
Pneumolysin Activates the NLRP3 Inflammasome and Promotes Proinflammatory Cytokines Independently of TLR4
Pneumolysin (PLY) is a key Streptococcus pneumoniae virulence factor and potential candidate for inclusion in pneumococcal subunit vaccines. Dendritic cells (DC) play a key role in the initiation and instruction of adaptive immunity, but the effects of PLY on DC have not been widely investigated. Endotoxin-free PLY enhanced costimulatory molecule expression on DC but did not induce cytokine secretion. These effects have functional significance as adoptive transfer of DC exposed to PLY and antigen resulted in stronger antigen-specific T cell proliferation than transfer of DC exposed to antigen alone. PLY synergized with TLR agonists to enhance secretion of the proinflammatory cytokines IL-12, IL-23, IL-6, IL-1β, IL-1α and TNF-α by DC and enhanced cytokines including IL-17A and IFN-γ by splenocytes. PLY-induced DC maturation and cytokine secretion by DC and splenocytes was TLR4-independent. Both IL-17A and IFN-γ are required for protective immunity to pneumococcal infection and intranasal infection of mice with PLY-deficient pneumococci induced significantly less IFN-γ and IL-17A in the lungs compared to infection with wild-type bacteria. IL-1β plays a key role in promoting IL-17A and was previously shown to mediate protection against pneumococcal infection. The enhancement of IL-1β secretion by whole live S. pneumoniae and by PLY in DC required NLRP3, identifying PLY as a novel NLRP3 inflammasome activator. Furthermore, NLRP3 was required for protective immunity against respiratory infection with S. pneumoniae. These results add significantly to our understanding of the interactions between PLY and the immune system.
Published in the journal:
Pneumolysin Activates the NLRP3 Inflammasome and Promotes Proinflammatory Cytokines Independently of TLR4. PLoS Pathog 6(11): e32767. doi:10.1371/journal.ppat.1001191
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001191
Summary
Pneumolysin (PLY) is a key Streptococcus pneumoniae virulence factor and potential candidate for inclusion in pneumococcal subunit vaccines. Dendritic cells (DC) play a key role in the initiation and instruction of adaptive immunity, but the effects of PLY on DC have not been widely investigated. Endotoxin-free PLY enhanced costimulatory molecule expression on DC but did not induce cytokine secretion. These effects have functional significance as adoptive transfer of DC exposed to PLY and antigen resulted in stronger antigen-specific T cell proliferation than transfer of DC exposed to antigen alone. PLY synergized with TLR agonists to enhance secretion of the proinflammatory cytokines IL-12, IL-23, IL-6, IL-1β, IL-1α and TNF-α by DC and enhanced cytokines including IL-17A and IFN-γ by splenocytes. PLY-induced DC maturation and cytokine secretion by DC and splenocytes was TLR4-independent. Both IL-17A and IFN-γ are required for protective immunity to pneumococcal infection and intranasal infection of mice with PLY-deficient pneumococci induced significantly less IFN-γ and IL-17A in the lungs compared to infection with wild-type bacteria. IL-1β plays a key role in promoting IL-17A and was previously shown to mediate protection against pneumococcal infection. The enhancement of IL-1β secretion by whole live S. pneumoniae and by PLY in DC required NLRP3, identifying PLY as a novel NLRP3 inflammasome activator. Furthermore, NLRP3 was required for protective immunity against respiratory infection with S. pneumoniae. These results add significantly to our understanding of the interactions between PLY and the immune system.
Introduction
Streptococcus pneumoniae is responsible for millions of deaths annually from pneumonia, meningitis and septicaemia while also causing other less serious infections, such as otitis media and sinusitis. Pneumolysin (PLY) is a major virulence factor that is expressed by virtually all clinical isolates of the bacterium. The toxin is a member of the cholesterol-dependent cytolysins, a family that includes perfringolysin O and streptolysin O, expressed by Clostridium perfringens and Streptococcus pyogenes, respectively. A classical feature of these toxins is their ability to create transmembrane pores in cholesterol-containing membranes and thereby cause cell lysis (reviewed in [1]).
The importance of PLY as a pneumococcal virulence factor is well established, with several studies showing reduced pathogenesis in mice infected with PLY-deficient strains of S. pneumoniae, compared to isogenic toxin-producing strains [2], [3], [4], [5]. Furthermore, application of PLY directly into the lungs of rats induced an acute inflammatory response similar to that observed during pneumococcal pneumonia [6]. At sublytic concentrations, the toxin has been reported to promote activation of host complement [7], potentiation of neutrophil activity [8], [9], activation and chemotaxis of CD4+ T cells [10] and enhanced production of proinflammatory cytokines in macrophages and monocytes [11], [12]. Studies have been carried out to address the mechanisms underlying the immunomodulatory effects of PLY and particularly the role of TLRs. It has been proposed that the ability of PLY to induce cytokine production [13] and apoptosis [14] in peritoneal macrophages was TLR4-dependent. In contrast, the activation of p38 mitogen-activated protein kinase in epithelial cells [15] and the activation of nuclear factor of activated T cells (NFAT) [16] by PLY were reported to be TLR4-independent. Furthermore, there are conflicting reports on the role of TLR4 in defence against pneumococcal infection. TLR4 was reported to be required for host defence against PLY-producing pneumococci, as mice lacking functional TLR4 were more susceptible to disease after nasopharyngeal challenge [13]. However, other studies have shown a more limited or indeed a redundant role for TLR4 in host resistance to pneumococcal disease, depending on the bacterial dose and the model of infection [17], [18]. Thus, the role of TLR4 in immune responses to the pneumococcus, and particularly to PLY, is unclear and must be resolved.
T cells play a key role in protection against pneumococcal infection [4], [10] so it is important to determine the factors underlying pneumococcus-induced T cell responses. In this regard it is noteworthy that studies in IFN-γ−/− mice indicate a protective role for IFN-γ during bacteremic pneumococcal pneumonia [19] and administration of recombinant IFN-γ to mice promoted protection from disease following intratracheal infection with S. pneumoniae [20]. Furthermore, an essential role was proposed for IL-17A in protection against pneumococcal nasopharyngeal colonization following intranasal immunization of mice with killed pneumococci and cholera toxin adjuvant, as protection was abrogated in mice deficient in the IL-17A receptor [21]. Thus, there is a strong rationale for the development of vaccination approaches that induce IFN-γ- and IL-17A-producing cells and for understanding the mechanisms by which pneumococci may either promote or evade such responses.
The activation and differentiation of naïve CD4+ T cells following immunization or infection depends on interactions with DC [22]. T cell activation requires antigen presentation on MHC class II molecules, as well as costimulatory signals provided by molecules including CD80 and CD86 on the DC surface. The differentiation of Th1 and Th17 cells requires polarizing cytokines which can be produced by DC. IL-12 and IL-18 are two of the key cytokines involved in Th1 cell differentiation, while IL-23, IL-1 and IL-6 promote Th17 cell development. To date, little is known about the interaction of PLY with DC. Therefore, in this study the effects of PLY on DC maturation and cytokine production and the role of TLR4 in these processes were determined. In addition to the key role of DC in dictating T cell differentiation and polarization, important roles for natural killer (NK) cells and γδ T cells have also been described. In particular, IFN-γ produced by NK cells plays a key role in the instruction of Th1 responses [23], while IL-17A derived from γδ T cells promotes Th17 responses [24].
We demonstrate that endotoxin-free PLY alone does not induce cytokine production by DC or macrophages but it can synergize with TLR agonists to enhance cytokine secretion. Furthermore, PLY promotes the secretion of cytokines including IFN-γ and IL-17A by splenocytes and is essential for S. pneumoniae to promote the production of IFN-γ by NK cells and IL-17A by γδ T cells in the lung following respiratory infection. In contrast to previous publications showing a role for TLR4 in PLY-induced immune responses, we show that the ability of the toxin to enhance cytokine secretion does not require TLR4. However, NLRP3 activation was required for PLY- and live S. pneumoniae-mediated enhancement of IL-1β secretion and NLRP3 was required for protection against respiratory infection with S. pneumoniae.
Results
PLY induces TLR4-independent secretion of proinflammatory cytokines by splenocytes
IFN-γ and IL-17 responses play an essential role in protective immunity against pneumococcal disease [19], [20], [21]. We therefore investigated the ability of PLY to promote these cytokines in vitro and the requirement for TLR4. In the absence of any stimulation with PMA or anti-CD3, PLY alone was unable to promote cytokine secretion by splenocytes (data not shown). However, we did find significant enhancing effects of PLY on IFN-γ secretion by splenocytes stimulated with heat-killed S. pneumoniae (HkSp) and PMA/ionomycin (Fig. 1A). This was true for spleen cells from both C3H/HeN and C3H/HeJ mice, indicating that the combination of PLY and HkSp is a potent stimulus for TLR4-independent IFN-γ secretion in spleen cells (Fig. 1A). The IFN-γ inducing ability of the toxin was not dependent on its cytolytic activity as a mutant of PLY (W433F) with greatly reduced cytotoxic activity was also capable of stimulating IFN-γ production by splenocytes (Fig. 1A). Indeed, concentrations of W433F from 0.32–200 ng/ml enhanced pneumococci-induced IFN-γ production, whereas the wild-type toxin only significantly augmented IFN-γ secretion at a concentration of 0.32 ng/ml (Fig. 1A). The pore-forming activity of the toxin may, therefore, interfere with its ability to stimulate IFN-γ production in splenocytes.

Furthermore, in the presence of anti-CD3, PLY significantly augmented IFN-γ secretion by splenocytes (Fig. 1B). We also examined the ability of PLY to induce the secretion of other cytokines by anti-CD3-stimulated splenocytes. At a concentration of 1.6 ng/ml, PLY significantly enhanced IL-5 (3-fold; P<0.05), IL-10 (3-fold; P<0.05) and IL-17A (2-fold; P<0.05), in addition to IFN-γ (14-fold; P<0.001), secretion by splenocytes from TLR4 hyporesponsive C3H/HeJ mice (Fig. 1B). These data demonstrate that PLY induces cytokine secretion by spleen cells in a TLR4-independent manner. PLY at 1.6 ng/ml was more effective at promoting cytokine secretion than at 40 ng/ml (Fig. 1B). However, a concentration of 40 ng/ml PLY may induce early apoptosis in splenocytes, as dual staining of these cells with AnnexinV and propidium iodide (PI) revealed slight increases in AnnexinV+/PI- cells following incubation with the toxin for 6 hours (Fig. S1A). These increases were more apparent following stimulation with 200 ng/ml PLY (Fig. S1A). In contrast, when splenocytes were incubated with 200 ng/ml PLY for 24 hours or 96 hours there was no increase in AnnexinV+/PI- cells (data not shown), although an increase in the percentage of PI+ cells was evident (Fig. S1B and S1C). Higher concentrations of PLY (1–5 µg/ml) induced the death of >25% of splenocytes from either C3H/HeN or C3H/HeJ mice after 24 hours (Fig. S1B) and this increased to >50% by 96 hours (Fig. S1C).
PLY is required for the induction of IFN-γ and IL-17A responses following pneumococcal infection
Having shown that PLY enhanced the secretion of cytokines, including IFN-γ and IL-17A by splenocytes stimulated in vitro (Fig. 1), we next determined the contribution of PLY to pneumococcus-induced cytokine production using two different murine models of pneumococcal infection in vivo. Using a model of acute pneumonia [4], we found that infection with a PLY-deficient strain of pneumococcus (PLN-A) induced significantly less IFN-γ in the lungs of mice compared to infection with its PLY-positive parental strain (Fig. 2A). As has been demonstrated previously in this pneumonia model [3], [4], [5], the PLY-deficient strain exhibited reduced virulence in the lungs compared to the wild-type strain (Fig. S2A). Furthermore, using a model of resolving pneumonia [25], [26], we found significantly lower concentrations of IL-17A in the lungs of mice infected with PLN-A compared to those infected with wild-type pneumococci, at both 24 and 48 hours post-infection (Fig. 2B). There were no significant differences in bacterial CFU at time 0 in the lungs of mice infected with wild-type bacteria compared to PLY-deficient bacteria and lung CFU of both pneumococcal strains were reduced at 24 and 48 hours post-infection (Fig. S2B).

We then investigated the cellular source of these cytokines by intracellular cytokine staining. NK cells were the principal source of IFN-γ in the lungs 48 hours following infection, although IFN-γ was also produced by γδ T cells and other T cells (Fig. 2C and 2E). In the case of IL-17A, the enhanced cytokine production was predominately by γδ T cells with limited IL-17A production in other T cells and NK cells (Fig. 2D and 2E).
PLY promotes dendritic cell maturation but not cytokine production
Having shown that PLY promotes IFN-γ and IL-17A production in vitro and in vivo, we next established the effects of the toxin on DC. DC play an important role in the activation and polarization of naïve T cells but to date the effects of PLY on DC have not been widely investigated. We therefore examined the effects of PLY on DC cytokine production and costimulatory molecule expression.
While the TLR2 agonist PAM3Csk4 promoted the secretion of proinflammatory cytokines from DC, endotoxin-free PLY, at concentrations of up to 1 µg/ml, did not induce the secretion of IL-6, IL-12p40 or TNF-α (Fig. 3A). Higher concentrations were not tested because PLY at 2 µg/ml caused some cell death in DC, as determined by PI staining (Fig. S3A). Indeed, concentrations of PLY ranging from 3 µg/ml to 6 µg/ml were highly toxic to DC, with PLY at 6 µg/ml inducing death in approximately 90% of DC from either C3H/HeN or C3H/HeJ mice after 24 hours (Fig. S3A and S3B). AnnexinV/PI staining of DC incubated with this higher concentration of the toxin for 6 hours indicated that PLY induced limited apoptosis (AnnexinV+/PI−), but greatly increased the percentage of dead AnnexinV+/PI+ cells (Fig. S3C).

Since previous studies demonstrated that PLY could directly induce cytokine production in monocytes and macrophages [12], [13], it was important to establish if the finding that endotoxin-free PLY does not promote cytokine secretion was specific to DC. Endotoxin-free PLY was also unable to induce secretion of significant concentrations of IL-6, TNF-α, IL-12p40 (Fig. 3B), IL-1β or IL-23 (data not shown) from murine bone-marrow derived macrophages (BMDM). These data indicate that PLY alone does not stimulate inflammatory cytokine production by DC or macrophages.
Since co-stimulation is required for DC to promote T cell activation, we assessed the effects of PLY on the expression of co-stimulatory molecules and MHC class II on DC and compared these effects to those induced by the TLR4 agonist LPS. Stimulation of DC with endotoxin-free PLY enhanced the expression of MHC class II and co-stimulatory molecules, particularly CD86. Interestingly, similar findings were observed with PLY-stimulated DC from both C3H/HeN and LPS-hyporesponsive C3H/HeJ mice, indicating that the enhancement of DC maturation by the toxin was TLR4-independent. In contrast, LPS strongly enhanced the expression of co-stimulatory molecules on DC from C3H/HeN mice only (Fig. 3C). Therefore, although endotoxin-free PLY does not induce cytokine secretion by DC, it can promote their maturation independently of TLR4.
To determine the functional significance of the direct effects of PLY on DC, cells were incubated with antigen (KLH) alone or KLH and PLY overnight and injected into mice. The exposure of DC to PLY increased their ability to promote antigen-specific T cell proliferation in splenocytes 7 days following DC transfer (Fig. 3D). We did not detect enhanced antigen-specific cytokine production by these cells (data not presented). However, the T cell proliferation data indicate that PLY can act directly on DC to enhance their T cell stimulatory activity.
Since exposure of DC to PLY enhanced their ability to promote adaptive immune responses, we determined the adjuvant properties of endotoxin-free PLY when co-injected with KLH. Injection of PLY with KLH did not enhance antigen-specific cellular immune responses (Fig. S4A). In contrast, antigen-specific antibody responses were significantly enhanced by co-immunization with antigen and PLY (Fig. S4B and S4C). The ability of PLY to promote antigen-specific antibody responses applied to both IgG1 and IgG2a subclasses and the toxin was equally effective in C3H/HeN and C3H/HeJ mice. Thus, the toxin has the ability to enhance antigen-specific immune responses independently of TLR4 signalling.
PLY synergizes with TLR agonists to enhance secretion of pro-inflammatory cytokines
Although PLY alone did not induce cytokine production by DC or macrophages, we investigated if the toxin could synergize with other stimuli to enhance cytokine secretion. We found that PLY (1 µg/ml) synergized with HkSp to significantly enhance the secretion of IL-6, IL-12p40, IL-23 and TNF-α by DC (Fig. 4A). Notably, this synergistic effect was observed in DC from both C3H/HeN and C3H/HeJ mice, indicating that the enhancement of cytokine secretion by PLY did not require the presence of functional TLR4. Furthermore, PLY was also able to synergize with HkSp to significantly enhance IL-6 and TNF-α production in BMDM (Fig. S5). PLY alone did not induce significant secretion of IL-1α or IL-1β by DC (Fig. 4B) or BMDM (data not shown). However, PLY synergized with HkSp to significantly enhance IL-1β secretion by DC (Fig. 4B). Importantly, the ability of PLY to promote IL-1β secretion was not limited to pneumococci as the toxin also significantly enhanced IL-1β secretion in response to a range of TLR and NLR agonists; PAM3Csk (TLR1/2 ligand), zymosan (TLR2/6 ligand), CpG (TLR9 ligand) and MDP (Nod2 ligand). Similarly, IL-1α secretion was significantly enhanced in DC stimulated with these TLR/NLR agonists and PLY compared to the agonists alone. Furthermore, by using DC from C3H/HeJ mice, we showed that PLY-induced enhancement of IL-1α and IL-1β secretion by TLR agonists was independent of TLR4 (Fig. 4B).
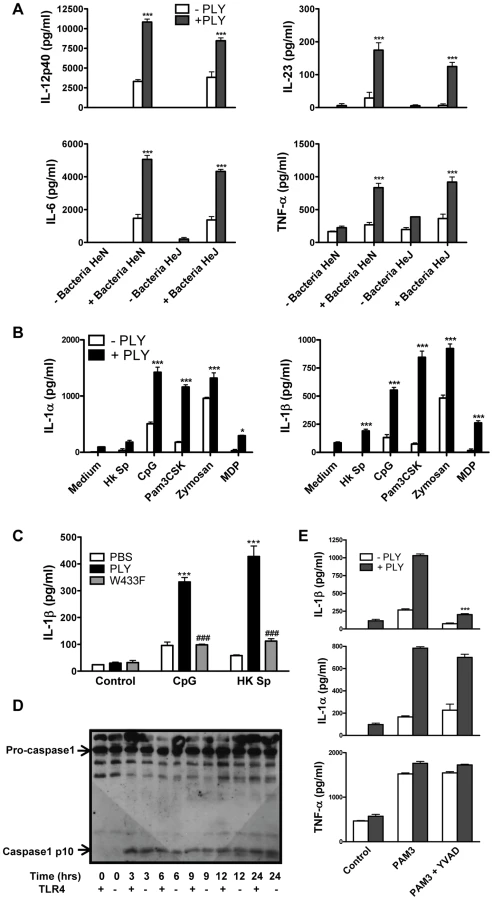
PLY activates the NLRP3 inflammasome
Since it has been shown that IL-1β is required for resistance to pneumococcal infection [27], we investigated the mechanism underlying PLY-induced IL-1β further. Firstly, to determine the importance of haemolytic activity in the promotion of IL-1β by PLY, DC were incubated with HkSp or CpG in the presence or absence of PLY or the W433F hemolysin mutant. PLY-driven IL-1β secretion was hemolysin-dependent as the ability of W433F to promote secretion of the cytokine was significantly less than wild-type PLY (Fig. 4C).
IL-1β is synthesised inside cells as a biologically inactive precursor that is processed by caspase-1 prior to its secretion. Caspase-1 is itself produced as an inactive zymogen that must be cleaved to generate the active p10 and p20 subunits. Since the secretion of IL-1β requires both the transcription of IL-1β and caspase-1 activation, we investigated how PLY promoted IL-1β secretion in DC. We found that PLY alone promoted caspase-1 activation in DC and that this was independent of TLR4, as processed (p10) caspase-1 was detected in DC lysates from both C3H/HeN and C3H/HeJ mice (Fig. 4D). Pre-treatment of DC with the caspase-1 inhibitor YVAD-fmk completely blocked the synergy between PLY and PAM3Csk for IL-1β secretion (Fig. 4E), confirming the importance of caspase-1 in PLY-induced IL-1β secretion. In contrast, the enhancement of IL-1α secretion by the toxin was not significantly affected, indicating that caspase-1 was not required (Fig. 4E). The importance of caspase-1 in PLY-induced IL-1β secretion was also confirmed using DC from caspase-1 knockout mice, as the synergy between PLY and either PAM3Csk or HkSp for IL-1β was dramatically reduced in caspase-1−/− DC compared to wild-type DC (Fig. 5A).

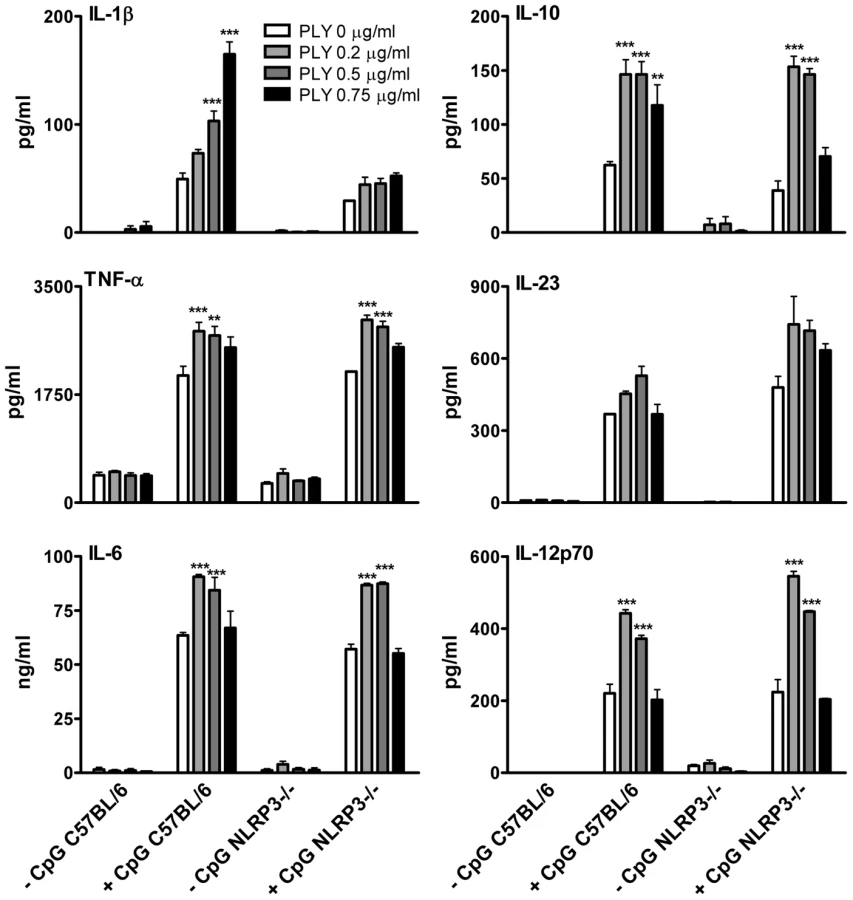
The activation of caspase-1 is regulated by the assembly of inflammasomes, cytoplasmic multiprotein complexes that contain a nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family member, such as NLRP3 or NLRC4 and caspase-1 [28]. We investigated the role of the NLRP3 inflammasome in PLY-driven IL-1β secretion and found that the ability of PLY to promote IL-1β secretion was compromised in cells from NLRP3−/− mice compared to wild-type mice (Fig. 5B), but not in DC from mice deficient in NLRP6 or NLRP12 (Fig. S6). In particular, the synergy between PLY and HkSp for IL-1β secretion was abrogated in DC from NLRP3−/− mice (Fig. 5B). This marked defect in the ability of NLRP3−/− DC to secrete IL-1β in response to stimulation with PLY was due to differences in IL-1β processing, as Western blot analysis did not reveal altered proIL-1β production in DC from NLRP3−/− mice compared to DC from wild-type mice (Fig. S7). In contrast to IL-1β secretion, PLY significantly enhanced bacteria-induced IL-1α secretion in DC from both wild-type and NLRP3−/− mice (Fig. 5B). The adapter protein ASC (apoptosis-associated speck like protein containing CARD) is necessary for caspase-1 activation via NLRP3. PLY-mediated enhancement of IL-1β secretion was also reduced in DC from ASC−/− mice, compared to wild-type mice (Fig. S8). DC from caspase-1−/− (Fig. 5A) or NLRP3−/− (Fig. 5B) mice secreted comparable concentrations of TNF-α to wild-type mice when stimulated with PLY and TLR agonist, indicating that the ability of these cells to produce inflammatory cytokines was not globally compromised. Indeed, the ability of PLY to promote the secretion of TNF-α, IL-6, IL-23, IL-12 and IL-10 was independent of NLRP3 (Fig. 6). CpG was used as the TLR for these experiments as it was the most effective TLR agonist to determine synergy with PLY for this panel of cytokines, particularly IL-12p70. The IL-10 data indicate that PLY can promote secretion of both pro- and anti-inflammatory cytokines, although the overall profile suggests that PLY is a highly pro-inflammatory factor.
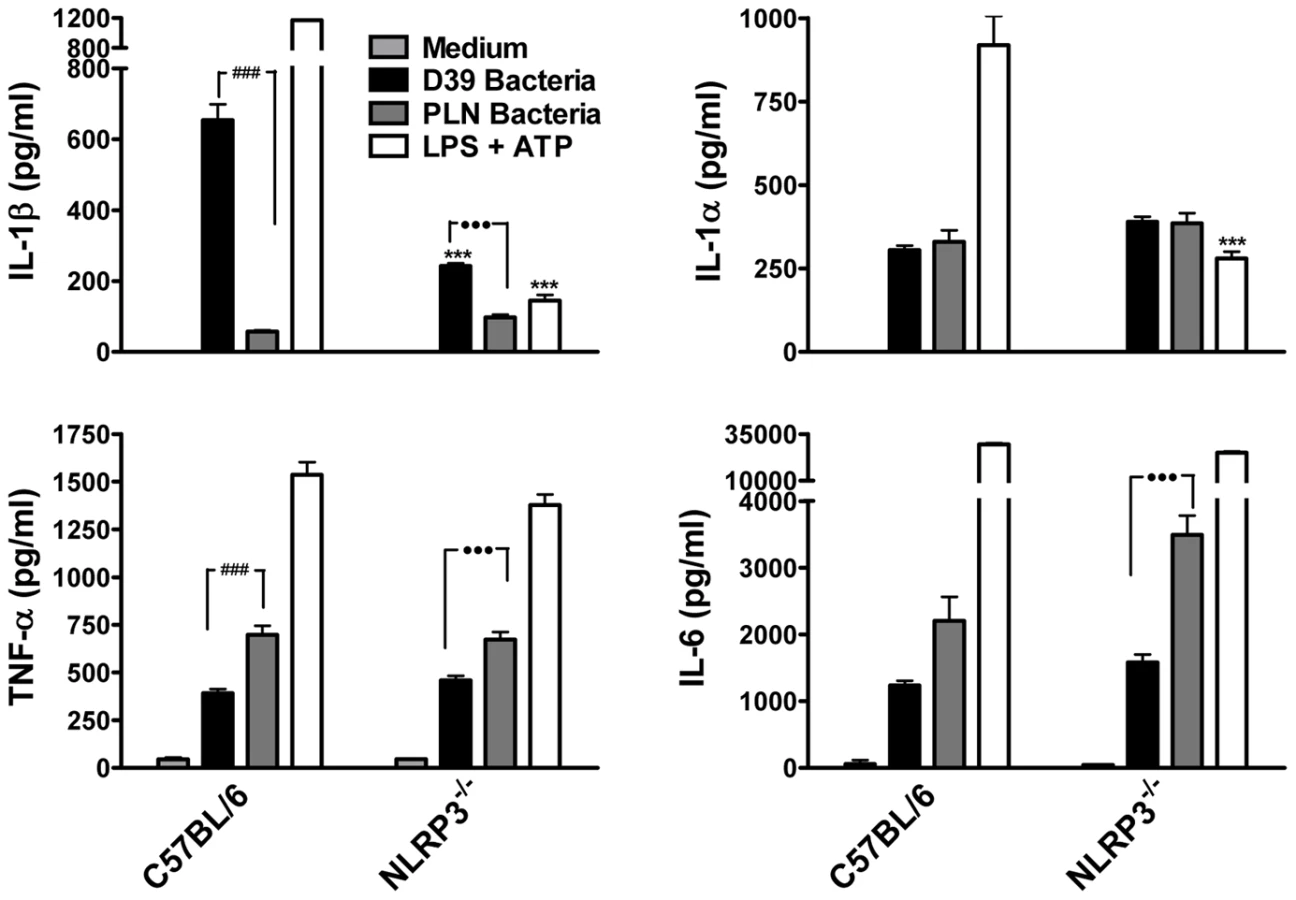
Having demonstrated that purified endotoxin-free PLY can promote IL-1β secretion via the NLRP3 inflammasome, we next investigated the role of NLRP3 in IL-1β production by DC in response to live S. pneumoniae. Wild-type S. pneumoniae induced robust IL-1β secretion by DC and this was dependent on PLY since the PLY-deficient strain induced very little IL-1β secretion (Fig. 7). In contrast, IL-1α, TNF-α and IL-6 secretion in response to the PLY-deficient strain was comparable to, or greater than, that induced by the wild-type bacteria. Thus, PLY appears to play a vital and selective role in pneumococcus-induced IL-1β secretion by DC. Furthermore, the induction of IL-1β secretion by the pneumococcus was strongly dependent on NLRP3 (Fig. 7).
In order to further investigate the mechanisms by which PLY may trigger caspase-1 activation and IL-1β secretion in DC, we examined the involvement of some known factors implicated in NLRP3 inflammasome activation such as reduced cytoplasmic potassium concentration. It has been reported that the pore forming toxins, Staphylococcus aureus α-toxin [29] and Aeromonas hydrophilia aerolysin [30] induce the secretion of IL-1β and the activation of caspase-1, respectively, by allowing the efflux of intracellular potassium (K+). We therefore assessed whether K+ efflux could also be the trigger for caspase-1 activation by PLY. The addition of extracellular KCl to the medium (50 mM) significantly inhibited IL-1β secretion by DC in response to PLY and PAM3Csk (Fig. 8A). Interestingly, IL-1α secretion was also significantly reduced in the presence of high potassium concentrations (Fig. 8A).
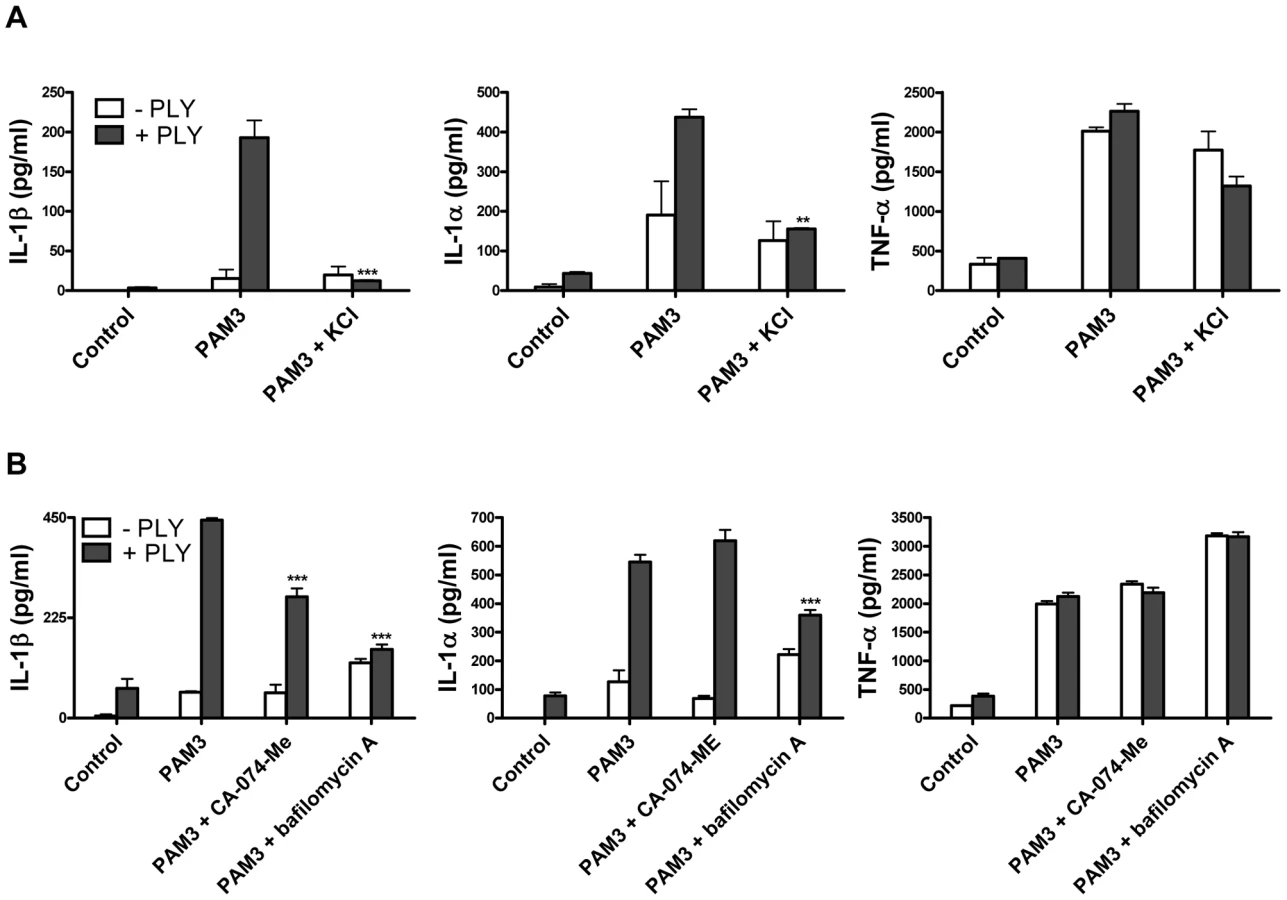
It has been proposed that the NLRP3 inflammasome may be activated by lysosomal damage and the subsequent release of cathepsin B into the cytoplasm of cells [31]. We investigated the effect of inhibiting cathepsin B on PLY-induced IL-1 secretion and found that inhibition with the specific cathepsin B inhibitor CA-074-Me resulted in a significant reduction in IL-1β, but not IL-1α, secretion by DC stimulated with PLY and PAM3CSk (Fig. 8B). Furthermore, treating DC with bafilomycin A, which inhibits the H+ ATPase system required for lysosomal acidification [32], significantly reduced the enhancement of IL-1β and IL-1α, but not TNF-α, by PLY (Fig. 8B).
The NLRP3 inflammasome is required for protection against S. pneumoniae infection
To investigate the significance of PLY and pneumococcal-induced inflammasome activation, NLRP3−/− and wild-type control C57BL/6 mice were intranasally infected with S. pneumoniae (strain D39). In contrast to control mice, which showed a significant reduction in lung CFU 24 hr post-infection, bacterial clearance was significantly compromised in NLRP3−/− mice and no reduction in bacterial CFU was evident in the lungs of these mice at this time-point (Fig. 9A).
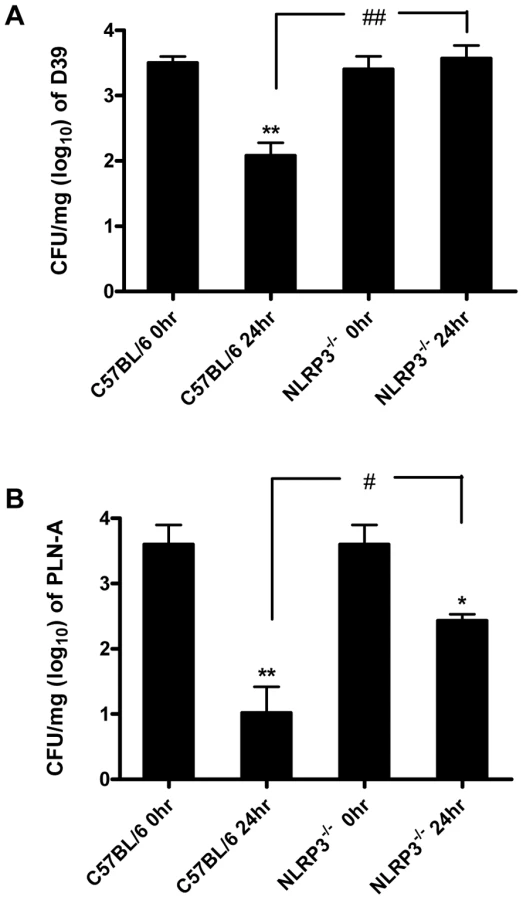
To determine the role of PLY, mice were also infected with a PLY-deficient isogenic mutant strain of D39 (PLN-A). Bacterial CFU of PLN-A in lungs of control C57BL/6 mice at 24 hr post-infection were significantly lower than in lungs of NLRP3−/− mice (Fig. 9B). In addition, although there were reductions in bacterial CFU of PLN-A in lungs of both NLRP3−/− and control mice over time, the reduction in control mice was significantly greater than observed in NLRP3−/− mice (Fig. 9B). Therefore, NLRP3 appears to play a particularly important role in infection with PLY-expressing S. pneumoniae, although the data with PLN-A suggest that even in the absence of PLY, NLRP3 may play a role in protective immunity.
These data indicate for the first time a role for NLRP3 in protection against S. pneumoniae infection and this is the first description of a role for NLRP3 in protection against a Gram-positive bacterial pathogen.
Discussion
In mice, the cytokines IL-1β [27], IFN-γ and IL-17A [19], [20], [21] have been shown to play important protective roles in immunity against pneumococci. Here we show that PLY strongly enhances the secretion of IFN-γ by splenocytes in vitro and is required for IFN-γ and IL-17A responses during pneumococcal infection in vivo. NK cells are the principal cellular source of IFN-γ in the lungs following infection while γδ T cells are the major producers of IL-17A. This supports recent findings that γδ T cells are a key source of IL-17A production in vitro and in vivo in response to IL-1 stimulation [24] and pathogen-derived products [33]. The latter study found that IL-17-producing γδ T cells share a number of characteristic features with Th17 cells. IL-1 and IL-23 play key roles in the induction of IL-17A production by γδ T cells and in the differentiation and expansion of Th17 cells [24]. Other cytokines, including IL-6 and TNF-α, have also been shown to be important in Th17 cell differentiation [34], [35], [36]. Importantly, our data show that PLY can synergize with TLR agonists to enhance the secretion of IL-1α, IL-1β, IL-23, TNF-α and IL-6 by DC, which can induce IL-17A production by γδ T cells and promote the differentiation and expansion of Th17 cells. The ability of Freund's complete adjuvant or LPS to induce antigen-specific Th17 cell responses in vivo requires IL-1R1, indicating a key role for IL-1 in adjuvant-driven Th17 cell responses [36].
Pneumolysin also strongly enhanced TLR agonist-induced IL-12 secretion, which together with cytokines such as IFN-γ and IL-18, is important for Th1 differentiation and stabilization [37]. Like IL-1β, active IL-18 also requires cleavage of its precursor form by caspase-1 and enhanced levels have been reported in macrophages co-stimulated with recombinant PLY and PLY-deficient S. pneumoniae [38]. Since IFN-γ produced by NK cells is important for the instruction of Th1 responses [23], our demonstration of NK cells producing IFN-γ in the lungs of infected mice could suggest subsequent promotion of pathogen-specific Th1 responses. Likewise, it has been suggested that IL-17A derived from γδ T cells can promote Th17 responses [24] and we demonstrate that there are IL-17A-producing γδ T cells in the lungs of mice infected with pneumococcus. Both of these effects were strongly dependent on pneumolysin, suggesting that the toxin is a key factor in pneumococcus-induced IFN-γ secretion by NK cells and IL-17A production by γδ T cells. Furthermore, since IL-23R signalling promotes the expansion and maintenance of γδ T cells in response to intracellular bacterial infection [39], the ability of pneumolysin to promote both IL-1 and IL-23 production is likely to contribute strongly to these γδ T cell responses.
During pneumococcal infection, PLY may synergize with pneumococcal or endogenous danger signals to induce the secretion of inflammatory cytokines. Reduced concentrations of plasma IL-6 have been reported from mice infected with PLY-deficient S. pneumoniae compared to isogenic wild-type pneumococci [40]. In addition, IL-1 and TNF-α are induced during murine pneumococcal disease [41] and elevated levels of both have been reported in patients with pneumococcal meningitis. Notably, studies using mice deficient in IL-1 receptor type 1 (IL-1R1−/−) [42] or IL-1β [27] have shown the importance of IL-1 in conferring resistance to pneumococcal meningitis and pneumonia respectively. The latter study found that IL-1β, but not IL-1α, plays a major role in resistance to pneumococcal infection. Therefore, the inflammasomes, which are required for IL-1β processing and secretion, are likely to be crucial components of the host defense to S. pneumoniae. Shoma et al. recently reported that recombinant PLY stimulated the secretion of IL-1α, IL-1β and IL-18 in a caspase-1 dependent manner in macrophages and that this was augmented by co-stimulation with pneumococci [38]. Importantly, we show here for the first time that the enhancement of IL-1β secretion by PLY is NLRP3-dependent. The ability of live pneumococci to promote IL-1β secretion by DC is also strongly dependent on NLRP3. One of the key observations in this study is that NLRP3 is required for protective immunity against pneumococcal infection. The first demonstration of a role for NLRP3 in defence against a Gram-negative pathogen, Salmonella typhimurium, was recently described [43], but our study is the first report to show that NLRP3 is required for protection against a Gram-positive bacterium.
NLRP3 activation by PLY required lysosomal damage and the release of cathepsin B, as inhibitors of these processes reduced PLY-induced IL-1β secretion. Phagosomal rupture has previously been shown to be important in NLRP3 inflammasome activation by other compounds including silica crystals, aluminium salts and microparticles [31], [32], [44] and a role for cathepsin B was recently reported in the promotion of IL-1β secretion by the pore-forming toxin tetanolysin O [45]. Furthermore, the ability of PLY to activate NLRP3 was dependent on the haemolytic activity of the toxin. Taken together, our data suggest that PLY-induced pore formation results in K+ efflux from DC and intracellular changes including lysosomal destabilization. The release of lysosomal products, such as cathepsin B, into the cytosol may promote the generation of danger signals, which are detected by NLRP3 or intermediary factors, resulting in inflammasome assembly and caspase-1 activation. Active caspase-1 could then process pro-IL-1β, which is generated in response to a second stimulus such as a TLR/NLR ligand, into mature IL-1β which is released from the cell.
Although a direct role for PLY in the induction of IL-1 or TNF-α during pneumococcal disease has not been established, the reduced inflammation and pathology in the lungs of mice infected with PLY-deficient pneumococci compared to wild type strains [4] suggests a diminished inflammatory cytokine response. In particular, lower levels of T cell infiltration and neutrophil recruitment into the lungs have been reported from mice infected with PLY-deficient S. pneumoniae compared to wild-type pneumococci [4]. TNF-α and IL-1β are two of the key cytokines involved in neutrophil recruitment in the lungs of mice infected intratracheally with wild-type S. pneumoniae [46]. Additionally, IL-17A plays a key role in neutrophil recruitment and its induction in vivo is strongly dependent on IL-1 [34], [36].
An additional major finding of this study is that the stimulatory effects of PLY on DC and splenocyte cytokine secretion are independent of TLR4. While we demonstrate that PLY can promote expression of costimulatory molecules and proinflammatory cytokines in DC, these effects are independent of TLR4. We also show that PLY exerts adjuvant effects, promoting antibody responses against a co-administered antigen in both wild-type and TLR4-defective mice, indicating that the immunostimulatory effects of PLY in vivo do not require TLR4. Therefore, further studies into the involvement of pathogen recognition receptors in sensing PLY in innate cells are warranted.
Streptococcus pneumoniae is a pathogen of significant clinical importance and understanding its interaction with the immune system is crucial. We propose that upon infection with S. pneumoniae, PLY, in synergy with pneumococcal PAMPs promotes the secretion of proinflammatory cytokines, particularly IL-1β, that promote an inflammatory response and mediate protective immunity. We identify PLY as a novel NLRP3 inflammasome activator and show that the ability of the live bacterium to promote IL-1β secretion is also strongly NLRP3-dependent. More importantly, NLRP3 is required for protective immunity against respiratory pneumococcal infection.
Materials and Methods
Ethics statement
Experiments on mice carried out at Trinity College Dublin were conducted under Irish Department of Health guidelines with ethical approval from the TCD ethics committee. Mice experiments carried out at the University of Leicester were done under UK Home Office guidelines with ethical approval from the UK Home Office.
Reagents
CpG ODN 1826 was from Oligos Etc. E. coli LPS, Serotype R515 was obtained from Alexis Biochemicals while all other TLR agonists were obtained from InvivoGen.
Recombinant PLY was expressed in E. coli and purified as previously described [47]. Unless otherwise stated the specific haemolytic activity of PLY was 100,000 HU/mg. The toxin was passed three times through an EndoTrap endotoxin removal column (Profos AG, Germany) after which LPS was undetectable using the PyroGene Recombinant Factor C assay (Lonza; detection limit 0.01 EU/ml). S. pneumoniae serotype 2, strain D39 (NCTC 7466), was obtained from the National Collection of Type Culture, London, UK. The pneumolysin-negative mutant, PLN-A, was made by insertion duplication mutagenesis as described previously [2]. Heat-killed S. pneumoniae (HkSp) D39 was obtained by boiling 1×108 CFU in phosphate buffered saline (PBS) for 20 minutes and checking for viability by colony counts and plate streaking. PLY W433F was a gift from The Netherlands Vaccine Institute.
Mice for culture of macrophages, DC or splenocytes
Female BALB/c, C57BL/6, C3H/HeN and C3H/HeJ mice were obtained from Harlan Olac (UK) and were used at 9–16 weeks old. NLRP3−/− mice were bred in the Bioresources Unit in Trinity College Dublin. Animals were maintained according to the regulations of the EU and the UK or the Irish Department of Health as appropriate. Caspase1−/− DC were kindly provided by Dr. Katherine Fitzgerald (UMass, USA).
Culture of DC and BMDM
DC and BMDM were prepared by culturing murine bone marrow cells using protocols adapted from Lutz et al. [48] and Davies et al. [49]. Briefly, bone marrow cells were flushed aseptically from the femurs and tibia of mice. For culture of DC, cells were grown in RPMI 1640 medium (Biosera) containing 8% v/v fetal calf serum (FCS; Biosera), 100 U/ml penicillin, 100 µg/ml streptomycin, and 100 mM L-glutamine (Gibco) and supplemented with supernatant from a granulocyte-macrophage colony-stimulating factor (GM-CSF)-expressing cell line (final concentration of 20 ng/ml GM-CSF). Macrophages were grown in Dulbecco's Modified Eagle's Medium containing 20 ng/ml M-CSF. Cultures were maintained in a humidified atmosphere (5% CO2) at 37°C, and medium was replaced on days 3, 6 and 8 for DC or days 2 and 4 for BMDM. On day 6 (BMDM) or day 10 (DC) of culture, cells were plated and stimulated with PLY and/or TLR agonists 24 hours later.
Effect of PLY on DC maturation and cytokine production
DC (6.25×105/ml) were cultured at 37°C for 24 h with medium, PLY or W433F alone, TLR and NLR agonists (PAM3Csk4, LPS, zymosan, MDP, CpG) alone, HkSp alone or PLY or W433F and TLR agonists or HkSp together. In certain experiments, DC were incubated with YVAD-fmk (Bachem), KCl (Sigma), CA-074-ME (Sigma) or Bafilomycin A (Sigma) 30 mins before the addition of PLY and/or TLR agonists. At the end of the incubation, supernatants were removed and cytokine concentrations were determined by ELISA using pairs of antibodies purchased from BD Biosciences (IL-6 and IL-12p40) or R&D Systems (IL-1α, IL-1β, TNF-α, IL-12p70, IL-10 or IL-23) according to the manufacturer's specifications. Alternatively, cells were recovered and used for immunofluorescence analysis or Western Blotting. The expression of DC surface markers was assessed using fluorochrome-labelled antibodies against murine CD80, CD86, I-A/I-E, CD11c and CD40 (BD Biosciences). After blocking for 10 minutes on ice with anti-mouse CD16/CD32 (BD Biosciences) followed by incubation with antibodies for 30 minutes on ice, cells were washed and immunofluorescence analysis was performed on a CyAN (Dako) flow cytometer using FlowJo software (Tree Star). For Western blot analysis, cells were lysed in Laemmli sample buffer, samples were boiled and proteins separated by SDS/PAGE. Caspase-1 p10 was detected as previously described [44].
Effect of S. pneumoniae on DC cytokine production
DC (6.25×105/ml) from C57BL/6 and NLRP3−/− mice were cultured at 37°C in RPMI 1640 medium (Biosera) containing 8% v/v FCS (Biosera) and 100 mM L-glutamine (Gibco) in the absence of antibiotics. Cells were stimulated with PBS or with wild-type (D39; 10 bacteria:1 DC) or PLY-deficient S. pneumoniae (PLN; 10 bacteria:1 DC) in PBS. Following 24 h incubation, supernatants were removed and analysed for cytokine production by ELISA.
Adoptive transfer of PLY and antigen-stimulated DC
DC (1×106/ml) were incubated overnight with medium alone or with KLH (10 µg/ml) with or without PLY (1 µg/ml). After extensive washing, cells (1×105/mouse) were injected subcutaneously into the footpads of mice. After 7 days, splenocytes were isolated and resuspended at 2×106 cells/ml in RPMI medium, supplemented with 8% v/v FCS. Cells were cultured in triplicate with KLH (2 or 50 µg/ml) or with medium only as a negative control. Proliferative responses were assessed after 4 days of culture by [3H]-thymidine incorporation.
Effect of PLY on cytokine production by spleen cells
Splenocytes were isolated from C3H/HeN or C3H/HeJ mice by passing spleens through a 70 µm cell strainer (BD Falcon). After lysing erythrocytes in 0.88% w/v NH4Cl solution for 5 minutes and washing, cells were incubated in 96-well plates (1×106/ml) in RPMI medium with 8% v/v FCS plus antibiotics. Splenocytes were stimulated for 72 hours with PLY in the presence of plate-bound anti-CD3 (BD Biosciences) or HkSp and in some cases for a further 24 hours with PMA (Sigma Aldrich) and ionomycin (Sigma Aldrich). Supernatants were removed after 72 or 96 hours and tested by ELISA for IL-10, IL-17A (R&D Systems), IFN-γ and IL-5 (BD Biosciences).
Intranasal infection of mice with wild-type and PLY-deficient pneumococci
The pneumococcal infection studies were done at the University of Leicester under UK Home Office guidelines or in Trinity College Dublin under Irish Department of Health guidelines. Wild-type S. pneumoniae serotype 2 strain D39, NCTC 7466 (NCTC, London, UK) was used. An isogenic PLY-negative mutant, PLN-A, has been described previously [2]. Pneumococci were cultured on blood agar base with 5% v/v horse blood, or in brain heart infusion broth (BHI; Oxoid, Basingstoke, UK) with 20% v/v foetal bovine serum (Gibco, Paisley, UK), supplemented with 1 µg/ml erythromycin (Sigma, Poole, UK) for PLN-A. Before use, pneumococci were passaged through mice, as described previously [4] and aliquots stored at −80°C. When required, the suspension was thawed at room temperature, and bacteria were harvested by centrifugation before resuspension in sterile PBS.
Female outbred MF1, BALB/c, C57BL/6 (Harlan, Bichester, UK) or NLRP3−/− (bred in the Bioresources Unit of Trinity College Dublin) mice (9–10 weeks old, 30–35 g) were used for infection studies. Mice were lightly anaesthetized, as described previously [4], and 50 µl PBS containing 1×106 CFU S. pneumoniae was administered into the nostrils. The inoculum dose was confirmed by viable count following infection. At pre-chosen time intervals following infection, mice were sacrificed and lungs removed, weighed, and homogenised with an Ultra-Turrax T8 homogeniser (IKA, Germany). CFU bacterial counts were determined by viable count on blood agar plates as described previously [4]. IFN-γ and IL-17A concentrations in lung homogenates were measured by ELISA (BD Biosciences).
Analysis of intracellular IL-17A and IFN-γ production in the lungs of infected mice
Forty-eight hours post-infection of BALB/c mice with D39 wild-type S. pneumoniae (as described in the previous section), mice were sacrificed, lungs removed and cells isolated as described previously [4]. For intracellular cytokine staining, lung cells were cultured for five hours at 5×105 cells/well in round-bottomed 96-well plates in RPMI 10% FCS supplemented with GolgiPlug (BD Biosciences), according to manufacturer's instructions, to block cellular secretion of cytokines. Media was further supplemented with 500 ng/ml ionomycin and 50 ng/ml PMA. Following culture, cells were washed and stained with antibodies against extracellular markers (anti-CD3, anti-CD4, anti-CD8, anti-NKp46, anti-NK1.1, anti-CD45 or anti-γδTCR; BioLegend or eBioscience). Cells were then incubated with Fix/Perm solution (BD Biosciences) for 20 minutes before washing in Perm/Wash buffer (BD Biosciences). Staining with antibodies against intracellular cytokines was performed with anti-IL17A and anti-IFN-γ antibodies diluted in Perm/Wash buffer. After staining, cells were washed and resuspended in PBS 3% (v/v) FCS prior to data collection.
Statistical analyses
Unless otherwise stated, data were compared by one-way ANOVA. The Tukey-Kramer multiple-comparison test was used to identify significant differences between individual groups.
Methods for supporting data
Methods for supporting data are described in Methods S1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. TwetenRK
2005 Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect Immun 73 6199 6209
2. BerryAM
YotherJ
BrilesDE
HansmanD
PatonJC
1989 Reduced virulence of a defined pneumolysin-negative mutant of Streptococcus pneumoniae. Infect Immun 57 2037 2042
3. CanvinJR
MarvinAP
SivakumaranM
PatonJC
BoulnoisGJ
1995 The role of pneumolysin and autolysin in the pathology of pneumonia and septicemia in mice infected with a type 2 pneumococcus. J Infect Dis 172 119 123
4. KadiogluA
GinglesNA
GrattanK
KerrA
MitchellTJ
2000 Host cellular immune response to pneumococcal lung infection in mice. Infect Immun 68 492 501
5. KadiogluA
TaylorS
IannelliF
PozziG
MitchellTJ
2002 Upper and lower respiratory tract infection by Streptococcus pneumoniae is affected by pneumolysin deficiency and differences in capsule type. Infect Immun 70 2886 2890
6. FeldmanC
MunroNC
JefferyPK
MitchellTJ
AndrewPW
1991 Pneumolysin induces the salient histologic features of pneumococcal infection in the rat lung in vivo. Am J Respir Cell Mol Biol 5 416 423
7. MitchellTJ
AndrewPW
SaundersFK
SmithAN
BoulnoisGJ
1991 Complement activation and antibody binding by pneumolysin via a region of the toxin homologous to a human acute-phase protein. Mol Microbiol 5 1883 1888
8. CockeranR
SteelHC
MitchellTJ
FeldmanC
AndersonR
2001 Pneumolysin potentiates production of prostaglandin E(2) and leukotriene B(4) by human neutrophils. Infect Immun 69 3494 3496
9. CockeranR
TheronAJ
SteelHC
MatlolaNM
MitchellTJ
2001 Proinflammatory interactions of pneumolysin with human neutrophils. J Infect Dis 183 604 611
10. KadiogluA
CowardW
ColstonMJ
HewittCR
AndrewPW
2004 CD4-T-lymphocyte interactions with pneumolysin and pneumococci suggest a crucial protective role in the host response to pneumococcal infection. Infect Immun 72 2689 2697
11. BraunJS
HoffmannO
SchickhausM
FreyerD
DagandE
2007 Pneumolysin causes neuronal cell death through mitochondrial damage. Infect Immun 75 4245 4254
12. HouldsworthS
AndrewPW
MitchellTJ
1994 Pneumolysin stimulates production of tumor necrosis factor alpha and interleukin-1 beta by human mononuclear phagocytes. Infect Immun 62 1501 1503
13. MalleyR
HennekeP
MorseSC
CieslewiczMJ
LipsitchM
2003 Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A 100 1966 1971
14. SrivastavaA
HennekeP
VisintinA
MorseSC
MartinV
2005 The apoptotic response to pneumolysin is Toll-like receptor 4 dependent and protects against pneumococcal disease. Infect Immun 73 6479 6487
15. RatnerAJ
HippeKR
AguilarJL
BenderMH
NelsonAL
2006 Epithelial cells are sensitive detectors of bacterial pore-forming toxins. J Biol Chem 281 12994 12998
16. KogaT
LimJH
JonoH
HaUH
XuH
2008 Tumor suppressor cylindromatosis acts as a negative regulator for Streptococcus pneumoniae-induced NFAT signaling. J Biol Chem 283 12546 12554
17. BentonKA
PatonJC
BrilesDE
1997 The hemolytic and complement-activating properties of pneumolysin do not contribute individually to virulence in a pneumococcal bacteremia model. Microb Pathog 23 201 209
18. BrangerJ
KnappS
WeijerS
LeemansJC
PaterJM
2004 Role of Toll-like receptor 4 in gram-positive and gram-negative pneumonia in mice. Infect Immun 72 788 794
19. RubinsJB
PomeroyC
1997 Role of gamma interferon in the pathogenesis of bacteremic pneumococcal pneumonia. Infect Immun 65 2975 2977
20. NakamatsuM
YamamotoN
HattaM
NakasoneC
KinjoT
2007 Role of interferon-gamma in Valpha14+ natural killer T cell-mediated host defense against Streptococcus pneumoniae infection in murine lungs. Microbes Infect 9 364 374
21. LuYJ
GrossJ
BogaertD
FinnA
BagradeL
2008 Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS Pathog 4 e1000159
22. SteinmanRM
HemmiH
2006 Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol 311 17 58
23. Martin-FontechaA
ThomsenLL
BrettS
GerardC
LippM
2004 Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol 5 1260 1265
24. SuttonCE
LalorSJ
SweeneyCM
BreretonCF
LavelleEC
2009 Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31 331 341
25. GinglesNA
AlexanderJE
KadiogluA
AndrewPW
KerrA
2001 Role of genetic resistance in invasive pneumococcal infection: identification and study of susceptibility and resistance in inbred mouse strains. Infect Immun 69 426 434
26. KadiogluA
AndrewPW
2005 Susceptibility and resistance to pneumococcal disease in mice. Brief Funct Genomic Proteomic 4 241 247
27. KafkaD
LingE
FeldmanG
BenharrochD
VoronovE
2008 Contribution of IL-1 to resistance to Streptococcus pneumoniae infection. Int Immunol 20 1139 1146
28. MartinonF
MayorA
TschoppJ
2009 The inflammasomes: guardians of the body. Annu Rev Immunol 27 229 265
29. WalevI
ReskeK
PalmerM
ValevaA
BhakdiS
1995 Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J 14 1607 1614
30. GurcelL
AbramiL
GirardinS
TschoppJ
van der GootFG
2006 Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126 1135 1145
31. HornungV
BauernfeindF
HalleA
SamstadEO
KonoH
2008 Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9 847 856
32. YoshimoriT
YamamotoA
MoriyamaY
FutaiM
TashiroY
1991 Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266 17707 17712
33. MartinB
HirotaK
CuaDJ
StockingerB
VeldhoenM
2009 Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity 31 321 330
34. IwakuraY
NakaeS
SaijoS
IshigameH
2008 The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev 226 57 79
35. StockingerB
VeldhoenM
2007 Differentiation and function of Th17 T cells. Curr Opin Immunol 19 281 286
36. SuttonC
BreretonC
KeoghB
MillsKH
LavelleEC
2006 A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med 203 1685 1691
37. ZhuJ
PaulWE
2008 CD4 T cells: fates, functions, and faults. Blood 112 1557 1569
38. ShomaS
TsuchiyaK
KawamuraI
NomuraT
HaraH
2008 Critical involvement of pneumolysin in production of interleukin-1alpha and caspase-1-dependent cytokines in infection with Streptococcus pneumoniae in vitro: a novel function of pneumolysin in caspase-1 activation. Infect Immun 76 1547 1557
39. Riol-BlancoL
LazarevicV
AwasthiA
MitsdoerfferM
WilsonBS
2010 IL-23 receptor regulates unconventional IL-17-producing T cells that control bacterial infections. J Immunol 184 1710 1720
40. BentonKA
EversonMP
BrilesDE
1995 A pneumolysin-negative mutant of Streptococcus pneumoniae causes chronic bacteremia rather than acute sepsis in mice. Infect Immun 63 448 455
41. BergeronY
OuelletN
DeslauriersAM
SimardM
OlivierM
1998 Cytokine kinetics and other host factors in response to pneumococcal pulmonary infection in mice. Infect Immun 66 912 922
42. ZwijnenburgPJ
van der PollT
FlorquinS
RoordJJ
Van FurthAM
2003 IL-1 receptor type 1 gene-deficient mice demonstrate an impaired host defense against pneumococcal meningitis. J Immunol 170 4724 4730
43. BrozP
NewtonK
LamkanfiM
MariathasanS
DixitVM
2010 Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207 1745 1755
44. SharpFA
RuaneD
ClaassB
CreaghE
HarrisJ
2009 Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A 106 870 875
45. ChuJ
ThomasLM
WatkinsSC
FranchiL
NunezG
2009 Cholesterol-dependent cytolysins induce rapid release of mature IL-1{beta} from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J Leukoc Biol
46. JonesMR
SimmsBT
LupaMM
KoganMS
MizgerdJP
2005 Lung NF-kappaB activation and neutrophil recruitment require IL-1 and TNF receptor signaling during pneumococcal pneumonia. J Immunol 175 7530 7535
47. GilbertRJ
RossjohnJ
ParkerMW
TwetenRK
MorganPJ
1998 Self-interaction of pneumolysin, the pore-forming protein toxin of Streptococcus pneumoniae. J Mol Biol 284 1223 1237
48. LutzMB
KukutschN
OgilvieAL
RossnerS
KochF
1999 An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 223 77 92
49. DaviesJQ
GordonS
2005 Isolation and culture of murine macrophages. Methods Mol Biol 290 91 103
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2010 Číslo 11
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Zn Inhibits Coronavirus and Arterivirus RNA Polymerase Activity and Zinc Ionophores Block the Replication of These Viruses in Cell Culture
- The Female Lower Genital Tract Is a Privileged Compartment with IL-10 Producing Dendritic Cells and Poor Th1 Immunity following Infection
- Crystal Structure and Size-Dependent Neutralization Properties of HK20, a Human Monoclonal Antibody Binding to the Highly Conserved Heptad Repeat 1 of gp41
- The Arabidopsis Resistance-Like Gene Is Activated by Mutations in and Contributes to Resistance to the Bacterial Effector AvrRps4