
Regulation of Contributes to the Lineage Potential of Neurogenin3+ Endocrine Precursor Cells in the Pancreas
During pancreatic development, transcription factor cascades gradually commit precursor populations to the different endocrine cell fate pathways. Although mutational analyses have defined the functions of many individual pancreatic transcription factors, the integrative transcription factor networks required to regulate lineage specification, as well as their sites of action, are poorly understood. In this study, we investigated where and how the transcription factors Nkx2.2 and Neurod1 genetically interact to differentially regulate endocrine cell specification. In an Nkx2.2 null background, we conditionally deleted Neurod1 in the Pdx1+ pancreatic progenitor cells, the Neurog3+ endocrine progenitor cells, or the glucagon+ alpha cells. These studies determined that, in the absence of Nkx2.2 activity, removal of Neurod1 from the Pdx1+ or Neurog3+ progenitor populations is sufficient to reestablish the specification of the PP and epsilon cell lineages. Alternatively, in the absence of Nkx2.2, removal of Neurod1 from the Pdx1+ pancreatic progenitor population, but not the Neurog3+ endocrine progenitor cells, restores alpha cell specification. Subsequent in vitro reporter assays demonstrated that Nkx2.2 represses Neurod1 in alpha cells. Based on these findings, we conclude that, although Nkx2.2 and Neurod1 are both necessary to promote beta cell differentiation, Nkx2.2 must repress Neurod1 in a Pdx1+ pancreatic progenitor population to appropriately commit a subset of Neurog3+ endocrine progenitor cells to the alpha cell lineage. These results are consistent with the proposed idea that Neurog3+ endocrine progenitor cells represent a heterogeneous population of unipotent cells, each restricted to a particular endocrine lineage.
Published in the journal:
Regulation of Contributes to the Lineage Potential of Neurogenin3+ Endocrine Precursor Cells in the Pancreas. PLoS Genet 9(2): e32767. doi:10.1371/journal.pgen.1003278
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003278
Summary
During pancreatic development, transcription factor cascades gradually commit precursor populations to the different endocrine cell fate pathways. Although mutational analyses have defined the functions of many individual pancreatic transcription factors, the integrative transcription factor networks required to regulate lineage specification, as well as their sites of action, are poorly understood. In this study, we investigated where and how the transcription factors Nkx2.2 and Neurod1 genetically interact to differentially regulate endocrine cell specification. In an Nkx2.2 null background, we conditionally deleted Neurod1 in the Pdx1+ pancreatic progenitor cells, the Neurog3+ endocrine progenitor cells, or the glucagon+ alpha cells. These studies determined that, in the absence of Nkx2.2 activity, removal of Neurod1 from the Pdx1+ or Neurog3+ progenitor populations is sufficient to reestablish the specification of the PP and epsilon cell lineages. Alternatively, in the absence of Nkx2.2, removal of Neurod1 from the Pdx1+ pancreatic progenitor population, but not the Neurog3+ endocrine progenitor cells, restores alpha cell specification. Subsequent in vitro reporter assays demonstrated that Nkx2.2 represses Neurod1 in alpha cells. Based on these findings, we conclude that, although Nkx2.2 and Neurod1 are both necessary to promote beta cell differentiation, Nkx2.2 must repress Neurod1 in a Pdx1+ pancreatic progenitor population to appropriately commit a subset of Neurog3+ endocrine progenitor cells to the alpha cell lineage. These results are consistent with the proposed idea that Neurog3+ endocrine progenitor cells represent a heterogeneous population of unipotent cells, each restricted to a particular endocrine lineage.
Introduction
The destruction or dysfunction of the insulin-producing beta cells of the pancreas contributes to a family of metabolic diseases known as diabetes mellitus. Given that the specification of the three major cell types in the pancreas, endocrine, exocrine and ductal cells, occurs in the embryo, understanding the normal course of pancreas development will ultimately facilitate the generation of insulin-producing beta cells from alternative cell sources for beta cell replacement therapies [1], [2], [3]. Single knockout mouse models have determined the relative importance of many transcription factors in the process of endocrine cell specification and differentiation. Of particular significance, deletion of the basic helix-loop-helix transcription factor Neurogenin3 (Neurog3; Ngn3) results in the loss of the hormone-producing cell types [4]. Subsequent lineage tracing experiments confirm that hormone-expressing endocrine cell types, including alpha cells (expressing glucagon), beta cells (insulin), delta cells (somatostatin), epsilon cells (ghrelin), and PP cells (pancreatic polypeptide), are Neurog3-derived [5], [6].
A recent study suggested that each Neurog3+ endocrine progenitor cell within the population is destined to become a single hormone+ cell type [7]. The idea that endocrine progenitor cells are unipotent implies that the transcription factor code responsible for the differentiation of each hormone+ cell type may be delineated before endocrine progenitors are specified. In support of this hypothesis, forced expression of factors within the Pdx1+ pancreatic progenitor cells can affect the resulting complement of differentiated endocrine cells [8], [9], [10]. Ultimately, the proper timing and location of transcription factor expression and function during pancreas development is essential for the appropriate differentiation of all the hormone-expressing endocrine cells.
The homeobox transcription factor Nkx2.2 is a particularly interesting pancreatic regulatory protein due to its dynamic expression pattern and cell-specific regulatory activities. Nkx2.2 is widely expressed throughout the early undifferentiated pancreatic epithelium, but gradually becomes restricted to beta cells and a large subset of alpha and PP cells [11], [12]. Despite its early and widespread expression, deletion of Nkx2.2 specifically affects later endocrine lineage specification: beta cells do not form, alpha and PP cell numbers are decreased, and there is a significant increase in the ghrelin cell population. Furthermore, while Nkx2.2 is expressed in both glucagon+ alpha cells and insulin+ beta cells [13] and the physical interaction of Nkx2.2 with the co-repressor Groucho3 (Grg3; Tle3) occurs in both cell types, the recruitment of a repressor complex to the promoter of the homeobox transcription factor Arx occurs in beta, but not alpha cells [14], presumably due to cell-specific and/or promoter-specific protein interactions. Disruption of the Nkx2.2/Grg3 interaction results in the mis-specification of islet cell types and the subsequent trans-differentiation of beta cells into alpha cells [14]. Studies of other developmental systems, including muscle and CNS, have also provided examples of how a single transcription factor can differentially regulate cell specification [15], [16], [17], [18]. Altogether these studies demonstrate that cell-specific transcription factor regulation plays a fundamental role in cell fate determination and the maintenance of cell identity.
While single knockout mouse models can uncover the role of a specific factor in the process of cell fate determination [19], [20], [21], compound deletion mutants demonstrate how multiple transcription factors work together to permit or restrict the differentiation of specific lineages. Whereas the deletion of Arx results in the loss of alpha cells and an increase in beta and delta cells [19], [22], deletion of Nkx2.2 affects all islet cell types in the pancreas except the delta cell population [12]. Interestingly, simultaneous deletion of these two factors revealed for the first time that Nkx2.2 was required to repress somatostatin in the ghrelin-expressing epsilon cell lineage [23], [24]. Furthermore, the simultaneous deletion of Nkx2.2 and the beta cell transcription factor Neurod1 identified an unexpected epistatic relationship between these factors that regulates the formation of the non-beta cell types [25]. While deletion of Neurod1 does not affect the formation of alpha or beta cells, alpha cells are reduced late in development and beta cells undergo catastrophic apoptosis by birth [26]. In contrast, the null mutation of Nkx2.2 results in a severe reduction in alpha cells, and beta cells are completely absent [12], [27]. Despite the expression of Nkx2.2 and Neurod1 in beta cells [13], [26], [28] and the severe phenotypes associated with beta cells in both single knockout mice [12], [26], the simultaneous deletion of Neurod1 and Nkx2.2 did not alter the beta cell phenotype but rather restored alpha cell and PP cell formation, while simultaneously reducing the ghrelin-expressing epsilon cells, which are over abundant in the Nkx2.2 null pancreas [25]. These examples demonstrate that deciphering the complex pancreatic gene regulatory network will provide valuable insight into the cellular processes required to generate each islet cell type, and will facilitate the in vitro differentiation of functional insulin-producing cells for therapeutic purposes.
The Nkx2.2−/−;Neurod1−/− (Nkx2.2null;Neurod1null) compound mutant provides a useful model for how two transcription factors coordinately regulate the specification of multiple endocrine cell types. Our study aimed to dissect the cooperative roles of Nkx2.2 and Neurod1, and determine specifically where and how these factors work together to permit endocrine cell formation in the pancreas. The result of this analysis demonstrated that in the absence of Nkx2.2, deletion of Neurod1 in the Pdx1+ pancreatic progenitors resulted in restoration of the alpha, PP and epsilon cells; however, deletion of Neurod1 from the Neurog3+ endocrine progenitor cells restored the PP and epsilon cells, but only a small population of alpha cells. Using in vitro reporter assays we also showed that Nkx2.2 repressed Neurod1 in certain cellular contexts. Consistent with the idea that Neurog3+ cells are unipotent [7], we hypothesize that Nkx2.2 must repress Neurod1 in the Pdx1+ pancreatic progenitors early in development to appropriately prime the Neurog3+ endocrine progenitor cells to become alpha cells.
Results
In the absence of Nkx2.2, Neurod1 deletion in Pdx1+ pancreatic progenitors recapitulates the Nkx2.2null;Neurod1null double-knockout phenotype
To determine the precise cell type in which the genetic interaction between Nkx2.2 and Neurod1 is required for endocrine cell specification, we conditionally removed Neurod1 from different pancreatic cell populations in the absence of Nkx2.2. We generated a pancreas-specific deletion of Neurod1 in the Nkx2.2 null background using Pdx1-cre [29] (Nkx2.2−/−;Neurod1flox/flox;Pdx1-cre, denoted as Nkx2.2null;Neurod1Δpanc). We first confirmed that the single deletion of Neurod1 in the Pdx1+ cells (Neurod1Δpanc) phenocopied the Neurod1null mouse (Figure 1B, 1F, 1J; Figure S1), displaying the expected reduction in insulin and glucagon mRNA levels at P0 (Figure 1M; Figure S1) [26], [30]. We also demonstrated that when Neurod1 was deleted from Pdx1+ cells in the absence of Nkx2.2, the pancreas phenotype was identical to the Nkx2.2null;Neurod1null mouse [25] (Figure S1). Specifically, all beta cells were absent, alpha and PP cells were restored, and epsilon cells, which were overabundant in the Nkx2.2null, were significantly reduced (Figure 1A–1L; Figure S1). The partial rescue of the epsilon cells is likely due to the inability of Neurod1 deletion to restore the balance between the epsilon and beta cell populations, similar to the Nkx2.2null;Neurod1null mice (Figure 1N; Figure S1; [25]). Hormone expression was quantified using real time PCR and cell numbers were determined with morphometric analysis; these analyses confirmed that the observed gene expression and cellular changes were equivalent between the Nkx2.2null;Neurod1Δpanc and the Nkx2.2null;Neurod1null (Figure 1M–1O; Figure S1). Moreover, we confirmed that Neurod1 was appropriately deleted in mutants and controls (Figure 1P). These data demonstrate that in an Nkx2.2 null background the deletion of Neurod1 in the pancreas progenitors phenocopies the Nkx2.2null;Neurod1null.
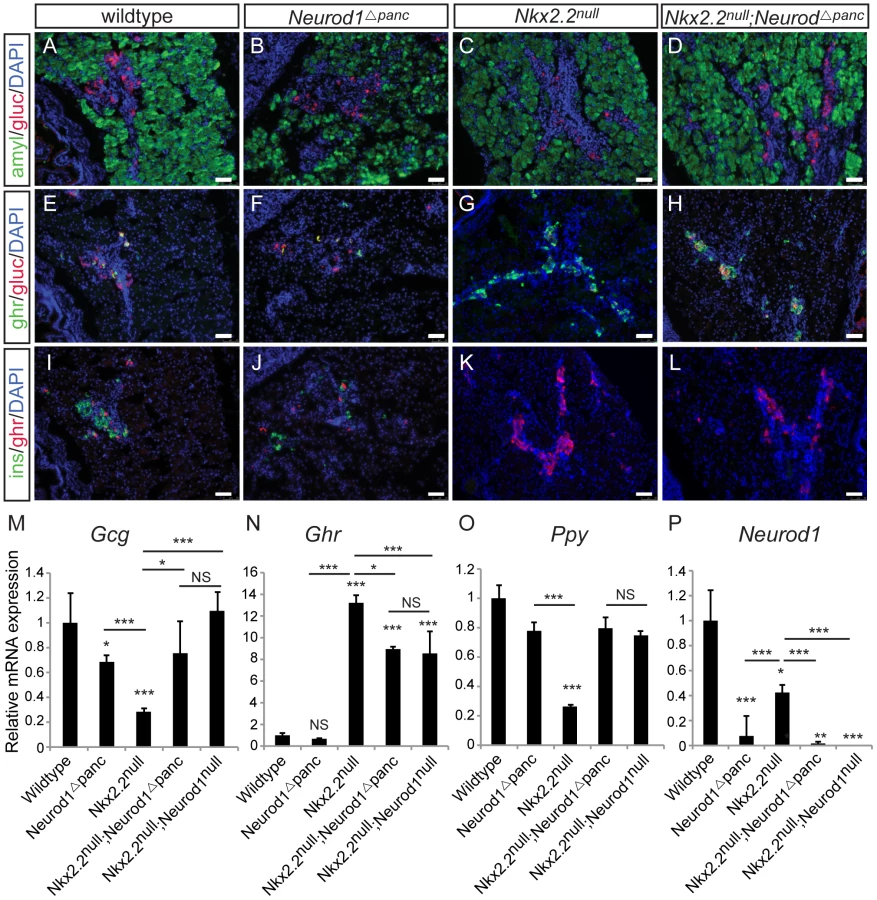
In mice lacking Nkx2.2, removal of Neurod1 from Neurog3+ endocrine progenitor cells restores relative ratios of PP and epsilon cells
Given that all hormone-producing endocrine cells are Neurog3-derived [4], [5], [6], we hypothesized that the genetic interaction between Nkx2.2 and Neurod1 would be required within the Neurog3+ endocrine progenitors to allow for the specification of particular hormone+ cell types. Using the Neurog3-cre allele [31], we generated an endocrine progenitor cell-specific deletion of Neurod1 in the Nkx2.2 null background (Nkx2.2−/−;Neurod1flox/flox;Neurog3-cre, denoted as Nkx2.2null;Neurod1Δendo), and assessed the pancreatic endocrine cell phenotype. To achieve optimal recombination in the Neurog3-expressing precursor population, we used the BAC-derived Neurog3-cre allele; Cre is highly co-expressed with Neurog3 in the embryonic pancreas and Cre activity is sufficient to lineage-label all pancreatic endocrine cells in the islet [31]. Importantly, despite the short half-life of Neurog3 protein, we can detect Cre activity in approximately 75% of Neurog3-expressing cells (Figure S2B). Similar to the Nkx2.2null;Neurod1Δpanc and Nkx2.2null;Neurod1null mice, we observed rescue of PP cells (Figure 2A, 2B), and a large reduction of ghrelin+ epsilon cells in the Nkx2.2null;Neurod1Δendo compared with the Nkx2.2null mice (Figure 2C–2H). As seen in the Nkx2.2null;Neurod1Δpanc and Nkx2.2null;Neurod1null mice, there was no rescue of the insulin-producing beta cell population (Figure 2C–2F; Figure S3). Given this similar phenotype between the Nkx2.2null;Neurod1null, Nkx2.2null;Neurod1Δpanc and Nkx2.2null;Neurod1Δendo we conclude that the genetic interaction between Nkx2.2 and Neurod1 is required in the Neurog3+ cells to permit specification of the PP and epsilon cell populations.
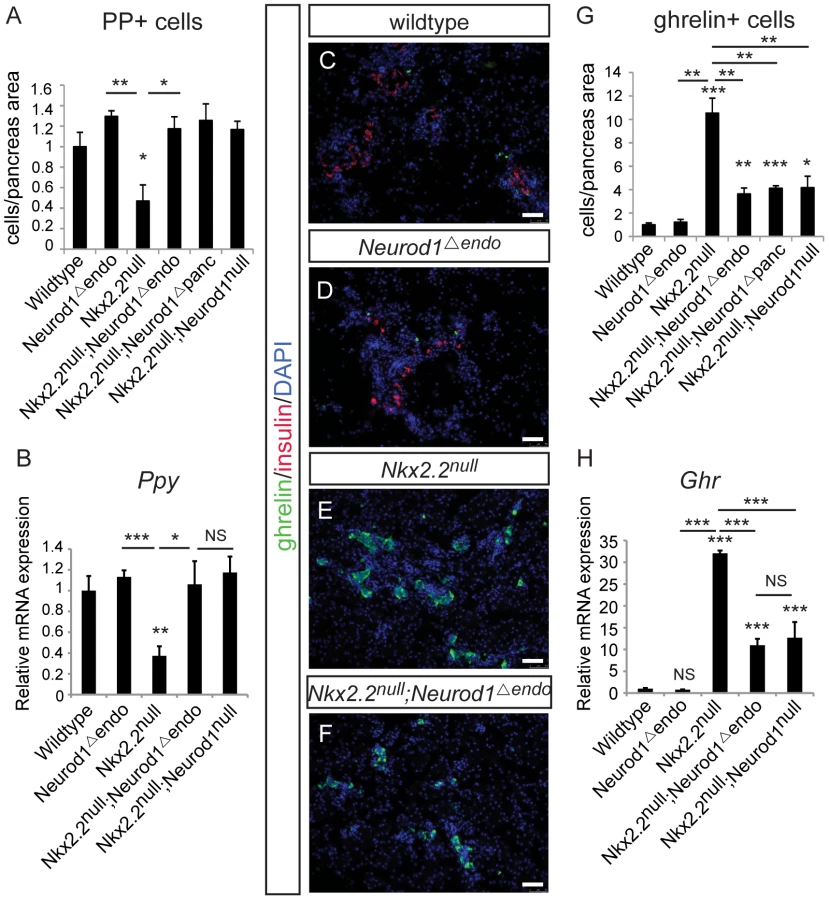
Neurod1 deletion from Neurog3+ endocrine progenitors, in an Nkx2.2 null background, is insufficient to restore the alpha cell population
Changes in the beta, PP and epsilon cell populations were identical when Neurod1 was deleted from either the pancreatic or endocrine progenitors in the absence of Nkx2.2. However, in contrast to the Nkx2.2null;Neurod1Δpanc and the Nkx2.2null;Neurod1null, the glucagon-expressing alpha cell population was only minimally restored in the Nkx2.2null;Neurod1Δendo (Figure 3A–3D). Morphometric analysis (Figure 3E) and real time PCR for glucagon expression (Figure 3F) confirmed this observation. We also established that the partial rescue was not due to incomplete deletion of Neurod1 by Neurog3-cre, as Neurod1 was reduced at an early stage of Neurog3 expression; becoming almost undetectable in the mutant pancreata by P0 (Figure 3G; Figure S4). Taken together, these data suggest that the genetic interaction between Nkx2.2 and Neurod1 in Pdx1+ progenitors, prior to Neurog3+ endocrine progenitor formation, is required for complete alpha cell formation.
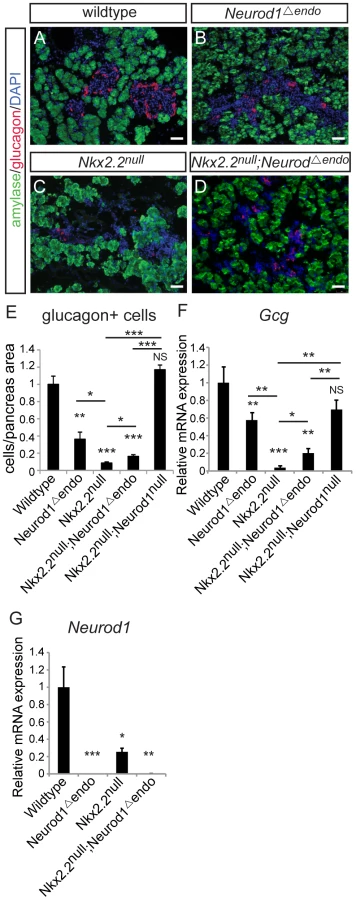
Alpha cells are not recovered with deletion of Neurod1 from the glucagon+ cells, in the absence of Nkx2.2
Data from the Nkx2.2null;Neurod1Δpanc and Nkx2.2null;Neurod1Δendo clearly demonstrate that Neurod1 must be deleted from the Pdx1+ progenitor population and not the Neurog3+ endocrine progenitor population to allow for complete rescue of alpha cell formation. Furthermore, the simultaneous loss of Nkx2.2 and Neurod1 was able to rescue even the earliest glucagon-expressing cell population; the number of glucagon-expressing cells was equivalent between the Nkx2.2null;Neurod1null and wildtype littermate controls at E10.5 (Figure 4A–4D; data not shown), Interestingly, the early glucagon-expressing cells are known to express low levels of Pdx1 (Figure S5; [24]). To determine whether the alpha cell restoration was due to deletion of Neurod1 specifically from this glucagon+ (Pdx1low) population in the absence of Nkx2.2, we deleted Neurod1 in the glucagon-expressing cells using Glu-cre [32] (Figure S2C, S2D). In the Nkx2.2−/−;Neurod1flox/flox;Glu-cre (denoted as Nkx2.2null;Neurod1Δalpha), the complement of all hormone-expressing cells in the pancreas was phenotypically identical to the Nkx2.2null, as determined by immunofluorescent analysis of islet cell markers (Figure 5A–5L; data not shown) and real time PCR for quantitative hormone expression (Figure 5M–5O; Figure S6). These results suggest that restoration of alpha cells requires the deletion of Neurod1 in Pdx1+ progenitors that have not yet committed to the glucagon-expressing lineage. We hypothesize that Nkx2.2 represses Neurod1 in the Pdx1+ cells to give rise to Neurog3+ endocrine progenitor cells that are primed to differentiate into the alpha cell fate.

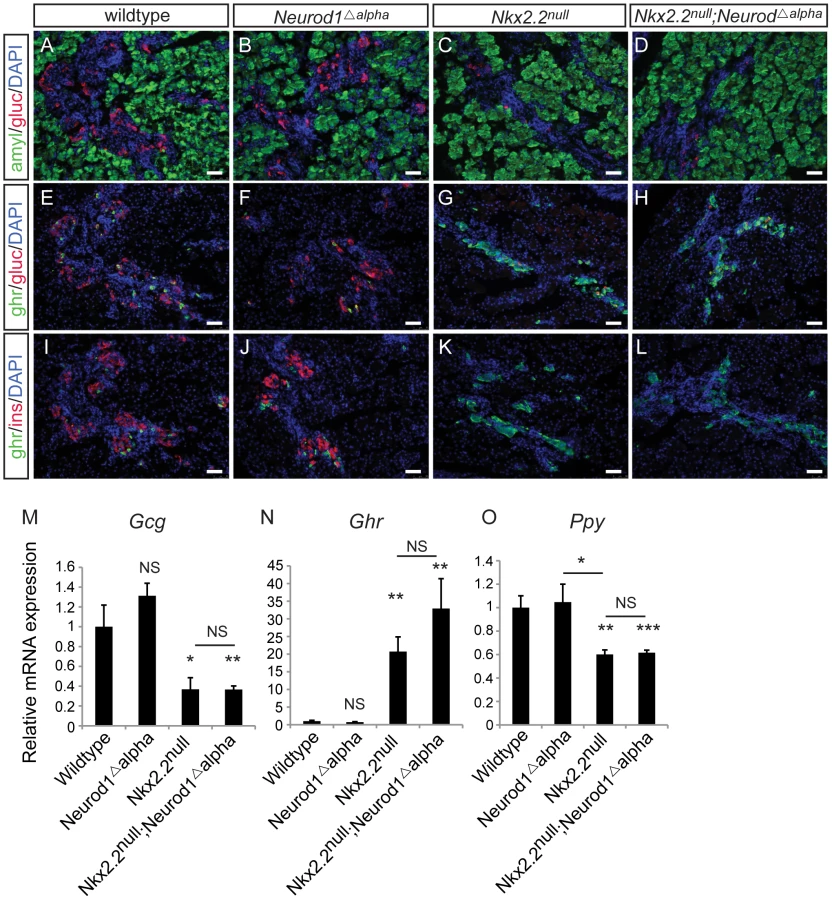
Neurod1 is expressed in a subset of Neurog3+ cells and glucagon+ cells
Since Neurod1 is a downstream target of Neurog3 [33], [34] and the Neurod1 single knockout phenotype does not manifest until the end of gestation [26], it was surprising that manipulation of Neurod1 within the Neurog3+ endocrine progenitors was not sufficient to rescue the alpha cell fate in the Nkx2.2 null background. To begin to reconcile these unexpected results, we re-examined when and where Neurod1 was expressed during pancreatic development. It was previously reported that Neurod1 is expressed at E9.5 in the earliest islet precursors, and is often co-expressed with glucagon [26]. Using the Neurod1 null mouse, which has a LacZ insertion into the Neurod1 locus [35], we confirmed the presence of Pdx1+/Neurod1(beta-gal+) cells and glucagon+/Neurod1(beta-gal+) cells in the earliest pancreatic domain (Figure 6A; Figure S7A); however, not all glucagon+ cells were Neurod1+ (Figure 6A, 6E). Consistent with previous reports [28], this pattern was also evident at E13.5 (Figure 6B, 6E) during the stage of pancreas development marked by a major wave of endocrine cell differentiation referred to as the “secondary transition” [36].

Neurod1 is expressed throughout the epithelial cord region, overlapping extensively with the Neurog3+ precursor cells (Figure S7B, S7C). We used expression of the Neurod1:LacZ allele to identify Neurod1 (beta-gal+) cells that co-expressed Neurog3 at E9.5 (Figure 6C) and at E13.5 (Figure 6D). Interestingly, the overlap of Neurog3 and Neurod1 was not exclusive at either age, and a subset of Neurog3+ cells did not express Neurod1 (Figure 6F). We also detected Neurod1 (beta-gal+) expression in a small population of Sox9low cells (Figure S7D–S7F), indicating that Neurod1 expression can be found in cells that are transitioning into Neurog3 precursor cells [37]. Taken together these expression analyses identified heterogeneous populations of Neurog3+ cells and glucagon+ cells based on their expression of Neurod1, and may suggest that the presence or absence of Neurod1 could influence downstream cell fate decisions.
Nkx2.2 represses Neurod1 in alpha cells
Our cumulative data suggest that Nkx2.2 may function to repress Neurod1 in a subset of Pdx1+ pancreatic progenitor cells to promote specification of the alpha cell fate. We had previously determined that Nkx2.2 directly activates the Neurod1 promoter in beta cells, which is consistent with the beta cell phenotypes of the single and double knockout mice [12], [26], [28] (Figure 7A). To determine whether Nkx2.2 could also repress Neurod1 expression in other (non-beta) cell contexts, we analyzed the effect of Nkx2.2 on Neurod1 expression in alpha cells in vitro. Utilizing previously described Neurod1 promoter deletion constructs [28] we determined that Nkx2.2 repressed the Neurod1 promoter in alphaTC1 cells, which express Nkx2.2 [28] (Figure 7). Specifically, the repressive activity of Nkx2.2 mapped to the proximal region of the Neurod1 promoter, which is retained in the NDΔ2 promoter construct (Figure 7B). We also determined that, similar to Nkx2.2-dependent activation of the Neurod1 promoter in beta cells, Nkx2.2 repression required the presence of at least one of the three Nkx2.2 binding sites; deletion of either region containing these consensus elements (promoter constructs NDΔ3, NDΔ4) resulted in a loss of Nkx2.2 repression (Figure 7B).

To begin to understand how Nkx2.2 mediates differential cell context-specific regulatory activities through the same set of promoter elements, we assessed the ability of Nkx2.2 to recruit specific cofactors and/or modified histones to the Neurod1 promoter in alpha versus beta cell lines. We previously demonstrated that Nkx2.2 preferentially recruits Grg3 and a large co-repressor complex to the inactive Arx promoter in beta cells, but this complex was not present on the same promoter region in alpha cells, where Arx was actively transcribed [14]. Surprisingly, neither Grg3 nor HDAC1 were recruited to the Neurod1 promoter in either alpha or beta cell lines (data not shown), suggesting that Nkx2.2 mediates Neurod1 regulation through an alternative mechanism. Interestingly however, we determined that histone H3K4me3 preferentially occupied the Neurod1 promoter in beta cells, and this differential binding was dependent upon the phosphorylation state of Nkx2.2 (Figure 7C). Histone H3K27me3 was not significantly present at the Neurod1 promoter in either alpha or beta cell lines (Figure 7D). These results suggest that while Nkx2.2 promotes activation of Neurod1 in beta cells [28], Nkx2.2 appears to prevent the activation of the Neurod1 promoter in alpha cells. This finding is consistent with the idea that Nkx2.2 is required to prevent expression of Neurod1 in a subset of Pdx1+ progenitor cells and then maintain this repression in “alpha-cell competent” Neurog3-expressing cells, and subsequently mature alpha cells.
Discussion
Single deletion mutants have identified the importance of a number of transcription factors for the process of endocrine cell differentiation (reviewed in [38]). Interestingly, very few factors when deleted affect only one islet cell type. Therefore we can deduce that each regulatory protein has multiple roles during development and it is likely that different combinations of these factors must be simultaneously present or absent within the endocrine progenitor cells to permit the specification of alpha, beta, delta, epsilon or PP cells. The generation of compound deletion mutants would assist in deciphering this combinatorial transcription factor code. One such example is the regulatory interaction between Nkx2.2 and the alpha cell transcription factor Arx; simultaneous deletion revealed that these factors differentially cooperate to affect the specification of several islet cell lineages [23], [24]. In this current study, we explore the relative roles of Nkx2.2 and the beta cell transcription factor Neurod1. The single deletion mutants for Nkx2.2 or Neurod1 display alterations in several islet cell types [12], [26]; however, these mutants are noted for their severe beta cell phenotypes. In particular, Nkx2.2 and Neurod1 are necessary for beta cell specification and maintenance, respectively [12], [26]. Interestingly, simultaneous deletion of Nkx2.2 and Neurod1 did not affect the respective beta cell phenotypes of the single mutants, but rather identified complex genetic interactions between these factors for the specification of alpha, PP and epsilon cells [25]. In this set of experiments, we have determined the cellular locations of the genetic interactions between Nkx2.2 and Neurod1, and have uncovered a possible mechanism for how these transcription factors contribute to the process of alpha cell specification. Given the increasing number of studies identifying transdifferentiation between alpha cells and beta cells [10], [14], [39], refining our understanding of alpha cell development may provide insight into the unique relationship between alpha and beta cells, and ultimately aid in understanding how beta cells develop in both the normal and diseased state.
Knowing that all endocrine cell types are derived from Neurog3-expressing cells [5], [6], we hypothesized that the genetic interaction between Nkx2.2 and Neurod1 would be required in the Neurog3+ endocrine progenitors to specify islet cell fates. In support of this hypothesis, deletion of Neurod1 from the Neurog3+ endocrine progenitor cells in an Nkx2.2 null background (Nkx2.2null;Neurod1Δendo) was sufficient to rescue the relative ratios of the ghrelin-expressing epsilon cells and pancreatic polypeptide-expressing PP cells when compared to the Nkx2.2 null phenotype. This demonstrates that the genetic interaction between Nkx2.2 and Neurod1 is required within the Neurog3+ endocrine progenitor population to permit appropriate specification of the PP and epsilon cell populations. In contrast, although alpha cells were completely rescued in the Nkx2.2null;Neurod1Δpanc, we observed only a minimal restoration of glucagon+ cells in the Nkx2.2null;Neurod1Δendo, suggesting that alpha cell recovery requires the genetic interaction between Nkx2.2 and Neurod1 to occur within the Pdx1+ pancreatic progenitors, prior to Neurog3+ endocrine progenitor cell formation. This finding would support the concept proposed by Degraz and Herrera [7] that the Neurog3+ endocrine progenitors represent a heterogeneous population of unipotential cells that are already committed to become a single hormone-producing cell fate.
If all Neurog3+ progenitors are indeed unipotent, then how do we explain rescue of the PP and ghrelin cell ratios that resulted from manipulating gene expression after the Neurog3+ cells are formed? It is possible that there are both unipotential and multipotential endocrine progenitor populations. Alternatively the “pro-PP” or “pro-ghrelin” Neurog3+ populations may retain more plasticity throughout development. The latter explanation is consistent with the findings of Johansson et al., [9], which demonstrated that as development proceeds the progenitor cells are less competent to produce alpha cells and instead favor the generation of other endocrine cell types. This would suggest that although the alpha cell fate decision can be made at multiple points during development, the ability to generate alpha cells is most robust in the earliest pancreatic progenitors and becomes restricted over time. Alternatively, it is possible that later born progenitors retain a certain degree of plasticity that accounts for their ability to respond to lineage manipulations after Neurog3+ cell specification has occurred.
The inability to rescue alpha cells by simultaneously removing Nkx2.2 and Neurod1 from the Neurog3+ precursor population, suggests that the genetic interaction between Nkx2.2 and Neurod1 is required in the Pdx1+ progenitor population, prior to acquisition of Neurog3 expression. However, it remains possible that there is a spectrum of Neurog3-cre activity within a Neurog3+ precursor cell, with Cre-based inactivation reaching its peak in the middle or late in the lifespan an individual cell. If this were the case, and the genetic interaction between Nkx2.2 and Neurod1 is required only early in the lifespan of a Neurog3+ precursor to rescue alpha cells, then Neurog3-cre activity may occur too late within this population to affect its differentiation potential. Although we are unable to resolve the kinetics of Cre activity in the lifespan of a single cell, we can demonstrate co-expression of Neurog3, Cre and R26R reporter activity, suggesting that although Neurog3 protein expression is transient, Cre is present and active in most of the Neurog3+ population during the time window when Neurog3 is expressed (Figure S2B). Furthermore, published lineage studies using this Neurog3-cre allele demonstrated that all endocrine cells of the islet, including the glucagon-expressing alpha cells, are labeled by a Cre-dependent R26R:LacZ reporter [31]. This would suggest that even if alpha cells can only be differentiated from “young” Neurog3+ precursors, there is sufficient Cre activity at this earliest stage during the lifespan of a Neurog3+ cell to genetically label the alpha cell population.
Our failure to recover alpha cells by deleting Neurod1 in a glucagon-expressing population may also be due to the inefficiency of the Glu-cre allele, especially in Nkx2.2null embryos that have a severe reduction in alpha cell numbers. However, we detected similar levels of Glu-cre activity in wildtype and Nkx2.2null pancreata, which should have been sufficient to permit any possible alpha cell rescue (Figure S2C–S2D; see Materials and Methods). Although caveats exist with the use of Cre/lox technologies, these are currently the best tools available to assess spatial and temporal protein function.
Interestingly, we do observe some rescue of alpha cells in the Nkx2.2null; Neurod1Δendo embryos. This could be due to deletion of Neurod1 in a subset of Neurog3+ progenitors that have not yet become restricted in their ability to differentiate into alpha cells. Alternatively, the glucagon-expressing cells recovered in the Nkx2.2null;Neurod1Δendo may represent alpha cells that form independent of Neurog3 function; such an alpha cell population has been previously documented [40], [41]. On the other hand, the recovered alpha cells may actually represent a distinct subpopulation of glucagon-expressing cells that express Neurod1, which would be consistent with our identification of a subpopulation of glucagon+/Neurod1+ cells. While these explanations are not mutually exclusive, the identification of unique alpha cell markers and the generation of genetic tools utilizing these markers, would be necessary to clarify the existence of subpopulations of alpha cells, as well as the factors involved in the generation of these distinct populations.
Our findings also suggest that Nkx2.2 must regulate Neurod1 differentially in the Pdx1+ progenitor population in the early pancreatic epithelium in order to initiate the specification of different populations of Neurog3-expressing cells. In particular, the prevention of Neurod1 activation by Nkx2.2 would result in alpha cell formation, while the activation of Neurod1 by Nkx2.2 results in beta cell formation (Figure 8). This is compatible with our discovery that not all Neurog3+ cells express Neurod1, and further supports the idea that the Neurog3+/Nkx2.2+/Neurod1+ cells most likely become beta cells, whereas Neurog3+/Nkx2.2+/Neurod1− cells would become alpha cells. Ideally, we would test this hypothesis by quantifying the increase in the number of Pdx1+/Neurod1+ pancreas progenitors and/or Neurog3+/Neurod1+ endocrine progenitors expected to be observed in the Nkx2.2null pancreas; however, this analysis is confounded by the simultaneous loss of the Neurod1+ pro-beta cell progenitor populations in the Nkx2.2null pancreas. Instead, we used an in vitro approach to determine whether it was possible for Nkx2.2 to differentially regulate the Neurod1 promoter in different cellular contexts. We had previously demonstrated that Neurod1 is activated by the cooperative binding of Nkx2.2 and Neurog3 specifically in beta cells [28]. Given the lack of availability of an appropriate pancreatic progenitor cell line, we reasoned that a genetic interaction between Nkx2.2 and Neurod1 that was initiated in a “pro-alpha cell” progenitor would be maintained in the mature alpha cell. We utilized alphaTC1 cells, which express Nkx2.2 [28], to demonstrate that Nkx2.2 prevents activation of Neurod1 in alpha cells. Highlighting the complexity of gene regulation, the cell type specific regulation of Neurod1 by Nkx2.2 appears to function through a mechanism that is different from Nkx2.2 regulation of the Arx gene [14]. This may reflect the mechanism by which Nkx2.2 functions as an activator and a repressor in the same cell type and/or the presence or absence of cell-specific co-regulatory proteins. As we gain the molecular tools to study transcriptional and epigenetic mechanisms in purified primary pancreatic cell populations, we hope to elucidate the complex regulatory interactions that are required to form and maintain appropriate islet-cell specific gene expression.

While the process of endocrine specification likely requires the concerted action of many factors, our data suggest a mechanism that involves the differential regulation of Neurod1 by Nkx2.2 in the Pdx1+ pancreatic progenitor cells to direct the subsequent endocrine progenitors to become specific islet cell types. The generation of tools to identify, separate and analyze different subpopulations of Neurog3+ progenitor cells would conclusively determine whether each hormone+ endocrine cell type is derived from a specific unipotent subpopulation of endocrine progenitor cells, each bearing a unique gene profile.
Using the pancreas as a model system, our study has provided a prime example of how lineage decisions are established in the developing epithelium. The cooperative action of multiple transcription factors within the early progenitor cells can dictate the fate of subsequent cell lineages. Altering the regulation or complement of this set of factors within the progenitor populations can ultimately skew cell lineage specification. These data have important implications for the current efforts to generate pancreatic cells in vitro for therapeutic use in diabetic patients. Understanding the cooperative transcription factor code will make it possible to initiate the appropriate program in the Pdx1+ pancreatic progenitor cells necessary to correctly prime the Neurog3+ endocrine progenitor cells and generate pools of functional, single hormone-expressing islet cell types in vitro.
Materials and Methods
Mice
All experiments involving mice were approved by the Columbia University Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health guidelines for the care and use of animals. All mouse strains were previously generated, and were bred and maintained on an outbred Black Swiss background (NTac:NIHBS, Taconic). Cell-specific Neurod1 null mice were generated by intercrossing Neurod1tm1Kan (Neurod1flox/flox; [42]) and either Tg(Ipf1-cre)1Tuv (Pdx1-cre; [29]), Tg(Neurog3-cre)C1Able (Neurog3-cre; [31]), or Glu-cre ([32]) mice. Neurod1flox/flox;Pdx1-cre and Neurod1flox/flox;Neurog3-cre mice died postnatal, similar to the Neurod1 null (data not shown; [30]). Certain experiments required the use of either Gt(ROSA)26Sortm9(CAG-tdTomato)Hze (R26R:Tomato; [43]) or Gt(ROSA)26Sortm1Sor (R26R:LacZ; [44]) reporter alleles. The Pdx1-cre will delete Neurod1 in all pancreatic progenitor cells; however, the Pdx1 expression domain also includes a portion of the stomach and the duodenum [45], [46]. We and others have previously reported the early and relatively non-mosaic activity of the Pdx1-cre allele ([29], [47]; Figure S2A). Previous characterization of the Neurog3-cre allele demonstrated almost complete co-expression of Neurog3 and Cre and sufficient Cre activity to lineage label all endocrine cells within an islet [31]. Consistent with this published analysis, quantification of cells co-expressing Neurog3 and the LacZ reporter in a Neurog3-Cre;R26R:LacZ E15.5 embryo indicated 74.82% Cre efficiency (268 Neurog3+ beta− gal+/349 total Neurog3+ cells; calculations were performed as described below (Figure S2B). Similar assessment of the Glu-cre mice demonstrated that the Glu-cre allele is active in approximately 30–35% of alpha cells; notably this degree of activity is unchanged in the Nkx2.2null background, despite the overall reduction in alpha cell numbers (Figure S2C, S2D).
The heterozygous mice (Neurod1flox/+;Pdx1-cre) were crossed to Nkx2-2tm1Jlr knock-in mice [12] to generate compound heterozygotes. Embryos were collected from timed matings between Nkx2.2+/−;Neurod1flox/+;Pdx1-cre and Nkx2.2+/−;Neurod1flox/flox or Nkx2.2+/−;Neurod1flox/+;Neurog3-cre and Nkx2.2+/−;Neurod1flox/flox or Nkx2.2+/−;Neurod1flox/+;Glu-cre and Nkx2.2+/−;Neurod1flox/flox mice. Noon on the day of appearance of a vaginal plug was considered embryonic day (E) 0.5. The experimental genotypes of wildtype, Nkx2.2−/− (Nkx2.2null), Neurod1flox/flox;Pdx1-cre (Neurod1Δpanc), Nkx2.2−/−;Neurod1flox/flox;Pdx1-cre (Nkx2.2null;Neurod1Δpanc), Neurod1flox/flox;Neurog3-cre (Neurod1Δendo), Nkx2.2−/−;Neurod1flox/flox;Neurog3-cre (Nkx2.2null;Neurod1Δendo), Neurod1flox/flox;Glu-cre (Neurod1Δalpha), and Nkx2.2−/−;Neurod1flox/flox;Glu-cre (Nkx2.2null;Neurod1Δalpha) were studied. Litters were assessed at postnatal day (P) 0. For expression studies, the Neurod1tm1Jle LacZ knock-in (Neurod1LacZ/+ or Neurod1null) [35] was used (also in combination with the Nkx2.2null thereby producing Neurod1null;Nkx2.2null double knockout embryos; DKO), and embryos were assessed at E9.5, E10.5, E13.5 and P0. All embryo dissections were carried out in cold PBS, using a dissecting microscope (Leica MZ8). A portion of each embryonic tail or yolk sac was detached from the embryo, digested with proteinase K, and DNA extracted for genotyping purposes. Genotyping was carried out with standard conditions and primers as previously described [12], [29], [31], [32], [35], [42].
Real-time PCR
Pancreas was dissected from each embryo and stored in RNAlater (Ambion) until RNA was extracted using the NucleoSpin RNAII Kit (Clontech). Subsequently, cDNA was made with equal amounts of RNA for each sample (Superscript III Kit, Invitrogen, CA). Real time PCR was performed using TaqMan gene expression assays (Applied Biosystems) for glucagon (Mm00801712_m1), ghrelin (Mm00445450_m1), somatostatin (Mm00436671_m1), insulin1 (Mm01950294_s1), insulin2 (Mm00731595_gH), pancreatic polypeptide (Mm00435889_m1) and Neurod1 (Mm01280117_m1). CyclophilinB was used as a control housekeeping gene, and was assayed using a probe and primer set previously described [25]. A standard two-step real time PCR program was used for all genes assessed, with an annealing temperature of 61°C and 40 cycles of amplification (CFX96 RealTime System C1000 Thermal Cycler, Biorad). All gene expression values were normalized to the internal control gene, cyclophilinB, and relative quantification was performed using a standard curve from embryonic age-matched cDNA. Statistical analyses were conducted with Prism Software (GraphPad Software, La Jolla, CA) using both the Mann-Whitney test and the Student t-test. Equivalent results were obtained; t-test results were reported in all Figures.
Immunofluorescence
Immunofluorescence was performed according to standard protocols, on E9.5, E10.5, E13.5, E15.5 and P0 whole embryos that were embedded in OCT, after fixation with 4% PFA and cryopreservation in 30% sucrose. Transverse frozen sections (8 µm) were cut and mounted on glass slides. Sections were stained with rabbit α-ghrelin (1∶800; Phoenix Pharmaceuticals, CA), goat α-ghrelin (1∶800; Santa Cruz), guinea pig α-glucagon (1∶1000; Linco/Millipore, MA), guinea pig α-insulin (1∶1000; Millipore), rabbit α-insulin (1∶1000; Cell Signaling Technology), rabbit α-somatostatin (1∶200; Phoenix Pharmaceuticals), rabbit α-pancreatic polypeptide (1∶200; Zymed), rabbit α-amylase (1∶1000; Sigma), rabbit α-Pdx1 (1∶1000; Millipore), guinea pig α-Pdx1 (1∶500; BCBC), rabbit α-Neurog3 (1∶500; BCBC), goat α-Neurog3 (1∶500; BCBC), goat α-FoxA (1∶1000; Santa Cruz), rabbit α-sox9 (1∶500; Chemicon), and chicken α-beta-galactosidase (1∶250; Abcam). Donkey α-guinea pig-Cy2, -Cy3 or -Cy5, α-rabbit-Cy2 or -Cy3, α-chicken-Cy3, and α-goat Cy2 or -Cy5 secondary antibodies were used (1∶400, Jackson ImmunoResearch). DAPI (1∶1000; Invitrogen) was applied for 30 minutes following secondary antibody incubation. Images were acquired on a Leica DM5500 or Leica 510 confocal microscope. Morphometric analysis was performed by immunostaining every 10th section throughout each embryo (N = 3 or 4 for each genotype). For quantification of individual hormone-expressing cells at P0, cell number was assessed versus total pancreas as defined by amylase area. For quantification of hormone-expressing cells at E10.5, cell number was assessed versus total pancreas as defined by Pdx1 area. Pancreas area was calculated using ImagePro software.
RNA in situ hybridization
RNA in situ hybridization was performed on 8 µm sections mounted on glass slides as previously described [25] using an antisense riboprobe transcribed from linearized plasmid. The riboprobe for Neurod1 was generated from the plasmid pCS2:MTmNeuroD1 (J. Lee). RNA in situ hybridization was performed on pancreas tissue sections from Neurod1Δendo and wildtype littermate controls at E10.5 and Neurog3-cre;R26RLacZ at E15.5.
Luciferase reporter assays
The Neurod1-2.2 kb minimal promoter was fused to the firefly luciferase open reading frame in the pGL3 Basic vector (Promega). The alphaTC1 cells were grown in 12-well plates. The design of all Neurod1 promoter deletion constructs and the transfection conditions were previously described [28]. Firefly luciferase readings were normalized to Renilla luciferase values. A Student t-test was performed to determine significance.
Chromatin immunoprecipitation
Point mutations were made to 3xmyc-tagged Nkx2.2 cDNA using the QuickChange II Site Directed Mutagenesis kit (Agilent Technologies) with the following primers S-11-A: (FWD) CAACACAAAGACGGGGTTTGCTGTCAAGGACATCTTGGAC, (REV) GTCCAAGATGTCCTTGACAGCAAACCCCGTCTTTGTGTTG; S-11-D: (FWD) CAACACAAAGACGGGGTTTGATGTCAAGGACATCTTGGAC, (REV) GTCCAAGATGTCCTTGACATCAAACCCCGTTTTGTGTTG. Wild type or mutated Nkx2.2 cDNA encoding a triple myc epitope tag (250 ng) was transfected into betaTC6 or alphaTC1 cells using X-treme gene HP (Roche) according to manufacturer's protocol. Chromatin was prepared using the ChIP-IT express kit (Active Motif). Immunoprecipitation protocol was modified from Tuteja et al. [48]. In brief, immunoprecipitation was performed using the isolated chromatin diluted in ChIP dilution buffer with 5 micrograms of either mouse anti-H3K27me3 (Abcam) or mouse anti-H3K4me3 (Abcam) antibodies while rotating overnight at 4°C. The following day antibody/chromatin complexes were pulled down using ChIP grade protein G magnetic beads (Cell Signaling). After washing, antibody/chromatin complexes were eluted from the beads and allowed to rotate at room temperature for 15 minutes. NaCl (5 micromolar) was added to the eluate and incubated at 65°C overnight. The following day Tris-HCl (1 M, pH 7.5), EDTA (0.5 M) and proteinase K (10 mg/mL) were added and allowed to incubate at 37°C for 1 hour. Samples were then purified using the QIAquick PCR purification kit (Qiagen). Quantitative analysis of ChIP products was performed using SYBR Green fluorescence with primers for Gapdh (FWD – CTCCACGACATACTCAGCACC; REV – TCAACGGCACAGTCAAGGC) or Neurod1 (FWD – AAAGGGTTAATCTCTCCTGCGGGT; REV - CATGCGCCATATGGTCTTCCCGGT).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. D'AmourKA, BangAG, EliazerS, KellyOG, AgulnickAD, et al. (2006) Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol 24: 1392–1401.
2. KroonE, MartinsonLA, KadoyaK, BangAG, KellyOG, et al. (2008) Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol 26: 443–452.
3. NostroMC, SarangiF, OgawaS, HoltzingerA, CorneoB, et al. (2011) Stage-specific signaling through TGFbeta family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells. Development 138: 861–871.
4. GradwohlG, DierichA, LeMeurM, GuillemotF (2000) neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A 97: 1607–1611.
5. GuG, DubauskaiteJ, MeltonDA (2002) Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129: 2447–2457.
6. HellerRS, JennyM, CollombatP, MansouriA, TomasettoC, et al. (2005) Genetic determinants of pancreatic epsilon-cell development. Dev Biol 286: 217–224.
7. DesgrazR, HerreraPL (2009) Pancreatic neurogenin 3-expressing cells are unipotent islet precursors. Development 136: 3567–3574.
8. CollombatP, Hecksher-SorensenJ, KrullJ, BergerJ, RiedelD, et al. (2007) Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J Clin Invest 117: 961–970.
9. JohanssonKA, DursunU, JordanN, GuG, BeermannF, et al. (2007) Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev Cell 12: 457–465.
10. YangYP, ThorelF, BoyerDF, HerreraPL, WrightCV (2011) Context-specific alpha -to-beta cell reprogramming by forced Pdx1 expression. Genes Dev 25.
11. JorgensenMC, Ahnfelt-RonneJ, HaldJ, MadsenOD, SerupP, et al. (2007) An illustrated review of early pancreas development in the mouse. Endocr Rev 28: 685–705.
12. SusselL, KalamarasJ, Hartigan-O'ConnorDJ, MenesesJJ, PedersenRA, et al. (1998) Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development 125: 2213–2221.
13. ArnesL, LeclercK, FrielJM, HipkensSB, MagnusonMA, et al. (2012) Generation of Nkx2.2:lacZ mice using recombination-mediated cassette exchange technology. Genesis
14. PapizanJB, SingerRA, TschenSI, DhawanS, FrielJM, et al. (2011) Nkx2.2 repressor complex regulates islet beta-cell specification and prevents beta-to-alpha-cell reprogramming. Genes Dev 25: 2291–2305.
15. DasenJS, TiceBC, Brenner-MortonS, JessellTM (2005) A Hox regulatory network establishes motor neuron pool identity and target-muscle connectivity. Cell 123: 477–491.
16. KitamuraT, KitamuraYI, FunahashiY, ShawberCJ, CastrillonDH, et al. (2007) A Foxo/Notch pathway controls myogenic differentiation and fiber type specification. J Clin Invest 117: 2477–2485.
17. LiH, de FariaJP, AndrewP, NitarskaJ, RichardsonWD (2011) Phosphorylation regulates OLIG2 cofactor choice and the motor neuron-oligodendrocyte fate switch. Neuron 69: 918–929.
18. MeadowsSM, MyersCT, KriegPA (2011) Regulation of endothelial cell development by ETS transcription factors. Semin Cell Dev Biol 22: 976–984.
19. CollombatP, MansouriA, Hecksher-SorensenJ, SerupP, KrullJ, et al. (2003) Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev 17: 2591–2603.
20. MellitzerG, BonneS, LucoRF, Van De CasteeleM, Lenne-SamuelN, et al. (2006) IA1 is NGN3-dependent and essential for differentiation of the endocrine pancreas. Embo J 25: 1344–1352.
21. SchafferAE, FreudeKK, NelsonSB, SanderM (2010) Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell 18: 1022–1029.
22. CollombatP, Hecksher-SorensenJ, BroccoliV, KrullJ, PonteI, et al. (2005) The simultaneous loss of Arx and Pax4 genes promotes a somatostatin-producing cell fate specification at the expense of the alpha- and beta-cell lineages in the mouse endocrine pancreas. Development 132: 2969–2980.
23. KordowichS, CollombatP, MansouriA, SerupP (2011) Arx and Nkx2.2 compound deficiency redirects pancreatic alpha- and beta-cell differentiation to a somatostatin/ghrelin co-expressing cell lineage. BMC Dev Biol 11: 52.
24. MastracciTL, WilcoxCL, PaneaC, GoldenJA, MayCL, et al. (2011) Nkx2.2 and Arx genetically interact to regulate pancreatic endocrine cell development and endocrine hormone expression. Dev Biol 359: 1–11.
25. ChaoCS, LoomisZL, LeeJE, SusselL (2007) Genetic identification of a novel NeuroD1 function in the early differentiation of islet alpha, PP and epsilon cells. Dev Biol 312: 523–532.
26. NayaFJ, HuangHP, QiuY, MutohH, DeMayoFJ, et al. (1997) Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev 11: 2323–2334.
27. PradoCL, Pugh-BernardAE, ElghaziL, Sosa-PinedaB, SusselL (2004) Ghrelin cells replace insulin-producing beta cells in two mouse models of pancreas development. Proc Natl Acad Sci U S A 101: 2924–2929.
28. AndersonKR, TorresCA, SolomonK, BeckerTC, NewgardCB, et al. (2009) Cooperative transcriptional regulation of the essential pancreatic islet gene NeuroD1 (beta2) by Nkx2.2 and neurogenin 3. J Biol Chem 284: 31236–31248.
29. HingoraniSR, PetricoinEF, MaitraA, RajapakseV, KingC, et al. (2003) Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4: 437–450.
30. GuC, SteinGH, PanN, GoebbelsS, HornbergH, et al. (2011) Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab 11: 298–310.
31. SchonhoffSE, Giel-MoloneyM, LeiterAB (2004) Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev Biol 270: 443–454.
32. HerreraPL (2000) Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 127: 2317–2322.
33. GasaR, MrejenC, LynnFC, Skewes-CoxP, SanchezL, et al. (2008) Induction of pancreatic islet cell differentiation by the neurogenin-neuroD cascade. Differentiation 76: 381–391.
34. HuangHP, LiuM, El-HodiriHM, ChuK, JamrichM, et al. (2000) Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Mol Cell Biol 20: 3292–3307.
35. MiyataT, MaedaT, LeeJE (1999) NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev 13: 1647–1652.
36. Pictet R, Rutter WJ (1972) Development of the embryonic endocrine pancreas; Steiner DF, Frenkel N, editors. Washington, DC: Williams and Wilkins. 25–66 p.
37. SeymourPA, FreudeKK, DuboisCL, ShihHP, PatelNA, et al. (2008) A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev Biol 323: 19–30.
38. PanFC, WrightC (2011) Pancreas organogenesis: from bud to plexus to gland. Dev Dyn 240: 530–565.
39. ThorelF, NepoteV, AvrilI, KohnoK, DesgrazR, et al. (2010) Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 464: 1149–1154.
40. MastracciTL, SusselL (2012) The Endocrine Pancreas: insights into development, differentiation and diabetes. WIREs Dev Biol doi:10.1002/wdev.44.
41. WangS, Hecksher-SorensenJ, XuY, ZhaoA, DorY, et al. (2008) Myt1 and Ngn3 form a feed-forward expression loop to promote endocrine islet cell differentiation. Dev Biol 317: 531–540.
42. GoebbelsS, BodeU, PieperA, FunfschillingU, SchwabMH, et al. (2005) Cre/loxP-mediated inactivation of the bHLH transcription factor gene NeuroD/BETA2. Genesis 42: 247–252.
43. MadisenL, ZwingmanTA, SunkinSM, OhSW, ZariwalaHA, et al. (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140.
44. SorianoP (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21: 70–71.
45. LarssonLI, MadsenOD, SerupP, JonssonJ, EdlundH (1996) Pancreatic-duodenal homeobox 1 -role in gastric endocrine patterning. Mech Dev 60: 175–184.
46. OffieldMF, JettonTL, LaboskyPA, RayM, SteinRW, et al. (1996) PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122: 983–995.
47. XuanS, BorokMJ, DeckerKJ, BattleMA, DuncanSA, et al. (2012) Pancreas-specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. J Clin Invest 122: 3516–3528.
48. TutejaG, JensenST, WhiteP, KaestnerKH (2008) Cis-regulatory modules in the mammalian liver: composition depends on strength of Foxa2 consensus site. Nucleic Acids Res 36: 4149–4157.
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2013 Číslo 2
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- Complex Inheritance of Melanoma and Pigmentation of Coat and Skin in Grey Horses
- Coordination of Chromatid Separation and Spindle Elongation by Antagonistic Activities of Mitotic and S-Phase CDKs
- Autophagy Induction Is a Tor- and Tp53-Independent Cell Survival Response in a Zebrafish Model of Disrupted Ribosome Biogenesis
- Assembly of the Auditory Circuitry by a Genetic Network in the Mouse Brainstem