myeloidních (mDC, CD11c+) dendritických buněk a B lymfocytů a NK buněk. Počet T lymfocytů byl většinou normální. Pokud došlo ke snížení počtu T buněk, pak proporčně výrazněji ubývaly CD4+ buňky, což vedlo k nižším hodnotám indexu CD4+/CD8+. U několika pacientů byla zjištěna přítomnost velkých atypických granulárních T lymfocytů (CD56+CD8-). Dále byl snížen počet T regulačních lymfocytů (Treg). I přes výrazný pokles cirkulujících B lymfocytů měli pacienti obvykle normální hladiny imunoglobulinů, protilátková odpověď na vakcinaci byla zachována. Jen v ojedinělých případech byla dokumentována nižší hladina IgA nebo podtříd IgG2 a IgG4. Při progresi do MDS či AML a sekundárním selhání kostní dřeně došlo k rozvoji anemie, neutropenie, respektive trombocytopenie, případně pancytopenie.
Kostní dřeň byla u pacientů většinou hypocelulární, často byla přítomna fibróza. Počty myeloidních progenitorů a prekurzorů B lymfocytů byly nižší, počty histiocytů a makrofágů (CD68+ a CD163+) byly však v kostní dřeni i v jiných tkáních překvapivě normální. Plazmatické buňky byly v kostní dřeni hojně zastoupeny, u některých pacientů byl jejich počet dokonce výrazně zvýšen (až 10 %), někdy byl patrný obraz Mottových buněk (hyperstimulované plazmatické buňky s cytoplazmatickými inkluzemi z imunoglobulinů), resp. aberantní exprese molekuly CD56 a absence CD19. U některých pacientů byl v kostní dřeni přítomen granulomatózní zánět, ojediněle byla detekovatelná hemofagocytóza. Z cytogetických abnormalit typických pro rozvinutý MDS byla u GATA-2 deficitu nejčastěji zjištěna monozomie 7. chromozomu, resp. delece jeho dlouhého raménka. Dále byly popsány trizomie 1., 8. nebo 21. chromozomu a dicentrický 6. chromozom.
Patogeneze GATA-2 deficitu
Vzhledem ke známé úloze GATA-2 v hematopoéze není překvapivé, že má jeho porucha závažné následky. V případě knock-out (gata-/-) myší je úplná absence GATA-2 letální ještě v embryonálním období, a to díky těžkému narušení primární hematopoézy. Myši se smíšenými populacemi buněk (chiméry) přežívají, ale dochází u nich k zástavě tvorby myeloidních i lymfoidních buněk, kterým GATA-2 chybí [3]. Na druhou stranu somatické mutace zvyšující aktivitu GATA-2 byly pozorovány u pacientů s chronickou myeloidní leukemií (CML) v průběhu akutní transformace leukemie, při tzv. blastické krizi [13].
Přestože se v případě pacientů s GATA-2 deficitem jedná o poškození kmenové buňky, není přesně jasné, zda je tento defekt patrný již od narození. Údaje od několika pacientů s normálním krevním obrazem v raném dětství a postupným nástupem cytopenií následované zvýšenou vnímavostí k infekcím naznačují postupnou pomalou progresi onemocnění. Buňky myeloidního kompartmentu (monocyty, mDC), respektive některé lym-foidní progenitory (B buňky, NK buňky, pDC), jsou k dysfunkci GATA-2 zřejmě vnímavější.
Překvapivým nálezem je přítomnost makrofágů v různých tkáních navzdory chybění progenitorů v kostní dřeni i v cirkulaci [12]. Pravděpodobně se jedná o buňky, které pochází z původních prekurzorů udržovaných lokální expanzí. Přestože jsou makrofágy v tkáních a v místech zánětu přítomny v normálních počtech, lze přepokládat, že nejsou funkčně zcela kompetentní. Méněcennost těchto buněk naznačuje vysoký podíl infekcí intracelulárními patogeny, jejichž likvidace probíhá v makrofázích, a je podpořen zjištěním, že GATA-2 hraje důležitou roli v procesu fagocytózy [14]. Významnou úlohu makrofágů v patogenezi GATA-2 deficitu podporuje také manifestace PAP. Kromě deficitu surfaktantu B [15] je nejčastější příčinou PAP poškození signalizační dráhy cytokinu GM-CSF, a to buď mutací v βc podjednotce jeho receptoru [16], nebo autoprotilátkami namířenými proti GM-CSF [17]. Možný příspěvek GATA-2 deficitu k patogenezi PAP je dán pravděpodobnou rolí tohoto transkripčního faktoru v signalizaci GM-CSF [18]. Na poškození plicní tkáně se může deficit GATA-2 podílet také díky roli v regulaci transkripce endoteliální syntetázy oxidu dusnatého (eNOS) [19].
Snížená schopnost zvládání některých virových infekcí je způsobena nízkým počtem DC a NK lymfocytů. Absence NK buněk hraje možná také roli ve zvýšené potratovosti dokumentované u několika pacientek. Zde by ovšem mohla hrát úlohu také exprese GATA-2 v placentě [2]. Nižší počet Tregs se může u některých pacientů podílet na vyšší incidenci autoimunitních projevů.
Inefektivní krvetvorba manifestující se chronickými cytopeniemi typicky přechází v plně vyjádřený MDS v mnohem mladším věku (medián 30 let) než v případě sporadického MDS (medián >70 let) [20]. Rozvoj MDS je nejspíš vyvolán chronickým drážděním kmenové buňky při recidivujících infekcích a stresem vyvolaným inefektivní krvetvorbou. K plnému rozvoji MDS, resp. AML, jsou nutné další genetické změny (např. recentně prokázaná mutace v genu ASXL1 spolu s monozomií 7. chromozomu) [21].
Přítomnost lymfedémů u pacientů s GATA-2 deficitem je vysvětlována expresí tohoto transkripčního faktoru v endotelu lymfatických cév v průběhu jejich diferenciace. Poškození GATA-2 vede k nedostatečnému vývoji chlopňového aparátu a následně k poruše transportu lymfy a jejímu hromadění v dolní části těla [9].
Genetika GATA-2 deficitu
Onemocnění se dědí autozomálně dominantně (mutace v zárodečné lini postihují pouze jednu alelu) s vysokou penetrancí a variabilní expresivitou. Zjištěné byly i de novo mutace. GATA-2 je lokalizován na 3. chromozomu, 6 exonů kóduje protein o velikosti 49 kDa obsahující 480 aminokyselin. Pro funkci proteinu jsou zásadně důležitě dvě domény mající strukturu zinc-finger (ZF). Doména ležící blíže N konci proteinu (ZF1) zprostředkovává vazbu s ostatními proteiny, doména blíže C konci proteinu (ZF2) zajišťuje interakci s cílovými geny. Mutace nalezené u pacientů se nachází především v oblasti ZF, respektive způsobují předčasné ukončení translace v počátečních úsecích genu. Je pravděpodobné, že různé fenotypy odráží různou transkripční aktivitu GATA-2, která je dána příslušnou mutací. Např. lymfedém se pravděpodobně rozvine spíše při haploinsuficienci (kompletní chybění produktu jedné alely) způsobené ztrátovou mutací, respektive delecí celého genu, než u bodové mutace mající dominantně negativní efekt na funkci GATA-2 [9].
Stanovení jasného vztahu mezi typem genetického poškození a projevy onemocnění je však v současné době obtížné, k dispozici jsou zatím údaje od relativně malého počtu pacientů. Publikované informace jsou zároveň zkresleny výběrem pacientů v popsaných kohortách, které je dáno zaměřením jednotlivých pracovišť a absencí standardního vyšetřovacího postupu.
Prognóza a léčba GATA-2 deficitu
Penetrance GATA-2 deficitu je vysoká a je málo pravděpodobné, aby zůstal klinicky němý. Mírnější průběh onemocnění s manifestací až v dospělosti však nelze vyloučit. Pravděpodobnost závažných infekcí, chronických plicních změn a především rozvoj myeloidní malignity s výraznou mortalitou je velmi vysoká. Kauzální terapií onemocnění je alogenní transplantace hematopoetických buněk. V roce 2011 publikovala skupina profesora S. Hollanda první zprávu o transplantaci 6 pacientů s GATA-2 deficitem [22]. Pacienti byli transplantováni z důvodu rozvinutého MDS s mediánem věku 33 let (rozmezí 15–46 let). Vzhledem k závažným infekcím byl u všech pacientů použit méně intenzivní (nemyeloablativní) přípravný režim, což je v případě MDS neobvyklé (vyšší riziko relapsu). Jedna pacientka vstupující do transplantace ve velmi vážném stavu (vyžadovala umělou plicní ventilaci a hemodialýzu) zemřela, u ostatních 5 pacientů došlo k rychlému přihojení dárcovského štěpu a vymizení maligního klonu; u dvou pacientů transplantace vedla i ke korekci plicní patologie (plicní hypertenze, PAP). Zdá se tedy, že vzhledem k imunodeficitnímu stavu pacientů a růstové výhodě dárcovských hematopoetických progenitorů je možné pacienty s GATA-2 deficitem léčit pomocí méně toxického nemyeloablativního přípravného režimu bez zvýšeného rizika relapsu onemocnění.
KAZUISTIKY
V České republice byli dosud diagnostikováni 2 pacienti s tímto onemocněním. Stručný přehled klinických a laboratorních nálezů u jednotlivých pacientů lze nalézt v tabulkách 2 a 3.
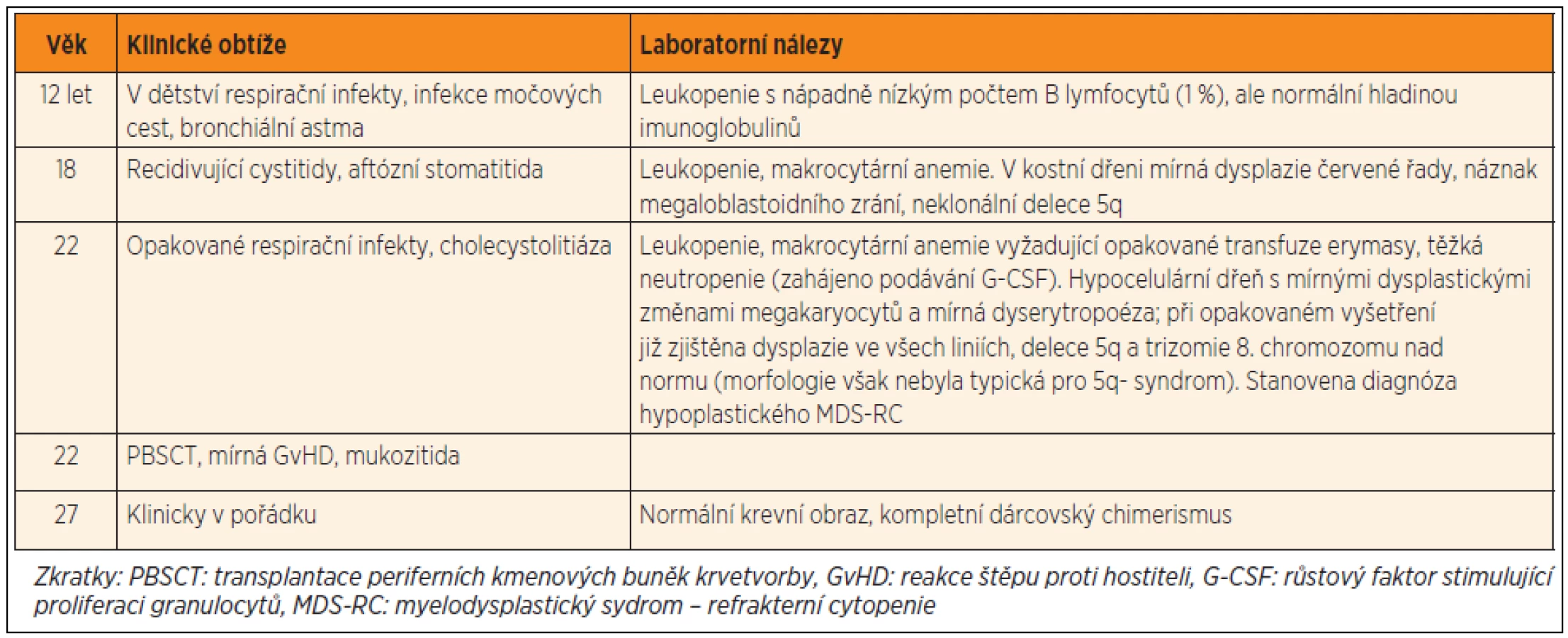

Kazuistika č. 1
Pacientka č. 1 se narodila v roce 1985, její perinatální anamnéza byla bez pozoruhodností, byla očkována podle kalendáře, bez komplikací, prospívala dobře. Od raného dětství trpěla na opakované infekty horních dýchacích cest, později se přidaly recidivující cystitidy a rozsáhlé herpetické kožní eflorescence. Dále byla sledována pro astma, chronické bolesti břicha a průjmy. Ve 12 letech byla náhodně zjištěna leukopenie (2,4–4,5 x 109/l) a nápadně nízký počet B lymfocytů (1 % z lymfocytů). Po celou dobu sledování byly hladiny imunoglobulinů normální, stejně jako množství postvakcinačních protilátek. Počty monocytů byly v normě (1–6,2 % z leukocytů, 100–220 x 106/l), NK buňky byly sníženy (2–4 % z lymfocytů, 0,03–0,06 x 109/l), DC nebyly vyšetřeny. Postupně došlo k rozvoji bicytopenie (leukopenie, makrocytární anemie). Vyšetření kostní dřeně v 18 letech věku ukázalo hypocelulární tkáň bez významnější dysplazie s normálním karyotypem hematopoetických buněk. V kostní dřeni bylo přítomno významné snížení B lymfoidních prekurzorů, prakticky zcela chyběly CD10 pozitivní prekurzory. V průběhu dalších let se krevní obraz postupně zhoršoval.
Ve věku 22 let si závažnost anemie a neutropenie vyžádaly intermitentní podávání erytrocytárních transfuzí a G-CSF. Nález v kostní dřeni progredoval do obrazu hypoplastického myelodysplastického syndromu typu refrakterní cytopenie (MDS-RC). Cytogenetickým vyšetřením byl zjištěn malý klon s delecí dlouhého raménka 5. chromozomu, který se postupně zvětšoval, následně přibyla i trizomie 8. chromozomu. Vzhledem k riziku progrese do pokročilého MDS byla indikována transplantace periferních kmenových buněk (PBSCT, transplantace periferních kmenových buněk krvetvorby) od HLA-identické zdravé sestry při použití myeloablativního přípravného režimu (Fludarabin/Busulfan). Přihojení štěpu bylo úspěšné, bez závažných potransplantačních komplikací. Po vysazení imunosuprese rok po transplantaci se manifestovala chronická kloubní GvHD s nutností další léčby. Čtyři a půl roku po transplantaci je přítomen kompletní dárcovský chimerismus, pacientka má normální krevní obraz a je klinicky v pořádku.
Retrospektivně byla u pacientky detekována heterozygotní mutace c.1187 G>A, p.R396Q v ZF2 doméně GATA-2, která byla již popsána u syndromu MonoMAC (rodina č. 18 v publikaci Hsu et al. [22]). U zdravé sestry (dárkyně štěpu) byl GATA-2 intaktní.
Kazuistika č. 2
Pacient č. 2 se narodil v roce 1995, jeho perinatální anamnéza byla bez pozoruhodností, v předškolním věku opakovaně stonal s nezávažnými infekcemi horních cest dýchacích. Očkován byl podle kalendáře, vždy bez komplikací. Kolem 8. roku věku prodělával opakované tonzilitidy, stav se později spontánně upravil.
V 17 letech byl chlapec hospitalizovaný pro oboustrannou pneumonii. Zobrazovací metody odhalily intersticiální plicní proces, zejména v horních plicních lalocích, místy cystické změny a trakční bronchiektazie. Bioptickým vyšetřením bylo zjištěno chronické resorptivně reparativní postižení s praktickou destrukcí plicního parenchymu s úseky nekróz se známkami výrazného postižení cévních stěn včetně organizující se vaskulitidy. Retrospektivní hodnocení rentgenového snímku hrudníku provedeného v jeho 13 letech pro bronchitidu potvrdilo, že intersticiální změny byly v plicní tkáni přítomny již v této době. Klinicky se plicní postižení manifestovalo postupným a nenápadným snižováním tolerance fyzické zátěže. V bronchoalveolární laváži byla zjištěna vysoká nálož EBV (22 000 kopií/10 000 genomických ekvivalentů, g.e.) při nízkém počtu EBV kopií v periferní krvi (288 kopií//10 000 g.e.) bez průkazu klonality. Vysoká nálož EBV DNA byla následně detekována také v biopsii plicní tkáně (2300 kopií EBV//10 000 g.e.). Díky několika měsícům trvající nestandardní sérologické odpovědi na EBV infekci (vysoká pozitivita anti-VCA IgM a anti-EA-D IgG protilátek po dobu šesti měsíců), stejně jako trvající detekci EBV DNA v periferní krvi, byla zvažována diagnóza atypické infekce virem EBV. Genom viru byl prokazatelný ve vysokých kvantitách 3428 kopií//10 000 g.e. v B lymfocytech a periferní krve, nicméně nebyl prokazatelný v T lymfocytech a NK buňkách. Přesto, že se v T a NK buňkách nepodařilo EBV genom prokázat a infekce tedy v tomto bodě atypickou není, délka trvání i významně zpožděná sérologická odpověď atypický průběh infekce jasně dokumentují. Přes výše uvedené nálezy byly obecné plicní funkce a saturace kyslíkem v normě.
Anamnesticky byla nápadná recidivující bolest varlat, zřejmě na podkladě intermitentních otoků nejasné etiologie, cholecystolitiáza a permanentní junkční reciproční tachykardie, pro kterou byla zvažována ablace.
V průběhu vyšetřování byla zjištěna pomocí ultrazvukového vyšetření nadhraniční velikost sleziny, dále hypergamaglobulinemie IgG a IgA, mírná pozitivita ANA, ANCA autoprotilátek a pozitivita HLA-B27. Odpověď na vakcinaci byla dostatečná. V krevním obraze byla nápadná monocytopenie. Chlapec byl intermitentně leukopenický, byly nalezeny velmi nízké hodnoty monocytů (0,2–1 % ze všech leukocytů, 9–38 x 106/l), nízké hodnoty B lymfocytů (1,9 a 3,9 % z lymfocytů, 0,15 x 109/l) a absence myeloidních i plazmacytoidních dendritických buněk. V rámci B lymfocytů byla zřejmá převaha paměťových forem B lymfocytů. Relativní zastoupení NK buněk v rámci lymfocytů bylo normální (10 a 11 %), ale v absolutním počtu bylo patrné jejich snížení (0,085 x 109/l).Cytologie kostní dřeně ukázala bilineární hematopoézu bez dysplastických změn. Při imunofenotypizaci bylo nápadné velmi nízké zastoupení monocytárních prekurzorů (0,2 %, CD14++CD45+SSCmed) a nízký počet CD34+ (0,38 %) a kit+ (0,76 %) kmenových buněk. Přibližně polovina CD34+ buněk exprimovala CD7 znak. B buňky byly ve dřeni zastoupeny 1,4 %, z toho téměř polovinu (42 %) tvořily plazmatické buňky, s částečnou aberantní expresí CD56. CD34+CD10+ prekurzory v aspirátu prakticky chyběly, CD20+CD10- tvořily 37 %. Při konvenčním a molekulárně cytogenetickém vyšetření nebyly nalezeny žádné chromozomové aberace.
Nápadné cytopenie vedly k indikaci vyšetření GATA-2. Molekulárně genetická analýza odhalila v doméně ZF2 heterozygotní mutaci c.1081 C>T, p.R361C v aminokyselině, jejíž defekt byl již popsán u pacienta s Embergerovým syndromem (pacient Emb-07 v práci Ostergaard et al.[8]).
V současné době je pacient 4 měsíce na terapii prednisonem a cotrimoxazolem, resp. nyní azitromycinem. Klinicky je stabilizovaný, hypergamaglobulinemie ustupuje.
DISKUSE
GATA-2 deficit je komplexní vrozená imunodeficience kombinující zvýšenou vnímavost k některým infekcím s poruchou krvetvorby s dispozicí ke vzniku myeloidních malignit. Změny laboratorních parametrů mohou o mnoho let předcházet klinické projevy.
V rámci diferenciální diagnostiky stavů asociovaných s GATA-2 deficitem je třeba zvážit některá další onemocnění. Z hlediska infekčního spektra se GATA-2 deficit překrývá s jinými imunodeficiencemi. Mykobakteriózy jsou typické pro defekty v signalizační dráze IFN-γ/IL-12 [23]. Infekce papilomaviry se vyskytují u veruciformní epidermodysplazie a syndromu WHIM (bradavice, hypogamaglobulinemie, a porucha vyplavování neutrofilů z kostní dřeně – myelokathexie), který je způsoben poškozením chemokinového receptoru CXCR4 [10]. Tyto nemoci ale nejsou doprovázeny alveolární proteinázou ani poruchou myeloidní diferenciace. Z hlediska familiárních syndromů spojených s manifestací MDS/AML je třeba zvážit například defekty RUNX1 nebo CEBPA či poruchy telomerázového komplexu. Plicní alveolární proteinóza se kromě primárních příčin (poruchy surfaktantu nebo signalizace GM-CSF) vyskytuje sekundárně i u jiných imunodeficitních stavů, infekcí a malignit [10]. Deficit monocytů a dendritických buněk je popisován u homozygotního poškození transkripčního faktoru IRF8. V případě této nemoci je ale v lymfatických tkáních patrná myeloidní hyperplazie a histiocytóza a v kost-ní dřeni jsou četnější osteoklasty. Vývoj B lymfocytů a NK buněk není narušen [24].
Zásadní roli, kterou hraje regulace přepisu DNA do mRNA v imunitním systému, demonstrují další imunodeficitní stavy spojené s postižením jiných transkripčních faktorů. Jmenujme například deficit FOXP3 u X-vázaného syndromu imunitní dysregulace, polyendokrinopatie a enteropatie (IPEX, MIM 304790) [25], poškození STAT3 u autozomálně dominantní varianty Hyper-IgE syndromu (MIM 147060) [26] nebo mutace ve STAT1 podmiňující chronickou mukokutánní kandidiázu a syndrom zvýšené vnímavosti k atypickým mykobakteriálním infekcím [27].
ZÁVĚR
Fenotypové spektrum GATA-2 deficitu je široké. Znalost příznaků je kromě hematologů, onkologů a imunologů důležitá také pro pneumology i všeobecné pediatry. Toto onemocnění by mělo být zváženo hlavně v případě chronické monocytopenie, B a NK lymfocytopenie, u opakovaných atypických mykobakterióz, rekurentních bradavic, herpetických infekcí, v případě plicní alevolární proteinózy, primárního lymfedému dolní poloviny těla a především při familiárním výskytu MDS a AML. Recentně probíhají analýzy incidence vrozené GATA-2 mutace u dětí a mladých dospělých s MDS. Lze očekávat, že u části pacientů bude tato mutace nalezena.
Odhalení defektu genu pro GATA-2 coby kauzální příčiny MDS a AML nás posunuje v chápání role transkripčních faktorů a komplexity vzniku těchto onemocnění. U obou našich pacientů bylo v kostní dřeni zřetelné snížení CD10 pozitivních B prekurzorů s vyšším podílem plazmatických buněk. Absence B prekurzorů je poměrně typická pro MDS, jak u dětí tak i dospělých. Pacienti s GATA-2 deficitem musí být pečlivě sledováni zejména po stránce poruchy krvetvorby a v případě známek klonální progrese včas indikováni ke kauzální léčbě transplantací alogenními hematopoetickými buňkami.
Poděkování
Práce byla částečně podpořena grantem GAUK 23010. Dr. Janda je stipendistou Fellowship programu Evropské společnosti pro imunodeficience (ESID) financované firmou Baxter.
Došlo: 7. 11. 2012
Přijato: 30. 12. 2012
MUDr. Aleš Janda, MSc., Ph.D.
Centre of Chronic Immunodeficiency (CCI)
University of Freiburg
Breisacher Str. 117
79106 Freiburg im Breisgau
Německo
e-mail: ales.janda@uniklinik-freiburg.de
Zdroje
1. Al-Herz W, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol 2011; 2: 54.
2. Bresnick EH, et al. Master regulatory GATA transcrip-tion factors: mechanistic principles and emerging links to hematologic malignancies. Nucleic Acids Res 2012; 40 (13): 5819–5831.
3. Rodrigues NP, et al. GATA-2 mediated regulation of normal hematopoietic stem/progenitor cell function, myelodysplasia and myeloid leukemia. Int J Biochem Cell Biol 2012; 44 (3): 457–460.
4. Hahn CN, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet 2011; 43 (10): 1012–1017.
5. Hsu AP, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 2011; 118 (10): 2653–2655.
6. Dickinson RE, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood 2011; 118 (10): 2656–2658.
7. Bigley V, Collin M. Dendritic cell, monocyte, B and NK lymphoid deficiency defines the lost lineages of a new GATA-2 dependent myelodysplastic syndrome. Haematologica 2011; 96 (8): 1081–1083.
8. Ostergaard P, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet 2011; 43 (10): 929–931.
9. Kazenwadel J, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood 2012; 119 (5): 1283–1291.
10. Vinh DC, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood 2010; 115 (8): 1519–1529.
11. Bigley V, et al. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med 2011; 208 (2): 227–234.
12. Calvo KR, et al. Myelodysplasia in autosomal dominant and sporadic monocytopenia immunodeficiency syndrome: diagnostic features and clinical implications. Haematologica 2011; 96 (8): 1221–1225.
13. Zhang SJ, Shi JY, Li JY. GATA-2 L359 V mutation is exclusively associated with CML progression but not other hematological malignancies and GATA-2 P250A is a novel single nucleotide polymorphism. Leuk Res 2009; 33 (8): 1141–1143.
14. Lasbury ME, et al. Effect of transcription factor GATA-2 on phagocytic activity of alveolar macrophages from Pneumocystis carinii-infected hosts. Infect Immun 2003; 71 (9): 4943–4952.
15. Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med 2002; 166 (2): 215–235.
16. Dirksen U, et al. Human pulmonary alveolar proteinosis associated with a defect in GM-CSF/IL-3/IL-5 receptor common beta chain expression. J Clin Invest 1997; 100 (9): 2211–2217.
17. Inoue Y, et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med 2008; 177 (7): 752–762.
18. Bert AG, et al. A modular enhancer is differentially regulated by GATA and NFAT elements that direct different tissue-specific patterns of nucleosome positioning and inducible chromatin remodeling. Mol Cell Biol 2007; 27 (8): 2870–2885.
19. German Z, et al. Molecular basis of cell-specific endothelial nitric-oxide synthase expression in airway epithelium. J Biol Chem 2000; 275 (11): 8183–8189.
20. Liew E, Owen C. Familial myelodysplastic syndromes: a review of the literature. Haematologica 2011; 96 (10): 1536–1542.
21. Bodor C, et al. Germ-line GATA2 p.THR354MET mutation in familial myelodysplastic syndrome with acquired monosomy 7 and ASXL1 mutation demonstrating rapid onset and poor survival. Haematologica 2012; 97 (6): 890–894.
22. Cuellar-Rodriguez J, et al. Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Blood 2011; 118 (13): 3715–3720.
23. Rosenzweig SD, Holland SM. Defects in the interferon-gamma and interleukin-12 pathways. Immunol Rev 2005; 203: 38–47.
24. Hambleton S, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med 2011; 365 (2): 127–138.
25. Chatila TA, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity – allergic disregulation syndrome. J Clin Invest 2000; 106 (12): R75–81.
26. Holland SM, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 2007; 357 (16): 1608–1619.
27. Liu L, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 2011; 208 (8): 1635–1648.
Štítky
Neonatológia Pediatria Praktické lekárstvo pre deti a dorastČlánok vyšiel v časopise
Česko-slovenská pediatrie

2013 Číslo 2
- Gastroezofageální reflux a gastroezofageální refluxní onemocnění u kojenců a batolat
- Léčba bolesti a horečky u dětí
Najčítanejšie v tomto čísle
- Telarché praecox (předčasný vývoj prsů)
- Adrenarché praecox (předčasné pubické a/nebo axilární ochlupení)
- Vývoj střevní mikroflóry a rizika používání probiotik u imunosuprimovaných dětí
- Dievča s karyotypom 46, XY