
The Cytotoxic Necrotizing Factor of (CNF) Enhances Inflammation and Yop Delivery during Infection by Activation of Rho GTPases
Some isolates of Yersinia pseudotuberculosis produce the cytotoxic necrotizing factor (CNFY), but the functional consequences of this toxin for host-pathogen interactions during the infection are unknown. In the present study we show that CNFY has a strong influence on virulence. We demonstrate that the CNFY toxin is thermo-regulated and highly expressed in all colonized lymphatic tissues and organs of orally infected mice. Most strikingly, we found that a cnfY knock-out variant of a naturally toxin-expressing Y. pseudotuberculosis isolate is strongly impaired in its ability to disseminate into the mesenteric lymph nodes, liver and spleen, and has fully lost its lethality. The CNFY toxin contributes significantly to the induction of acute inflammatory responses and to the formation of necrotic areas in infected tissues. The analysis of the host immune response demonstrated that presence of CNFY leads to a strong reduction of professional phagocytes and natural killer cells in particular in the spleen, whereas loss of the toxin allows efficient tissue infiltration of these immune cells and rapid killing of the pathogen. Addition of purified CNFY triggers formation of actin-rich membrane ruffles and filopodia, which correlates with the activation of the Rho GTPases, RhoA, Rac1 and Cdc42. The analysis of type III effector delivery into epithelial and immune cells in vitro and during the course of the infection further demonstrated that CNFY enhances the Yop translocation process and supports a role for the toxin in the suppression of the antibacterial host response. In summary, we highlight the importance of CNFY for pathogenicity by showing that this toxin modulates inflammatory responses, protects the bacteria from attacks of innate immune effectors and enhances the severity of a Yersinia infection.
Published in the journal:
The Cytotoxic Necrotizing Factor of (CNF) Enhances Inflammation and Yop Delivery during Infection by Activation of Rho GTPases. PLoS Pathog 9(11): e32767. doi:10.1371/journal.ppat.1003746
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003746
Summary
Some isolates of Yersinia pseudotuberculosis produce the cytotoxic necrotizing factor (CNFY), but the functional consequences of this toxin for host-pathogen interactions during the infection are unknown. In the present study we show that CNFY has a strong influence on virulence. We demonstrate that the CNFY toxin is thermo-regulated and highly expressed in all colonized lymphatic tissues and organs of orally infected mice. Most strikingly, we found that a cnfY knock-out variant of a naturally toxin-expressing Y. pseudotuberculosis isolate is strongly impaired in its ability to disseminate into the mesenteric lymph nodes, liver and spleen, and has fully lost its lethality. The CNFY toxin contributes significantly to the induction of acute inflammatory responses and to the formation of necrotic areas in infected tissues. The analysis of the host immune response demonstrated that presence of CNFY leads to a strong reduction of professional phagocytes and natural killer cells in particular in the spleen, whereas loss of the toxin allows efficient tissue infiltration of these immune cells and rapid killing of the pathogen. Addition of purified CNFY triggers formation of actin-rich membrane ruffles and filopodia, which correlates with the activation of the Rho GTPases, RhoA, Rac1 and Cdc42. The analysis of type III effector delivery into epithelial and immune cells in vitro and during the course of the infection further demonstrated that CNFY enhances the Yop translocation process and supports a role for the toxin in the suppression of the antibacterial host response. In summary, we highlight the importance of CNFY for pathogenicity by showing that this toxin modulates inflammatory responses, protects the bacteria from attacks of innate immune effectors and enhances the severity of a Yersinia infection.
Introduction
Enteropathogenic Yersinia species such as Y. enterocolitica and Y. pseudotuberculosis initially infect the terminal ileum and colonize the Peyer's patches (PPs) within several hours of infections. Bacteria are subsequently transported to the mesenteric lymph nodes (MLNs) and can also spread systemically to reach liver and spleen via the bloodstream. The infections typically result in enteritis, enterocolitis and mesenteric lymphadenitis where the infected tissues show formation of microabscesses or granuloma-like lesions with central necrosis [1].
Enteropathogenic yersiniae have been shown to secrete exotoxins and/or inject effector proteins by specialized secretion machineries to manipulate host cell functions, including cytoskeletal rearrangements, to prevent immune responses and to establish a successful infection. They encode a type III secretion system (T3SS) on a 70 kb virulence-associated plasmid (pYV) that is essential for their defense against the host immune system [2]–[4]. The Yersinia T3SS has been shown to form a syringe-like apparatus with a thin needle-like surface exposed projection [5]. It is used to insert a translocation channel (composed of YopB and YopD) within the host membrane to inject the effector proteins YopE, YopH, YopJ/YopP, YopK/YopQ, YopM, YopO/YpkA, and YopT into the cell's cytoplasm. Yops target different cell signaling molecules and processes, in particular cytokine production and actin dynamics, often resulting in the inhibition of phagocytosis [6].
YopH is a tyrosine phosphatase that dephosphorylates proteins of the focal adhesion complex [7]–[10]. The effectors YopE, YopT and YopO/YpkA manipulate the regulation of Rho GTPases, which control the formation of lamellipodia, filopodia and stress fibers [2], [11]. YopJ/YopP promotes cell death of macrophages by inactivating the counterregulators of the Toll-like receptor 4-triggered apoptotic pathway, the mitogen-activated protein kinase kinases (MEKs) and the inhibitor κB kinase β (IKK β) [12]–[16]. YopM forms a complex with RSK and PRK kinase isoforms, traffics to the nucleus, and is important for Yersinia to persist in liver and spleen with a contextual decrease of several proinflammatory cytokines, including IL-1β, IL-12, IL-18, interferon γ, and TNF-α, and depletion of NK cells [17]–[21]. The effector YopK/YopQ seems to play a role in orchestrating the translocation of effector proteins by modulating the ratio of the pore-forming proteins YopB and YopD. This appears to prevent unintended Yop delivery and neutrophil death, which would enhance the inflammatory response possibly favoring the host [22]–[24].
Insertion of the YopB/D translocation channel allows Yop delivery while maintain the host cell membrane intact. The YopB/D complex results in activation of Rho GTPases, actin polymerization and pore-formation. However, pore formation is usually prevented by the GTPase-downregulating function of YopE and YopT. Yet, expression of constitutively active forms of Rac1 and RhoA leads to a loss of membrane integrity and results in increased pore formation even when YopE and YopT are expressed [25]. In addition, signaling pathways triggered by high affinity-binding of the main Yersinia adhesins YadA and InvA to β1 integrin receptors and YopB/D signaling were shown to induce activity of Rho GTPases and actin polymerization which are crucial for efficient translocation of the Yop effectors [26].
Another Yersinia factor shown to activate the small GTPase RhoA is the cytotoxic necrotizing factor-Y (CNFY) [27], [28]. CNFY is prevalent in some Y. pseudotuberculosis isolates, e.g. the widely used Y. pseudotuberculosis strain YPIII. All these strains belong to the serogroup III, but other isolates of this serogroup do not express CNFY and contain deletions within the corresponding cnfY gene [27].
On the amino acid level, CNFY is highly similar (>68%) to the CNF toxins found mainly in E. coli strains isolated from patients and domestic animals with extraintestinal infections (CNF1-3) [29], [30]. CNF1 is the best-characterized toxin of this class of bacterial toxins and is transferred to host cells through outer membrane vesicles (OMVs) [31]–[34]. The CNF1 toxin is a single-chain A-B toxin with an N-terminal delivery domain including subdomains for receptor binding, pore formation and proteolytic cleavage, and a C-terminal deamidase domain [35], [36]. Internalization of the toxin into target cells occurs through receptor-mediated endocytosis, which appears to be independent of clathrin and lipid rafts (sphingolipid/cholesterol rich microdomains) [37], [38]. After uptake, the 55 kDa C-terminal deamidase domain is autocatalytically cleaved off in the late endosome, and delivered into the cytoplasm in a pH-dependent manner [39].
CNF1 deamidates Gln-61/-63 of RhoA, Rac1 and Cdc42 to Glu-61/-63 resulting in Rho GTPases with a blocked GTP hydrolase activity. Deamidated Rho GTPases induces polymerization of F-actin at focal contacts, increase cell-matrix adhesion, and promote formation of stress fibers, lamellipodia and filopodia, which led to the classification as ‘constitutively active’ [40]–[45]. Cytoskeletal rearrangements attributed to CNF lead to multinucleated cells due to inhibited cytokinesis with ongoing cell cycle progression [46]. Additionally, CNF1 has been reported to (i) induce phagocytosis in epithelial cells and reduce CR3-mediated phagocytosis in monocytes [47], [48], (ii) promote bacterial cell entry [49], (iii) decrease the barrier function of intestinal tight junctions [40], [50], (iv) decrease transmigration of polymorphonuclear leukocytes across a T84 monolayer [51], and (v) induce apoptosis of bladder cells [52].
The overall amino acid sequence of CNFY of Y. pseudotuberculosis is very similar to CNF1. However, CNFY is not recognized by neutralizing antibodies against CNF1 [27]. Moreover, CNFY seems to bind to different cell receptors and preferentially deamidates RhoA (over Rac1 and Cdc42) in cultured epithelial cells [28], [38]. Although CNFY and certain Yop effectors alter the cytoskeleton by affecting the activity of the Rho GTPases, little is known about the interplay, cooperation and joint role of these toxins in the pathogenic lifestyle of Y. pseudotuberculosis. Here, we provide evidence that CNFY is an important virulence factor of Y. pseudotuberculosis YPIII. CNFY is shown to enhance Yop protein delivery, which is crucial for pathogenicity. Furthermore, the toxin was found to induce inflammatory responses and increase the severity of a Yersinia infection.
Results
CNFY is expressed in all infected tissues and organs throughout the infection
Since many Y. pseudotuberculosis isolates as well as Y. pestis contain deletions within the cnfY gene [27], we first tested whether the intact cnfY toxin gene in the Y. pseudotuberculosis wild-type strain YPIII is expressed and induced under virulence-relevant growth conditions. A cnfY-lacZ transcriptional fusion was only slightly expressed when Y. pseudotuberculosis was grown at 25°C, but its expression was strongly induced at 37°C and reached its maximum during stationary phase (Fig. S1A). High cnfY transcription was generally observed in complex media, in particular BHI, whereas only low expression levels were detected in all tested minimal media (Fig. S1B, data not shown). In summary, cnfY is predominantly expressed at 37°C in a nutrient rich environment, resembling conditions found in the mammalian intestinal tract.
This result prompted us to test expression of the toxin during infection. BALB/c mice were orally infected with 2×108 bacteria of the Y. pseudotuberculosis wild-type strain YPIII expressing a cnfY-luxCDABE fusion, and the bioluminescent signal was monitored in the mice for six days using an in vivo imaging system. Only very low luciferase activity was measured in the bacterial culture before infection (data not shown) and in the intestinal tract directly after oral ingestion (1 h, Fig. 1). However, a very strong bioluminescent signal of the cnfY-luxCDABE fusion was detectable during the entire following course of the infection. The most intensive signals were detected two days post infection in the intestine and associated lymphoid tissues (Fig. 1). No light emission was monitored in mice infected with bacteria carrying the promoterless luxCDABE operon in the identical expression system (data not shown).
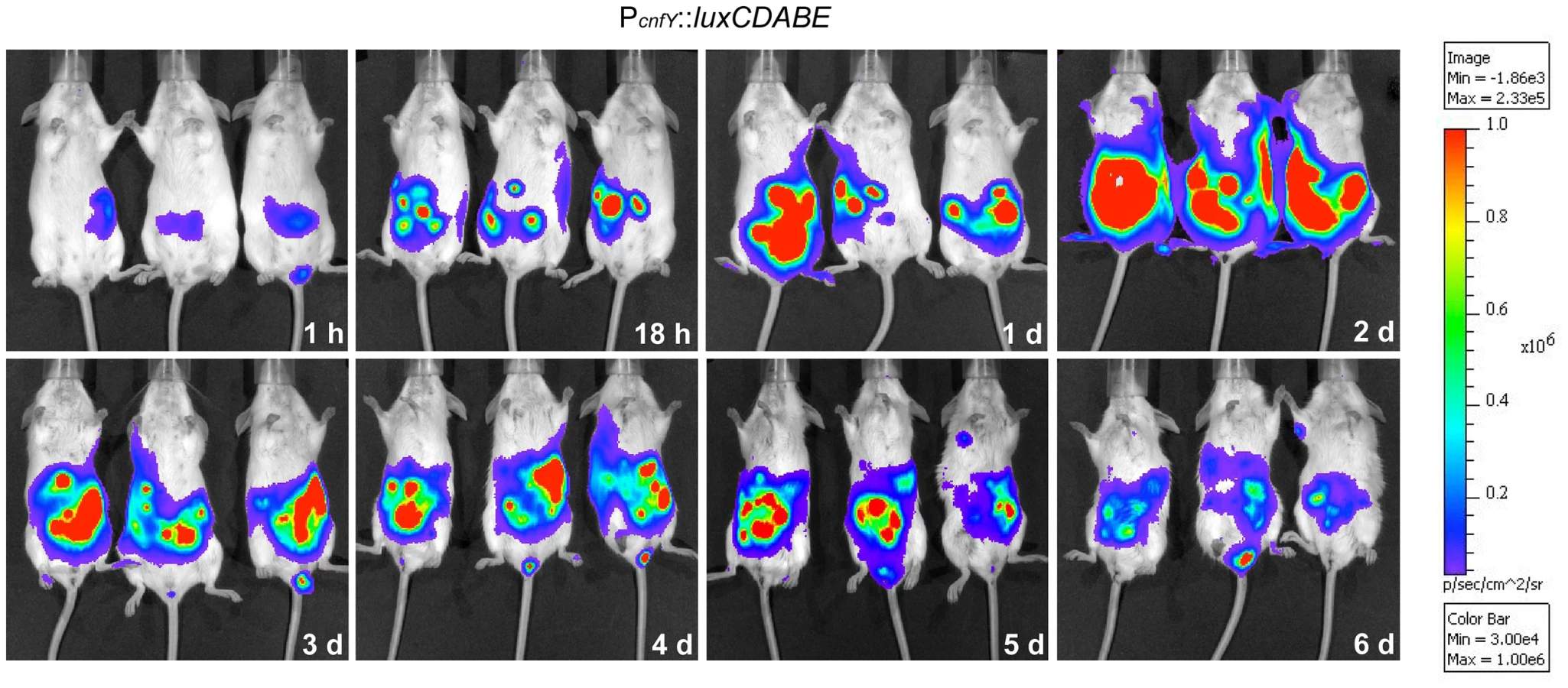
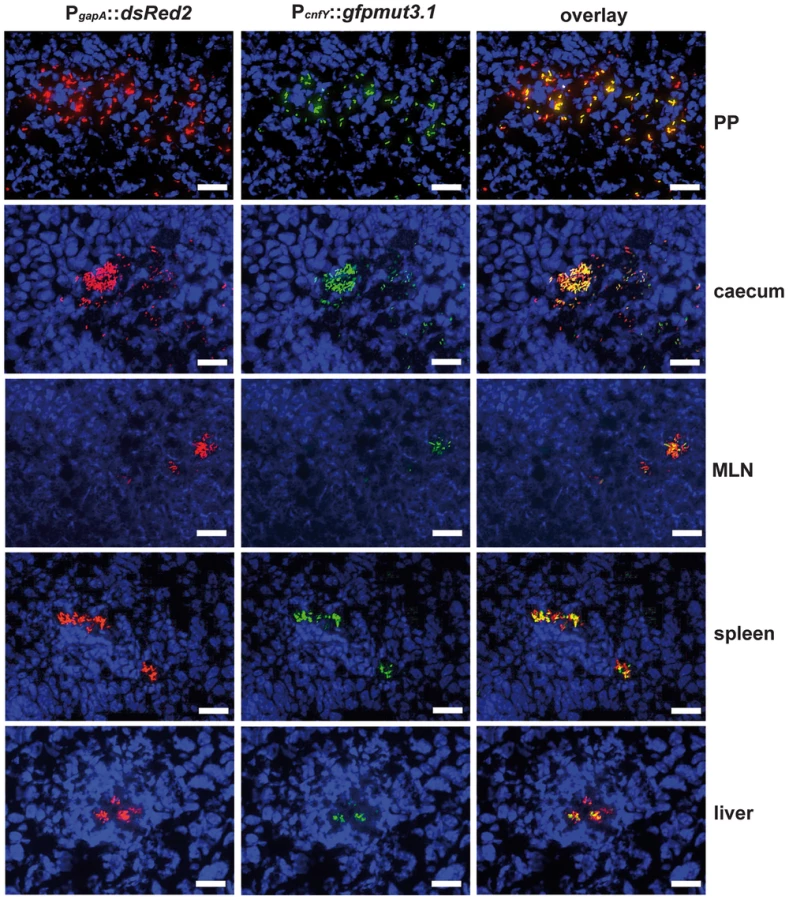
In order to study cnfY expression in the individual infected tissues, we used a set of established fluorescent fusion vectors for in vivo expression analysis. To do so, Y. pseudotuberculosis YPIII harboring a plasmid-encoded constitutive PgapA::dsred2 reporter construct and a compatible PcnfY::gfpmut3.1 fusion was used to infect BALB/c mice. Five days post infection, the small intestine, caecum, colon, PPs, MLNs, spleen and liver were isolated and cryosections were prepared. The bacteria in the tissues were visualized by monitoring dsRed2, and then tested for PcnfY::gfpmut3.1. As shown in Fig. 2, the PcnfY::gfpmut3.1 fusion was expressed in all tested organs. In summary, a temperature shift to 37°C, but most likely no tissue-specific signals are required to induce toxin expression in infected tissues.
CNFY is crucial for virulence of Y. pseudotuberculosis YPIII
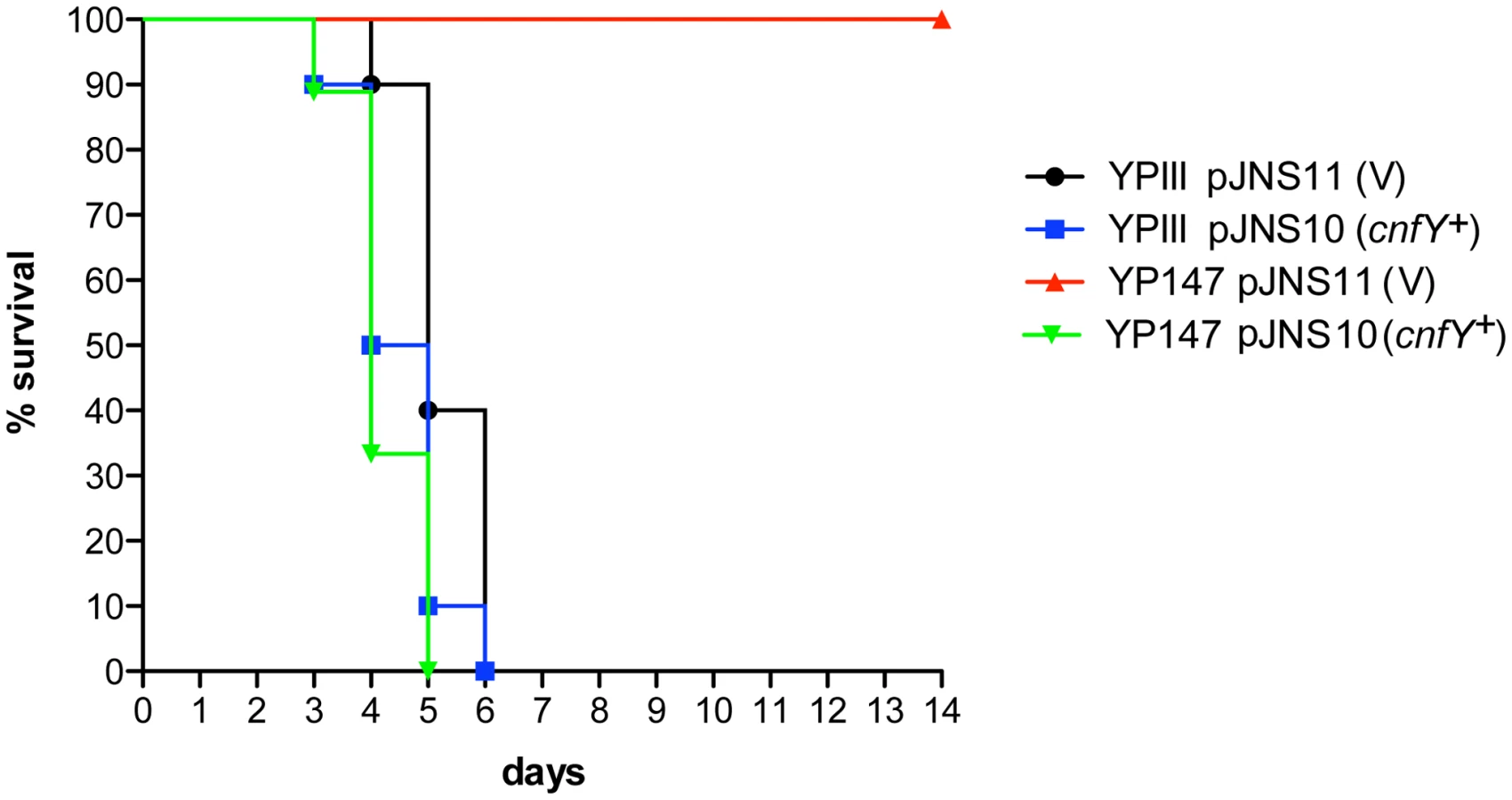
Absence of a functional toxin gene in other Y. pseudotuberculosis clinical isolates, may suggest that CNFY only adds another potential virulence factor to the variety of effector proteins and toxins that are produced by this pathogen. However, high expression of cnfY during the entire course of an infection also indicates that presence of this toxin may enhance the pathogenicity of Y. pseudotuberculosis. To first assess the impact of CNFY on pathogenesis, the potential of the Y. pseudotuberculosis wild-type strain YPIII and the isogenic cnfY-deficient strain to cause lethal infections was compared. BALB/c mice were orally infected with 2×109 bacteria of the cnfY mutant (YP147) and the wild-type strain (YPIII) harboring the empty vector (pJNS11) or a cnfY-encoding plasmid (pJNS10). Survival and weight of the mice were monitored over two weeks and date of death was recorded (Fig. 3, S2). Mice infected with YPIII showed signs of the infection, e.g. weight loss, piloerection and lethargy, and succumbed to infection between day four and day six. Strikingly, none of the mice infected with YP147 developed severe disease symptoms and all mice were still alive 14 days post infection. Monitoring of body weight demonstrated that also mice infected with the cnfY knock-out strain YP147 showed a slight reduction in weight, but they recovered quickly and regained weight (Fig. S2). Presence of the cnfY-encoding low-copy number plasmids reverted the avirulent phenotype of the cnfY mutant and reduced the average day of death of the wild-type strain YPIII by one day, most likely due to the overexpression of the toxin. The Y. pseudotuberculosis YPIII isolate, unlike other Y. pseudotuberculosis strains, is unable to replicate in murine macrophages due to a defective allele of phoP [53]. To exclude that CNFY influence on virulence is only visible in a phoP-deficient derivative with an overall lower pathogenicity, the inability to grow in macrophages was complemented by an exchange of the allele against the phoP ORF from Y. pseudotuberculosis IP32953. However, when mice were challenged with 2×109 CFU of the equivalent phoP+ strains, 100% of the mice infected with the CNFY-positive strain died during the observation period, while 80% of the mice infected with the isogenic cnfY-deficient strain survived and regained weight (Fig. S3).
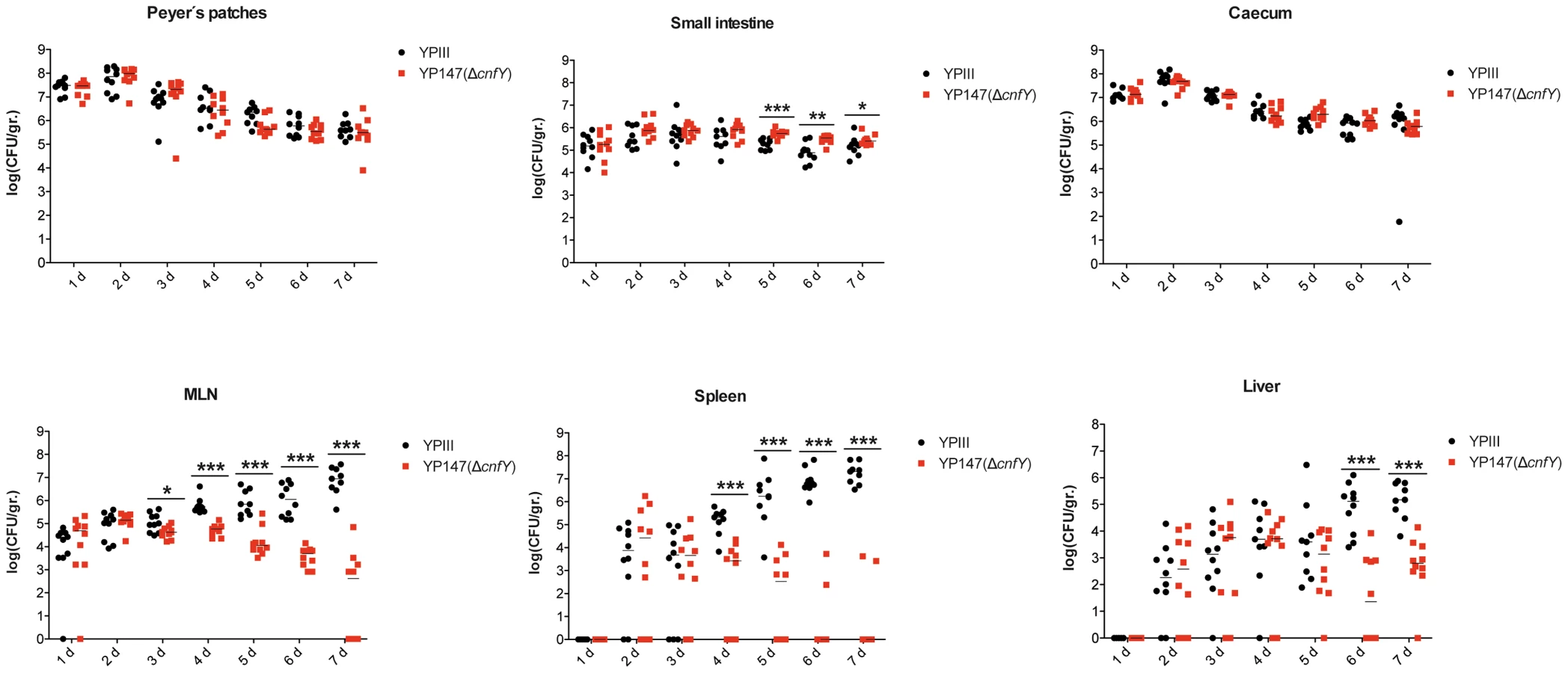
To gain a deeper insight into the differences in the infection process of CNFY-positive and -negative strains, we determined the number of bacteria that colonized the small intestine, caecum, PPs, MLNs, liver and spleen of BALB/c mice at different time points after oral infection with 2×108 bacteria (Fig. 4). Comparable amounts of wild-type (YPIII) and the mutant strains (YP147) were recovered from PPs and caecum during infection, and only a very small increase of bacterial counts was observed with the cnfY mutant in the small intestine at days 5–7 post infection (Fig. 4). However, significantly reduced numbers of YP147 were recovered from MLNs and spleen (Fig. 4). The number of cnfY-positive and -negative bacteria in these organs was almost identical up to day three post infection, but the cnfY mutant was eliminated very rapidly later during the infection. At day seven, none or only few mutants were recovered from MLNs and spleen, whereas 108–109 bacteria of the wild-type strain were recovered per gram of both organs. The effect was less pronounced in the liver, but the strongly reduced number of mutant bacteria relative to the wild-type bacteria six and seven days after infection clearly indicated that the presence of CNFY is also advantageous for the colonization of the liver (Fig. 4). This demonstrated that loss of CNFY, resulting in avirulence of Y. pseudotuberculosis YPIII, is reflected by a fast elimination of the bacteria from MLNs, liver and spleen.
Within the first week after infection with wild-type strain YPIII the size of the spleen and liver decreased two-fold, whereby changes of the organ size were first visible at day three post infection (Fig. S4A,B). In contrast, infection with the isogenic cnfY mutant strain YP147 had no effect on the size of the liver and induced a considerable increase of the size of the spleen. In addition, mice infected with wild-type strain YPIII had significantly shorter intestines (30%) at day six and seven post infection than mice infected with the cnfY mutant (Fig. S4C). The shortening of the intestine is a sign of marked intestinal inflammation. This indicated that CNFY not only affects colonization of systemic organs, but has also a strong influence on the host's inflammatory response against the bacterial infection.
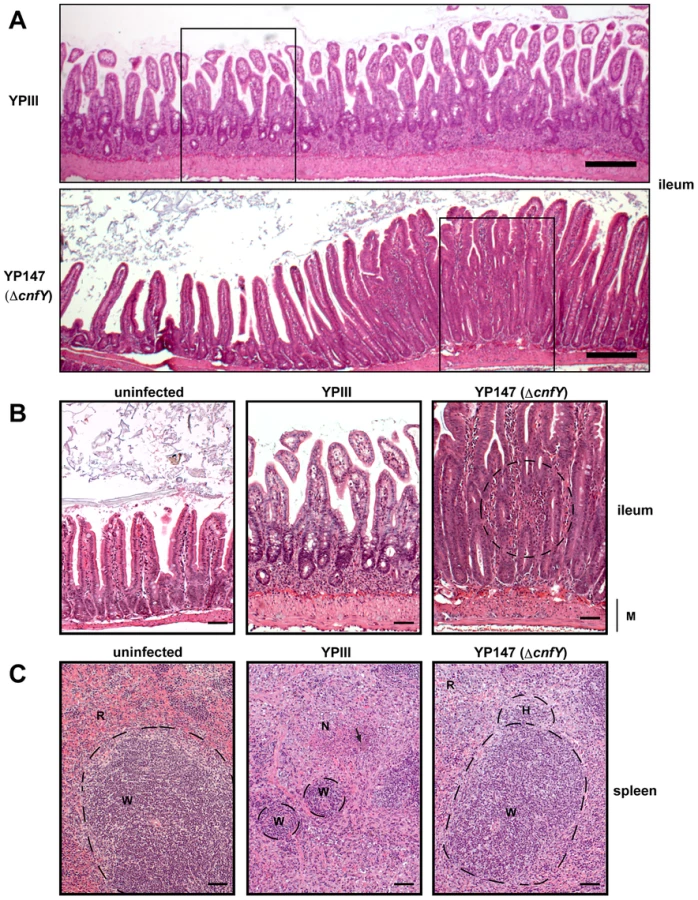
Histopathological examination of the infected host tissues demonstrated marked differences of the overall inflammatory reaction, which was stronger in YPIII-infected animals, especially in the small intestine and spleen compared to YP147-infected mice. In the intestine, inflammation was most prominent in the ileum and caecum in both groups. However, in YPIII-infected mice inflammation was diffuse affecting the entire ileum at day six (Fig. 5A, upper panel, 5B middle panel). In YP147-infected mice inflammation was locally restricted to multifocal lesions characterized by the presence of inflammatory cells from the muscular layer up to the epithelial cells (Fig. 5A, lower panel, 5B right panel). In these areas inflammation led to epithelial cell hyperplasia (increased proliferation) resulting in an increase of the villi length. However, this lesion is only locally restricted and adjacent tissue remains unaltered. In addition, inflammation was more generalized in YPIII- compared to YP147-infected organs. In mice infected with YP147, no bacterial foci (diffuse patches of bacteria) could be detected microscopically in hematoxylin and eosin (H & E) stained sections of the spleen at day six post infection, whereas in the majority of YPIII-infected mice bacterial foci were visible in the histological sections (Fig. 5C). YPIII infections were accompanied by a more severe inflammation of the spleen, where presence of the bacteria resulted in necrotizing spleenitis leading to splenic atrophy with marked depletion of the white pulp. YPIII caused multifocal necrosis in spleen, whereas in YP147-infected spleens, only mild hyperplasia of the white pulp and increased erythropoiesis were found (Fig. 5C). Taken together, CNFY has a significant influence on the number of microcolonies in the tissues and leads to a more severe and widespread inflammation in the small intestine, liver and spleen.
CNFY modulates the innate immune response
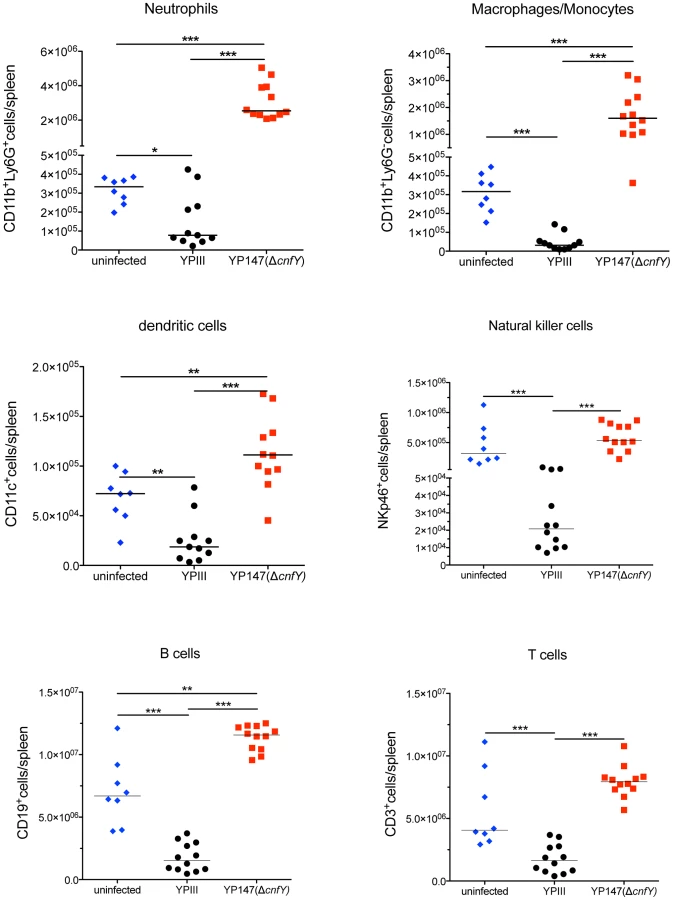
Because of the strong influence of CNFY on the colonization of bacteria in MLNs, spleen and liver, it was hypothesized that the toxin might counteract host immune defenses. To test this hypothesis, we infected BALB/c mice with 2×108 bacteria of the wild-type or the cnfY mutant strain, and immune cell composition in the spleen was analyzed by multi-color flow cytometry three days and six days post infection. The spleen was chosen since here the most pronounced CNFY-triggered pathological effects had been observed. Cell suspensions of isolated tissues were prepared and cells were stained with fluorescently labeled antibodies to distinguish neutrophils from macrophages/monocytes, dendritic cells (DCs), natural killer (NK) cells, B cells, and T cells (Fig. S5). All alterations of immune cell populations seen at day six (data not shown) were already visible at day three post infection, when the bacterial load is still similar and the overall health status of YPIII-infected mice is only slightly and not severely reduced as at day six. A very pronounced variation of the immune cell population between the YPIII- and YP147-infected mice was observed (Fig. 6). All types of immune cells were significantly decreased in the spleen three days after infection with YPIII when the spleen started to shrink, but the most severe changes were observed with cells of the innate immune system. In particular, numbers of macrophages, monocytes and NK cells were significantly reduced; whereas reduction of neutrophils and conventional DCs was less pronounced. In contrast, no reduction of immune cells was detectable in spleens of YP147-infected mice (Fig. 6). In contrast, a significant higher influx of neutrophils and macrophages/monocytes was observed, which is consistent with the rapid clearance of mutant bacteria from the spleen upon triggering of the immune response. To determine whether CNFY affects the steady-state level of certain cell populations, the population percentage was also compared and further confirmed a significant expansion of neutrophils and macrophages/monocytes in YP147-infected spleens (Fig. S6). These and the histopathological data strongly suggest that the CNFY toxin reduces influx and/or causes rapid cell death of invading immune cells in the spleen.
CNFY enhances Yop delivery into macrophages
Our infection experiments clearly demonstrated that absence of the CNFY toxin renders the bacteria completely avirulent, resulting in the clearance of the bacteria in MLNs, liver and spleen. A similar attenuation in mouse models of oral infection was observed (i) when the virulence plasmid, encoding the T3SS and the Yop effectors is cured from Y. pseudotuberculosis YPIII, (ii) when multiple yop genes were deleted or (iii) when the regulator LcrF that controls expression of the T3SS/Yops is absent [54], [55]. Moreover, a significant influx of neutrophils was observed in the spleen of mice infected with a yopM mutant strain of Y. pestis, while the numbers of neutrophils decreased during infection with the parental strain [20], [21]. In addition, YopJ translocation has been shown to promote cell death of professional phagocytes [13], [15]. This suggested that the CNFY toxin is important for the efficient injection of the Yop effectors into host cells during the infection process. In fact, recent work by Mejia et al. [26] demonstrated that efficient translocation of the Yop effectors requires Rho activation – a process that has been shown to be stimulated by the CNFY toxin [28], [38].
To address whether CNFY-mediated activation of Rho GTPases influences Yop-translocation into professional phagocytes, we first tested the influence of recombinant CNFY toxin on non-activated and PMA-activated macrophages, thus mimicking its effect on unstimulated and stimulated macrophages during infection. Intoxification of murine macrophages (J774A.1) led to activation of all three Rho GTPases, RhoA, Cdc42 and Rac1 (Fig. 7A). CNFY further induced a marked increase in cell size with some giant multinucleated cells (Fig. 7B). These CNFY effects occurred independently of macrophage stimulation with PMA. This indicates that CNFY controls actin dynamics in macrophages through deamidation of Rho GTPases.
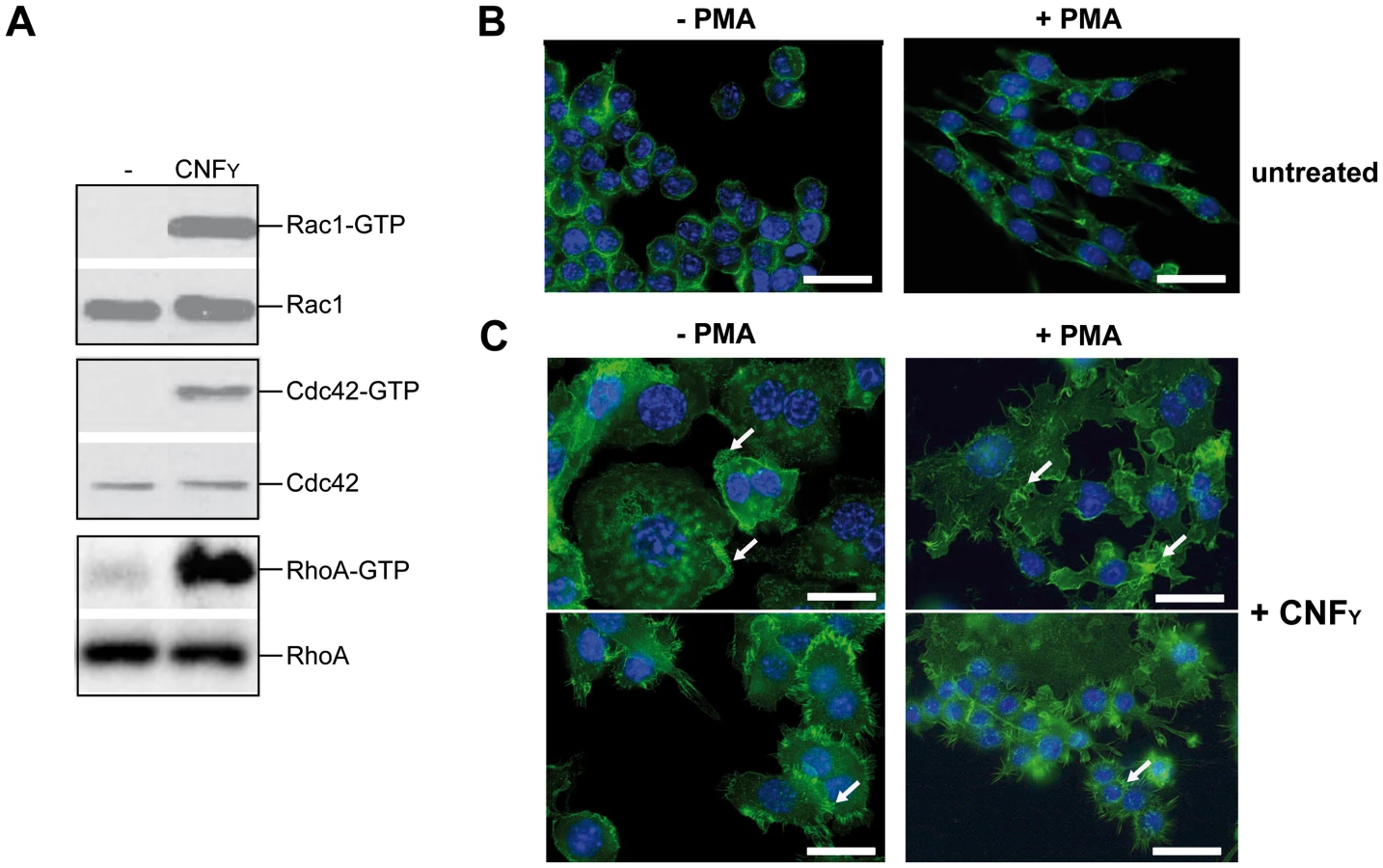
Since host actin polymerization by Rho activation plays a role in Yop translocation by Y. pseudotuberculosis [26] we also tested the influence of CNFY on Yop delivery. To do so, we generated Y. pseudotuberculosis strains expressing a YopE-β-lactamase reporter fusion [56], namely YP173 (YPIII-ETEM), YP174 (YP101ΔsycS-ETEM), and YP217 (YP147ΔcnfY-ETEM), and used these strains to infect host cells treated with the dye CCF4-AM. CCF4-AM consists of coumarin and fluorescein conjugated by a lactam ring and is modified by cellular esterases, whereby the dye becomes green fluorescent and is trapped inside the cell. If the β-lactam ring is cleaved by β-lactamase the dye changes its fluorescence from green to blue [57], [58]. The green to blue conversion allows identification of host cells in which the YopE-β-lactamase fusion protein has been successfully injected. We first used this fluorescence-based system to monitor translocation of the chimeric protein into HEp-2 cells, and determined the number of green and blue fluorescent cells by fluorescence microscopy and flow cytometry. Efficient translocation of YopE-β-lactamase into epithelial cells was observed upon infection with YP173 (YPIII-ETEM), but not with the secretion-deficient control strain YP174 (YP101ΔyscS-ETEM) (Fig. 8A,B). YopE-β-lactamase translocation by the cnfY-deficient strain YP217 (YP147ΔcnfY-ETEM) was significantly reduced compared to YP173 (YPIII-ETEM), whereas preincubation of the host cells with CNFY increased translocation of the fusion protein (Fig. 8A, B), indicating that CNFY enhances effector delivery. Since Y. pseudotuberculosis predominantly injects the Yops into professional phagocytes in vivo [59], we also tested CNFY influence on YopE-β-lactamase translocation into murine macrophages, and found that pretreatment with CNFY also boosts Yop delivery into these phagocytes (Fig. 8C).
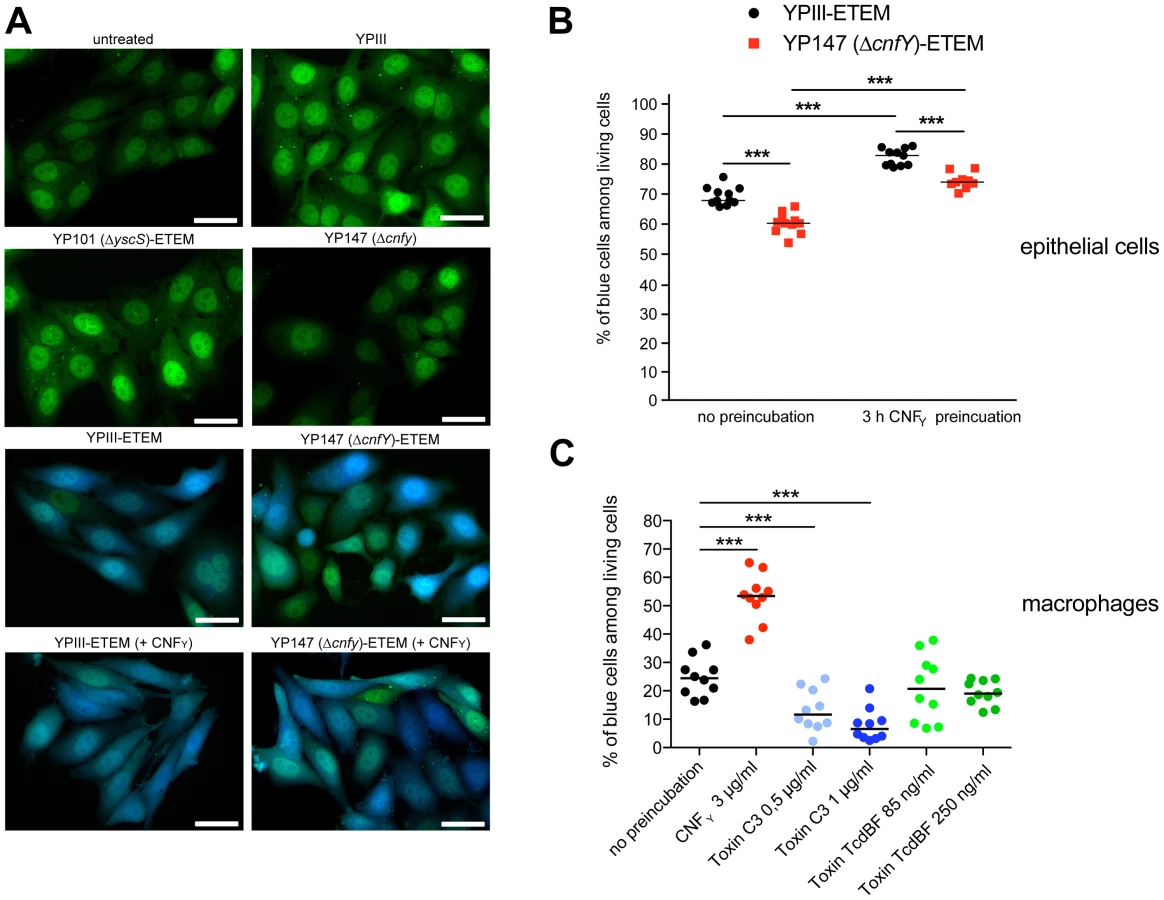
Stimulation of Rac1 through YadA and invasin-bound β1-integrins was shown to be essential for Yersinia uptake into epithelial cells [11], but neither internalization nor activation of Rac1 was required for Yop translocation by Y. pseudotuberculosis into HeLa cells [26]. This suggested that CNFY-mediated stimulation of Yop delivery into macrophages might preferentially be caused by activation of RhoA. To validate this assumption, we pretreated macrophages with the Clostridium botulinum C3 toxin, an ADP-ribosylating protein that specifically inhibits RhoA, B and C, or with toxin B from variant Clostridium difficile serotype F strain 1470 (TcdBF), which specifically inhibits Rac but not RhoA/B/C [60], [61]. Treatment with the toxins induced actin cytoskeleton rearrangements and cell morphology changes, but had no effect on the viability of the macrophages and the number of associated bacteria (data not shown). As shown in Fig. 8C, the RhoA/B/C inhibitor reduced the percentage of blue macrophages significantly, whereas the Rac inhibitor had no influence on YopE-β-lactamase translocation. These findings indicated that the CNFY toxin enhances Yop delivery into murine macrophages, and in particular activation of RhoA seems to play a role in the processes that stimulate Yop translocation into these professional phagocytes.
It has been reported that translocated effector YopE of Y. pseudotuberculosis YPIII is a GTPase-activating protein (GAP) for Rac1 and RhoA and this function appears important to regulate Yop translocation and modulate host defenses crucial for virulence [62]–[65]. This raised the question how YopE and CNFY contribute to RhoA-GTP and Rac1-GTP levels and Yop translocation. To address this, we analyzed RhoA and Rac1 activation and Yop translocation in the presence and absence of YopE in untreated or CNFY-pretreated murine macrophages. As shown in Fig. S7, only low amounts of active Rac1 and RhoA could be detected in uninfected macrophages. Addition of the wild-type strain YPIII pregrown at 37°C to mimic the situation prior to host cell contact induced activation of RhoA and Rac1. Absence of YopE resulted in a small additional increase in RhoA-GTP, but had no or only a slight influence on Rac1-GTP levels. Furthermore, it had no or only a very small stimulatory effect on the translocation of YopD and YopH without or after pretreatment of the macrophages with CNFY (Fig. S7). This indicates that under these conditions intracellular YopE is unable to efficiently counteract CNFY-mediated RhoA/Rac1 activation and reduce Yop translocation into murine macrophages.
We next analyzed whether the CNFY toxin affects Yop translocation into host cells in the original tissue environment. MLNs were harvested from uninfected mice and filtered to disrupt the tissue architecture and generate single-cell suspensions. Single cell suspensions were infected with a multiplicity of infection (MOI) of 10, incubated with CCF4-AM, and then analyzed by flow cytometry. As shown in Fig. S8, significantly higher numbers of blue cells with translocated YopE-β-lactamase were measured after infection with YPIII, indicating that Yop delivery into host cells can be enhanced by the toxin through activation of Rho GTPases.
CNFY enhances Yop delivery into phagocytes during infection
It has been previously reported that Y. pseudotuberculosis selectively targets Yops to professional phagocytes in the PPs, MLNs and spleen during the oral route of infection [59]. To analyze whether the CNFY toxin also affects YopE-β-lactamase delivery in the course of an infection, we orally infected mice with 2×109 bacteria YP173 and the isogenic cnfY mutant strain YP217. The T3SS-deficient yscS mutant, encoding the YopE-β-lactamase, and YPIII without the fusion were used as negative controls. At day three post infection mice were sacrificed, the PPs, MLNs, and spleen were harvested, and the translocation of Yops into various immune cell subsets was analyzed by flow cytometry (Fig. S9).
Following infection with the YopE-β-lactamase expressing wild-type strain 4.5% of all living cells within PPs were affected by Yop translocation. In contrast, only 1.5% of all living cells in the PPs contained the fusion protein after infection with the cnfY-deficient strain (Fig. S10). Yop translocation efficiency was still significantly reduced in tissues infected with the cnfY-deficient strain when the percentage of translocated blue cells was normalized to the bacterial load of the tissue/organ (Fig. 9). This excludes that lower bacterial numbers account for this effect, but it also assumes that bacteria are infecting different cell types at the same MOI, which is unknown. Yop delivery was also significantly lower in the absence of the CNFY toxin in MLNs and spleen in which the total number of targeted cells was reduced compared to PPs (Fig. 9, S10).
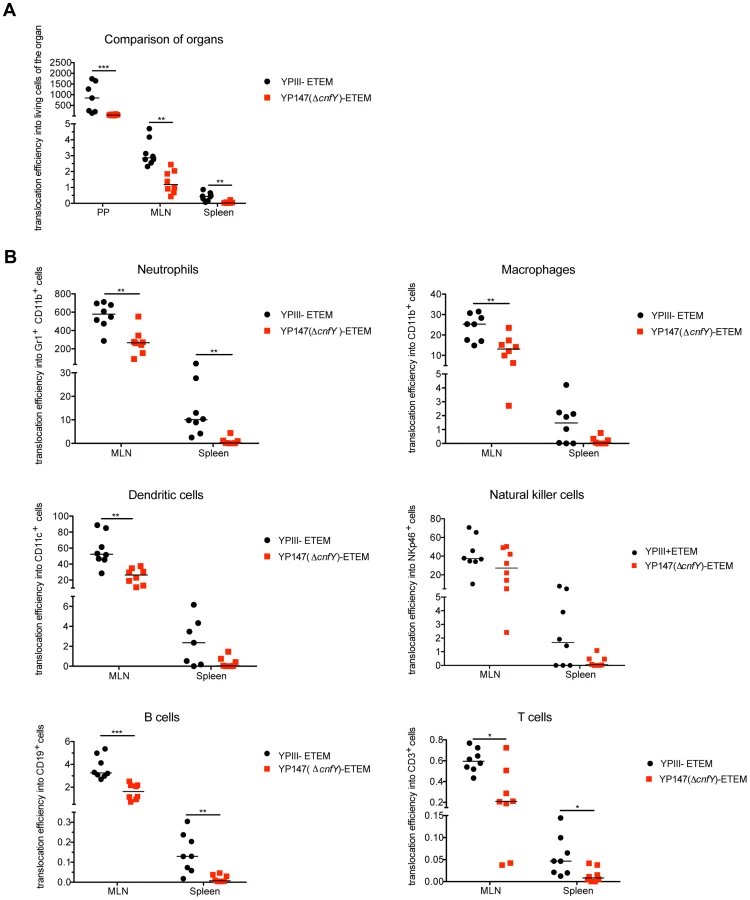
We further determined whether CNFY-mediated stimulation of Yop translocation affected specific immune cells more frequently than others. Translocation of YopE-β-lactamase into each immune cell type was compared in MLNs and spleen from mice infected with YP173 (YPIII-ETEM) or the cnfY mutant derivative YP217 (YP147-ETEM) (Fig. 9B, S10). In general, all immune cells analyzed were targeted by Y. pseudotuberculosis. However, Yop-injected neutrophils were significantly enriched in the MLNs and the spleen, indicating that this cell population is preferentially targeted in the tissues. In addition, DCs, NK cells and macrophages were well represented in the blue population, while B and T cells remained underrepresented (Fig. 9B, S10). This is in full agreement with previous studies demonstrating that translocated YopH of Y. pseudotuberculosis strain IP2666 is enriched in neutrophils, macrophages and DCs in MLNs and spleen [59].
We further found that the apparent enhanced targeting to professional phagocytes, in particular neutrophils, macrophages and DCs and to a smaller extent also translocation into B and T cells was reduced in the absence of CNFY in the MLNs and spleen three days post infection (Fig. 9B, S10). Yop translocation into NK cells was also somewhat reduced in the spleen. Since Y. pseudotuberculosis induces host cell death [66], which may be reflected in the strong reduction of professional phagocytes in the spleen (Fig. 6), the actual amount of Yop translocation in this organ is probably underestimated. Taken together, these results demonstrate that the CNFY toxin plays a critical role during the infection, facilitating targeting of Yops to host immune cells, in particular professional phagocytes.
Discussion
Many bacterial toxins and translocated effector proteins target Rho GTPases, which control crucial eukaryotic signal transduction pathways involved in the organization of the cell cytoskeleton, cell cycle progression, genetic information processing, and host defense processes to promote invasion, survival and replication of pathogens within their hosts [29], [67]. In this study we investigated the Rho-activating cytotoxic necrotizing factor CNFY of Yersinia. Although much progress has been made unravelling the molecular mechanism of this toxin, the functional consequences for host-pathogen interaction and pathogenesis were largely unknown. Using a murine model for gastrointestinal tract infections we provide evidence that this Rho-activating protein is crucial for virulence of the naturally toxin-expressing Y. pseudotuberculosis strain YPIII. The importance of CNFY for pathogenesis was first established by the analysis of the expression and the role of the toxin during the infection of mice. We show that cnfY is strongly expressed in all infected tissues during pathogenesis in mice, and is crucial for virulence, in particular for the dissemination of the bacteria into the MLNs, spleen and liver.
Histological analysis and immune cell composition of the infected tissues suggest that CNFY contributes significantly to the acute characteristics of the inflammatory response and host tissue damage during infection. Histo-pathologic evaluation underlines the finding that CNFY induces apoptosis, as focal necrosis was not seen in YP147-infected animals. Cell death leads to atrophy of the spleen in YPIII-infected mice. Moreover, a restriction of the inflammation to small foci could be observed in the intestine of YP147-infected animals, whereas the entire ileum was affected by a diffuse inflammation in YPIII-infected animals, explaining the shortening of the intestine. Hyperplasia of the white pulp seen in YP147-infected mice displays the immune response triggered by the infection. The infection is restricted to small foci in the intestine and is reversible, whereas the infection in YPIII infected animals is generalized and most probably leads to death by endotoxiemia. This inflammatory necrotizing phenotype is reminiscent of earlier studies analyzing the effect of CNF1 of E. coli using subcutaneous injections as well as animal models of urinary tract and prostatitis infection [31], [68], [69].
Infections of the gastrointestinal tract by enteropathogenic Yersiniae lead to a biphasic inflammatory process in which bacterial adhesion and transmigration through the intestinal epithelial layer triggers an initial antibacterial defense response with little inflammation, e.g. expression of IL-8 by epithelial cells, which is followed by an acute infiltration and activation of neutrophils, cytokine production and tissue necrosis [70]. First recognition of Y. pseudotuberculosis occurs through contact of the bacterial LPS with TLR4 on naïve host macrophages and this leads to proinflammatory cytokine production through activation of MAPK and NF-kB. However, translocation of YopJ inhibits activation of MAPK and NF-κB and induces an apoptotic signaling pathway including activation of initiator caspase-8, and the executioner caspase-3, -7, and -9 [70], [71]. Apoptotic macrophages are eliminated and this process also triggers production of anti-inflammatory cytokines such as IL-10 and TGF-β [72], [73]. However, induction of apoptosis is probably not always fully immunologically silent, e.g. phagocytosis of apoptotic cells by other phagocytes, can prime other immune responses such as activation of CD8+ T cells [71]. During the course of the infection, the number of activated macrophages increases whereas the number of naïve macrophages declines. In activated macrophages Yersinia causes cell death by inflammatory pyroptosis. This occurs through activation of a multiprotein complex, called the inflammasome, which forms a platform for the autoprocessing and activation of the cysteine protease caspase-1. Activation of caspase-1 results in the secretion of the inflammatory cytokines such as IL-1α, IL1-β, and IL-18, and triggers cell death [66]. LPS, the T3SS and the translocated YopJ protein of Yersinia were shown to induce caspase-1 activation and pyroptosis [71], [74].
Induction of pyroptosis (inflammatory death) in activated macrophages corresponds to later stages of the infection with Y. pseudotuberculosis, where inflammation and necrosis is evident from histopathology. Based on our results it is very likely that CNFY supports Yersinia-induced pyroptosis of activated phagocytes in the spleen during later stages of the infection. CNFY was shown to manipulate the number of immune cells and induce inflammatory responses. The number of macrophages, monocytes and neutrophils decreased strongly (4- to 15-fold) in all lymphoid organs three days post infection. In contrast, infection with the cnfY mutant resulted in no reduction, but rather in an increase of phagocytes when compared to uninfected control mice, and the overall inflammation of the infected tissues was considerably reduced. Similar to CNF1 of E. coli [34], it is possible that CNFY is transported by OMVs, which act as long-range toxin delivery vectors, and is then able to reduce chemotaxis and influx of professional phagocytes by constitutive Rho GTPase activation. We further demonstrate that CNFY enhances Yop delivery into phagocytes during infection. This strongly suggests that increased translocation of YopJ could stimulate cell death in the spleen. Consistent with this assumption, YopJ and CNFY promote systemic dissemination following oral infection. Work by Monack et al. [75] showed that a yopJ mutant is deficient for spread from the PPs to other lymphoid tissues (MLNs, spleen), similar to the cnfY mutant investigated in this study. Moreover, wild-type Yersinia induce apoptosis of macrophages from infected spleens [75], implying that mainly YopJ is used to eliminate immune cells in the spleen to dampen the immune response against Yersinia during infection. YopJ was also shown to subvert the NOD2/RICK/TAK1 pathway, activate caspase-1 and induce IL-1β secretion within PPs, which is associated with increased barrier permeability [76]. This suggests that CNFY action also enhances YopJ-dependent intestinal barrier disruption and promotes the dissemination of Yersinia by exploiting the mucosal inflammatory response. In addition, CNFY seems to contribute to depletion of NK cells in the spleen. It was observed that Y. pestis but not an isogenic yopM deficient mutant caused a significant global decrease in NK cell numbers [21], indicating that NK cell depletion is enhanced by CNFY-mediated activation of YopM translocation.
Here, we observed that RhoA, Rac1 and Cdc42 are activated in CNFY-intoxicated macrophages, which is reflected by the high content of actin cables/stress fibres, the formation of lamellipodia and filopodia, pronounced cell spreading and inhibited cytokinesis. Previous work demonstrated that CNFY predominantly activates RhoA in epithelial cells [27], [28]. However, a very recently published study also reported CNFY-mediated activation of Rac1 and Cdc42 in HeLa cells [77]. Use of different toxin concentrations, incubation times and cell types (human epithelial cells versus murine macrophages) in which the CNF toxins may display a different selectivity and different efficiencies of cell toxifications are likely to account for these variations. In fact, CNFY-mediated Rho GTPase activation pattern varies during intoxication whereby RhoA activation is generally more pronounced than Rac1 and Cdc42 2–3 h after toxin addition (J. Schweer, unpublished results, [77]). This suggests that at very early time points and/or under low toxin concentrations predominantly RhoA might be activated. A previous report demonstrates that Y. pseudotuberculosis selectively modulates RhoA activity (e.g. by signals triggered by the YopB/D translocon and/or from engagement of β1-integrin receptors) to induce cellular changes that control T3SS pore formation and effector translocation [26]. Here, we strengthen this observation, as CNFY-mediated stimulation of Yop delivery of Y. pseudotuberculosis was sensitive to the Rho inhibitor C3-transferase of C. botulinum, but insensitive to Rac1 inhibition by TcdBF toxin of C. difficile. In contrast, new experiments addressing the influence of CNFY on Yop translocation of Y. enterocolitica demonstrated that CNFY also stimulates effector delivery by this pathogen, although this process seemed entirely dependent on Rac and not on Rho GTPases [77]. Different YadA/InvA-promoted signalling events, differences in Yop protein abundance (e.g. RhoA-inactivating YopT is absent in YPIII) and differences in the regulation of Yop delivery by Rho GTPases between the different species may be responsible for this discrepancy.
Some effector proteins, in particular YopE, were shown to inhibit Yop delivery by inactivation of RhoA and Rac1 most likely as part of an intra-cellular control mechanism which measures and adapts the amount of protein translocated by Yersinia during infection. This is reflected by elevated levels of Yop effector translocation into epithelial cells by yopE-deficient strains [11], [63]–[65], [78]. Our analysis demonstrated that absence of YopE caused no or only a small increase in Rac1/RhoA activation and Yop translocation during infection of murine macrophages with Y. pseudotuberculosis YPIII with or without treatment with CNFY. This indicates that intracellular YopE is not able to counteract CNFY in these phagocytes. Recently published work showing that none of the Rho inhibiting effectors (YopE, YopT and YopO) could reduce the effect of CNFY on Yop translocation by Y. enterocolitica into human epithelial cells supports this observation [77]. However, we cannot exclude the possibility that other conditions (e.g. conditions which enhance (i) deamidation and subsequent ubiquitin-dependent degradation of the modified Rho GTPases or (ii) YopE translocation and activity) allow counterregulation.
Very recently, it has also been reported that NOD1, a pattern recognition receptor that senses cytosolic microbial products similar to NOD2, monitors the activation state of all three Rho GTPases. Activation of Rho GTPases triggered the NOD1 signalling cascade with consequent RIP2-mediated induction of NF-κB-dependent inflammatory responses [79]. NOD1 activation was triggered by activation of Rac1 and Cdc42 by the Salmonella effector SopE. In line with this, all three Rho GTPases, Rac1, RhoA and Cdc42 were previously shown to activate the NF-κB pathway [80] and particular Rac1 has been reported to contribute to NF-κB activation by CNF1 of E. coli by clustering the NF-κB inhibitor IκBa and components of the IκBα E3-ubiquitin ligase into membrane ruffles [79].
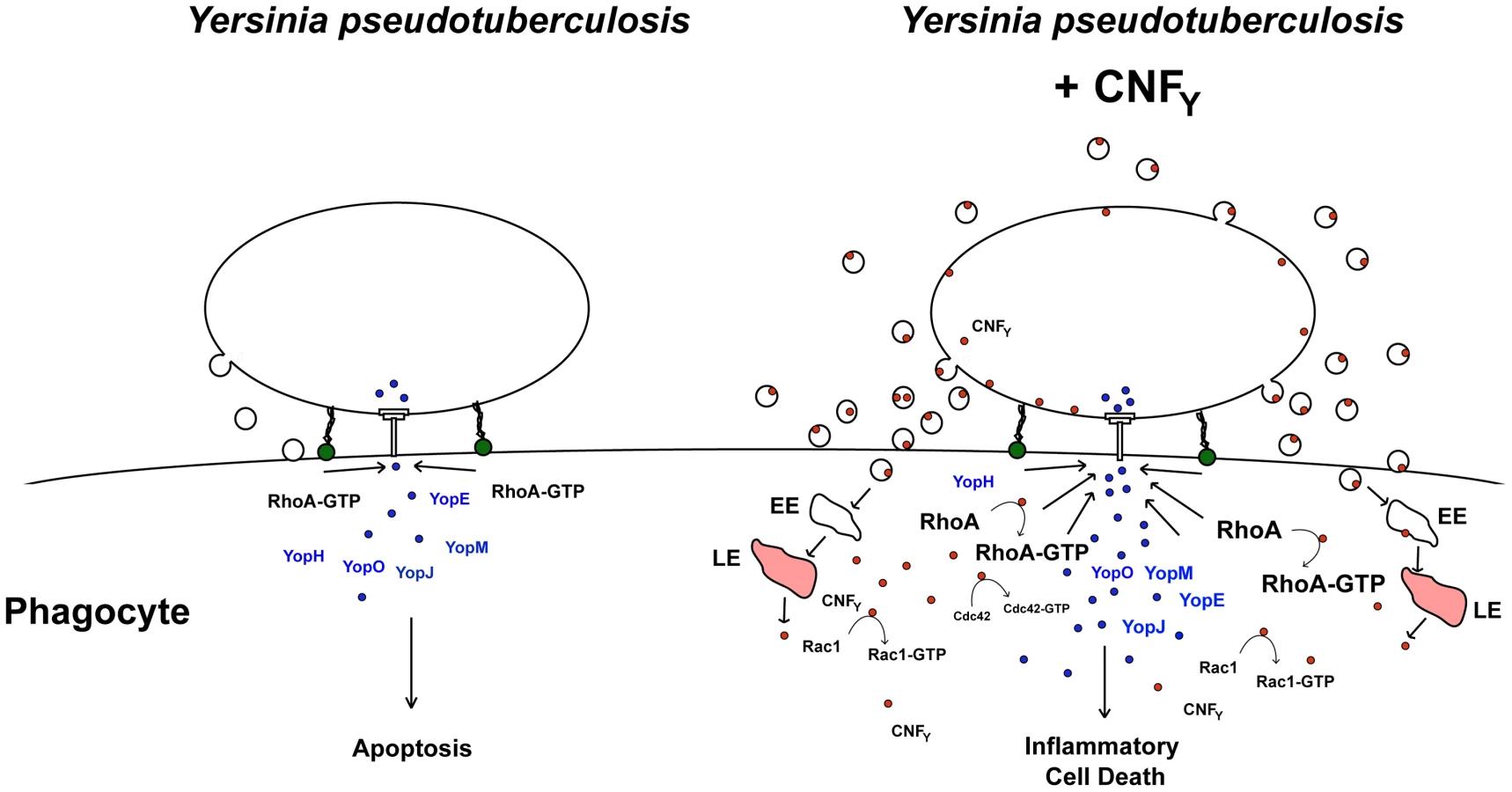
Based on our current knowledge we envision a model in which the CNFY toxin exerts its function in a multi-step process (Fig. 10). The first step corresponds to the uptake of the CNFY toxin by infiltrating innate immune cells (e.g. neutrophils, macrophages, DCs) in the early phase of the infection process. This triggers activation of the Rho GTPases, in particular RhoA, in the phagocytes. Induced actin polymerization resulting from Rho GTPase activation enhances Yop delivery into host cells to counteract innate and adaptive immune responses. As a consequence, invading immune cells are inhibited and undergo apoptosis leading to uncontrolled proliferation of the pathogens. Higher CNFY toxin concentrations by replicating pathogens potentiate activation of RhoA, Rac1 and Cdc42 which triggers inflammatory responses e.g. via the NOD1-RIP2 signalling cascade. In addition, interaction of Yersinia with increased numbers of activated macrophages causes cell death by inflammatory pyroptosis leading to strong inflammation and necrosis of the organs during later phases of the infection process. In summary, our data identify CNFY as an important Rho GTPase-activating toxin which is instrumental for Yersinia to amplify crucial virulence factor functions which determines the success of the infection and the severity of the associated disease.
Material and Methods
Ethics statement
All animal work was performed in strict accordance with the German Recommendations of the Society for Laboratory Animal Science (GV-SOLAS) and the European Health Recommendations of the Federation of Laboratory Animal Science Associations (FELASA). The animal protocol was approved by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit: animal licensing committee permission no. 33.9.42502-04-055/09. Animals were handled with appropriate care and welfare, and all efforts were made to minimize suffering.
Bacterial strains, cell culture, media and growth conditions
The strains used in this study are listed in Table 1. Overnight cultures of E. coli were routinely grown at 37°C, Yersinia strains were grown at 25°C or 37°C in LB (Luria-Bertani) broth. The antibiotics used for bacterial selection were as follows: carbenicillin 100 µg/ml, chloramphenicol 30 µg/ml, kanamycin 50 µg/ml, and gentamicin 50 µg/ml. For infection experiments, bacteria were grown at 25°C or 37°C, washed and diluted in PBS prior to infection. For invasion assays and mouse infections, bacteria were grown to stationary phase, washed and resuspended in PBS. For the in vitro Yop delivery assay, bacteria were grown in LB medium at 37°C.
Human HEp-2 cells were cultured in RPMI 1640 media with an alternative to L-glutamine with increased stability (Invitrogen) supplemented with 7.5% newborn calf serum (Sigma Aldrich). Murine J774A.1 macrophages were cultured in the same medium supplemented with 5% fetal calf serum (PAA). All cell lines were cultivated at 37°C in the presence of 5% CO2.
DNA manipulations and construction of plasmids
All DNA manipulations, PCR, restriction digestions, ligations and transformations were performed using standard techniques as described previously [81], [82]. Plasmids used in this study are listed in Table 1. Plasmid pJNS01 (cnfYhis6) was constructed by amplification of cnfY (ypk_2615) from genomic DNA of Y. pseudotuberculosis YPIII with primers II795/II795 and integrated into the XhoI/NheI sites of pET28a (Novagen).
For construction of the luxCDABE, gfpmut3.1 and lacZ reporter gene fusions encoded on plasmids pJNS2-4, the promoter region of cnfY (primer pair II896/II898) was amplified and ligated into the BamHI/SalI sites of pFU54, pFU58 and pFU68.
Plasmid pJNS10, containing the cnfY promotor region with the cnfY gene, was constructed by the insertion of a PCR fragment amplified with primers II896 and IV16 from chromosomal DNA of Y. pseudotuberculosis YPIII into the BamHI/NotI sites of pFU234. All clones were confirmed by sequencing and restriction.
Construction of mutant strains
For generation of the mutant strain YP147, a cnfY::KanR mutation encoded on plasmid pJNS05 was constructed. To do so, the kanamycin resistence gene was amplified using the kan primers (I661/I662) and plasmid pACYC177 as template. Next, Y. pseudotuberculosis YPIII genomic DNA was used as a template to amplify 500-bp regions flanking the target gene cnfY. The upstream fragment was amplified with the primer pair III710/715 of which the reverse primer contained 20 nt at the 5′-end which are homologous to the start of the kanamycin resistance gene. The downstream fragment was amplified with primer pair III712/III714 of which the forward primer contained additional 20 nt at the 3′-end which are homologous to the end of the kanamycin resistance gene. A PCR reaction was performed with the forward primer and the reverse primer using the upstream and downstream PCR products of the target gene and the kanamycin gene fragment as templates and cloned into pAKH3. The resulting plasmid pJNS05 was integrated into the cnfY locus of YPIII via conjugation as described [83]. Chromosomal integration of the fragments was selected by plating on LB supplemented with kanamycin. Excision of the plasmid including the defective cnfY allele of YPIII was obtained by plating of the strain on 10% sucrose and generated strain YP147 was analyzed by PCR and DNA sequencing. For the construction of strain YP56 (ΔphoPQ), a phoPQ::KanR PCR fragment was generated. For this purpose, the kanamycin resistance cassette was created using the primers 360/361 and pACYC177 as template. The primers contain homology regions (20 nt) to the upstream or downstream region of the phoPQ gene. A fragment including sequences of the phoPQ upstream region was generated by PCR using the primers 538/539, a fragment including sequences of the phoPQ downstream region was amplified using the primers 540/541. Primer 539 and 540 contain 20 nt of the kanamycin resistance cassette. A PCR fragment consisting of these three fragments was amplified using the primers 538/541. The product was transformed into Y. pseudotuberculosis YPIII pKOBEG-sacB and a phoPQ::KanR mutant (YP56) was generated and selected for as described [84]. For the construction of YP149, the phoPQ gene of strain IP32953 was amplified by PCR with primer III926 and III927, creating SacI and XhoI restriction sites. The fragment was cloned into plasmid pDM4, generating vector pVP1. Integration of the plasmid was obtained through conjugation of strain S17λpir pVP1 with the YP56 as described [83]. Excision of the plasmid including the defective phoPQ allele was obtained by plating of the strain on 10% sucrose. PCR and DNA sequencing proved presence of the intact phoPQ allele. To generate strain YP188 the cnfY gene was destroyed as described above for YP147 (ΔcnfY). Strains YP173, YP174 and YP217 were constructed by chromosomal integration of the YopE-β-lactamase (ETEM) fusion plasmid pSR47s-E-TEM1 into the yopE locus. Integration was obtained through conjugation of E. coli K-12 strain S17λpir pSR47s-E-TEM1 with the Y. pseudotuberculosis strains YPIII, YP101 (ΔyscS) and YP147 (ΔcnfY) as described [83]. For generation of the mutant strain YP275, a yopE::KanR mutation encoded on plasmid pJNS13 was constructed as described above for YP147. Primer V553/V554 and V555/V556 were used to amplify 500-bp regions flanking the target gene yopE. Subsequently, plasmid pJNS13 was integrated into the yopE locus of YPIII via conjugation as described [83].
β-galactosidase assays
β-galactosidase activity was determined of three independent cultures of bacteria harboring the cnfY-lacZ fusion as described [83]. The activities were calculated as follows: β-galactosidase activity OD420 • 6.75 OD600−1 • Δt (min)−1 • vol (ml)−1.
Purification of CNFY-His6
For overexpression of CNFY E. coli strain BL21λDE3 was transformed with the cnfY expression plasmid pJNS01 and grown at 37°C in LB medium to an OD600 of 0,6. Subsequently, Plac-driven expression was induced upon addition of 250 µM IPTG and grown at 17°C overnight. CNFY-His6 production was tested by westernblot analysis using an antibody directed against the His-tag (Qiagen). For purification of CNFY, cells were harvested, resuspended in 50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 10 mM imidazole and lysed with a French press (120.000 psi). The soluble CNFY-His6 extract was separated from insoluble cell material by centrifugation at 25.000 g. The CNFY-His6 protein was then purified by affinity chromatography on Ni-NTA agarose (Qiagen). The column was washed with three column volumes of 50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 20 mM imidazole and eluted with 50 mM NaH2PO4, pH 8.0, 300 mM NaCl containing 250 mM imidazole.
Visualization of the actin cytoskeleton
In order to study the influence of the recombinant CNFY protein on actin cytoskeleton rearrangements, 1×105 J774A.1 cells were incubated with purified CNFY toxin (10 nM) or PBS for 24 h. Subsequently, cells were fixed with 4% paraformaldehyde (in PBS) for 10 min at room temperature, washed with PBS and permealized with 0,1% Triton X-100 in PBS for 5 min. The actin cytoskeleton was stained with Phalloidin-FITC (0,5 µg/ml PBS; Invitrogen) for 15 min at room temperature. Cells were washed in PBS, and the nuclei were stained with DAPI (1 µg/ml in TBST) for 5 min at room temperature. Cells were visualized using a fluorescence microscope (Axiovert II with Axiocam HR, Zeiss, Germany) and the AxioVision program (Zeiss, Germany).
Rho GTPase activation assay
Activation of RhoA was tested using the Rho activation assay kit 17-294 (Millipore, Billerica, MA, USA) and activation of Rac1 and Cdc42 was determined with the Rho/Rac/Cdc42 Activation Assay Combo Kit (Cell Biolabs, San Diego, CA, USA). Approximately 1×106 cells of the macrophage cell line J774A.1 were starved for at least 20 h in RPMI 1640 without FCS, and incubated with PBS or 25 nM (3 µg/ml) recombinant CNFY for 2–3 h. To test the influence of YopE on Rac1 and RhoA activation, macrophages were subsequently infected for 20 min with wild-type strain YPIII or the isogenic yopE mutant YP275 with an MOI of 100. Cells were lysed and activation of small Rho GTPases was tested and visualized by western blotting according to the manufacturer's protocol.
Yop delivery assay
The Yop delivery assay was performed as described previously [85]. 5×104 (for fluorescence microscopy) or 1×106 (for flow cytometry) HEp-2 or J774A.1 cells were incubated with recombinant CNFY (25 nM/3 µg/ml), exoenzyme C3 transferase from C. botulinum (CT04, Cytoskeleton. Inc) (0.5 µg/ml, 1 µg/ml), C. difficile toxin TcdBF (85 ng/ml, 250 ng/ml) [60], [61], or the same amount of PBS before the cells were infected with bacteria with a MOI of 10. After 1 h cells were washed and dyed with CCF4-AM according to the manufacturer's protocol using the LiveBLAzer-FRET B/G Loading Kit from Life Technologies. Yop translocation was visualized by a fluorescence microscope (Axiovert II with Axiocam HR, Zeiss, Germany) using the AxioVision program (Zeiss, Germany) or detected with an LSR Fortessa cell analyzer (BD Bioscience). Acquired data of flow cytometry were then analyzed with FlowJo software (Treestar).
To compare Yop translocation of YPIII and YP275 (ΔyopE), both strains were pregrown at 37°C and added with an MOI of 100 to approximately 1×106 cells of murine macrophages incubated with PBS or 25 nM recombinant CNFY for 3 h. One hour post infection, cells were washed with PBS, resuspended in SDS sample buffer and separated on 12% SDS polyacrylamide gels. Proteins were blotted onto a membrane and intracellular Yops were visualized with an antiserum directed against all secreted Yops (α-Yop).
For the in vitro analysis of Yop delivery into primary cells, mesenteric lymph nodes from uninfected 6- to 8-week-old BALB/c mice were removed. To generate single-cell suspensions the cells were pressed through a 70 µm cell strainer. Harvested cells were counted using an Accuri C6 flow cytometer (BD Bioscience). Bacteria were cultured overnight at 25°C in LB medium, inoculated 1∶20 in fresh LB and grown for 3 h at 37°C prior to infection. Cells were infected for 1 h at 37°C with the wild-type (YP173), the cnfY (YP217) or the yscS (YP174) mutant encoding the YopE-β-lactamase fusion – ETEM with an MOI of 10. Infected cells were washed twice with RPMI 1640 medium supplemented with 20 mM HEPES (pH 7.0), 0.4% BSA and 50 µg/ml gentamicin to kill bacteria. Subsequently, 2×106 cells were labeled with LiveBLAzer-FRET B/G Loading Kit from Life Technologies. After staining for 1 h at room temperature, cells were prepared for flow cytometry and analyzed as described above.
Mouse infection
BALB/c female mice aged between 6- and 8-week-old were purchased from Janvier (Saint Berthevin Cedex, France) and housed under specific pathogen-free conditions according to FELASA recommendations in the animal facility of the Helmholtz Centre for Infection Research, Braunschweig. For the survival assays, mice were infected orally with approximately 2×109 bacteria of each strain. The infected mice were monitored for 14 days every day to determine the survival rate, the body weight and health status.
Bacteria used for organ burden experiments were grown over night in LB medium at 25°C, washed and resuspended in PBS. Groups of 7–10 animals were orally infected with approximately 2×108 bacteria of Y. pseudotuberculosis strains YPIII and YP147 (ΔcnfY) using a gavage needle. At specific time points after infection, mice were euthanized by CO2 asphyxiation. PPs, small intestine, caecum, MLNs, liver and spleen were isolated. The ileum was rinsed with sterile PBS and incubated with 100 µg/ml gentamicin in order to kill bacteria on the luminal surface. After 30 min, gentamicin was removed by washing with PBS. Subsequently, all organs were weighted and homogenized in PBS at 30.000 rpm for 30 sec using a Polytron PT 2100 homogenizer (Kinematica, Switzerland). To determine the bacterial load of the organs serial dilutions of the homogenates were plated on LB plates with and without antibiotics. The colony forming units (cfu) were counted and are given as cfu per g organ/tissue.
Yop delivery assay during mouse infection
BALB/c mice were infected intragastrically with 2×109 bacteria of strain YPIII-ETEM (YP173) and the isogenic cnfY mutant YP147-ETEM (YP217), wild-type YPIII and YP101-ETEM (YP174). Infection was allowed to proceed for three days. Subsequently, the infected lymphatic tissues (PPs, MLNs, and spleen) were isolated and single cell suspensions were generated in PBS by pressing the cells through a cell strainer (70 µm, Falcon). To eliminate erythrocytes, spleen cells were incubated for 3 min in lysis buffer (7.8 mM NH4Cl, 10 mM KHCO3, 100 µM EDTA). All cells were resuspended in PBS containing 0.2% BSA and total cell number was determined using an Accuri C6 flow cytometer (BD Bioscience). For flow cytometry analysis 1×106 cells were transferred per tube and FcγR was blocked using CD16/CD32 (BioXCell; anti-mouse CD16/CD32) antibody for 15 min at 4°C. Immune cells were first stained for 15 min at 4°C using a biotin-conjugated antibody against CD19. Subsequently, other cellular surface marker for innate immune cells or T cell panel were stained for 20 min at 4°C in FACS buffer (PBS+0.2% BSA) using the following antibodies: SA-PerCP-Cy5.5, CD11c-APC, CD11c-PE-Cy7, Gr1-A750, CD3-PE, CD4-APC-Cy7, CD3-PE-Cy7, NKp46-PE and CD25-APC. Samples were washed twice in FACS buffer and labeled with 1 µg/ml CCF4-AM using the LiveBLAzer-FRET B/G Loading Kit (Life Technologies) for 1 hour at 20°C in the presence of 1.5 mM probenecid (Sigma) and 50 µg/ml gentamicin. Cell subsets were defined as following: B cells (CD19+ CD3−), T cells (CD19− CD3+), NK cells (CD19− CD3− NKp46+), neutrophils (CD19− CD3− CD49b− Ly6G+ CD11b+), macrophages/monocytes (CD19− CD3− CD49b− Ly6G− CD11b+), and DCs (CD19− CD3− CD49b− Ly-6G− B220− F4/80− CD11c+). Cells were analyzed in a LSR Fortessa cell analyzer (BD Bioscience). Acquired data were analyzed with FlowJo software (Treestar). Cells from tissues that were not treated with CFF4-AM and/or antibodies were used as negative controls.
CNFY influence in the host immune response
To characterize the host immune response induced upon infection with the wild-type strain YPIII or the isogenic cnfY mutant strain YP147, mice were orally infected with approximately 2×108 bacteria of Y. pseudotuberculosis strains YPIII or YP147 (Δ cnfY). Three and six days after infection, PPs, MLNs and spleen were isolated. Single cell suspensions were obtained by mechanical disruption of the organs through a cell strainer. To eliminate erythrocytes, spleen cells were also incubated for 3 min in erythrolysis buffer (7.8 mM NH4Cl, 10 mM KHCO3, 100 µM EDTA). All cells were resuspended in FACS buffer and total cell number was determined using an Accuri C6 flow cytometer (BD Bioscience). Amounts of 1–2×106 cells were transferred per tube. To exclude dead cells from the analysis live/dead staining (Invitrogen; Live/dead fixable blue dead cell stain kit, UV excitation) was performed for 30 min. FcγR and IgG were blocked by 15 min incubation with CD16/CD32 (BioXCell; anti-mouse CD16/CD32) and ratIgG (Jackson ImmunoResearch; ChromPure Rat IgG, whole molecule) antibodies.
Cellular surface markers for either lymphoid or myeloid panel were stained for 15 min at 4°C in PBS-BSA (0.2%) using the following antibodies: CD3-APC, CD4-PerCP-Cy5.5, CD8-eFluor450, CD335-PerCP-Cy5.5, CD11b-PacificBlue, CD19-Biotin, CD45R-PerCP-Cy5.5, F4/80-PE and CD11c-APCeFluor780 from BD Bioscience, and CD19-FITC, CD49b-Biotin, and Ly-6C-APC from BioLegend. All antibodies were titrated for optimal staining conditions. Biotin-conjugated antibodies were incubated with streptavidin for 15 min at 4°C. After staining cells were fixed with the Foxp3 staining buffer set from eBioscience. Cells were then washed twice and resuspended in 200 µl FACS buffer. Samples were loaded into an LSR Fortessa cell analyzer (BD Bioscience). Acquired data were analyzed with FlowJo software (Treestar).
In vivo expression of CNFY
YPIII harboring a PcnfY::gfpmut3.1 fusion (pJNS03) and a PgapA::dsRed2 expression construct (pFU228) were grown in LB medium at 25°C overnight. Mice were infected orally with 2×108 bacteria. After five days mice were sacrificed by CO2 asphyxiation. For cryosections, the small intestine, colon, caecum, MLNs, spleen and liver were frozen in Tissue-Tek OCT freezing medium (Sakura Finetek) on dry ice. Sections of 8–10 mm were prepared using a Zeiss cryostat Hyrax C50 and mounted on SuperFrost Plus slides (Thermo Scientific). Air-dried sections were fixed for 10 min in ice-cold acetone and washed twice with PBS. For visualization of the nuclei in the fixed tissue, samples were stained with 49,6- diamidino-2-phenylindole (DAPI, Sigma) for 3–5 min, air-dried and mounted with 80% glycerol in PBS. Localization of yersiniae in the infected tissues and expression of the PcnfY::gfpmut3.1 fusion of these bacteria were visualized by a fluorescence microscope (Axiovert II with Axiocam HR, Zeiss, Germany) using the AxioVision program (Zeiss, Germany).
To detect the cnfY gene expression during the infection process Y. pseudotuberculosis wild-type strain YPIII harboring the PcnfY::luxCDABE fusion vector pJNS02 or the empty vector pFU54 were grown in LB medium at 25°C overnight. About 2×108 luminescent bacteria were used for oral infection. For in vivo imaging mice were anesthesized with isoflurane and the bacterial infection was followed daily using the IVIS Lumina system (Xenogen). To ensure maintenance of the plasmids during the course of infection, the bacteria were isolated from the small intestine, colon, caecum, MLNs, spleen and liver and tested for the presence of the plasmid.
Histology
MLNs, spleen, liver, small intestine, caecum and colon were analyzed histopathologically of three mice per group. According to standard histology procedures, organs were fixed in 4% neutrally buffered formaldehyde for 24 to 48 h, embedded in paraffin and 3 µm sections were stained with hematoxylin-eosin (H & E). Sections were evaluated by light-microscopy blinded to the experimental groups.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Carniel E, Autenrieth A, Cornelis G, Fukushima H, Guinet F (2002) Y. enterocolitica and Y. pseudotuberculosis. In: Dworkin M, Falkow S, Rosenberg E, Scheifer KH, Stackebrandt E, editors. The Prokaryotes: An evolving electronic resource for the microbiological community. New York: Springer.
2. ViboudGI, BliskaJB (2005) Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol 59: 69–89.
3. CornelisGR, BolandA, BoydAP, GeuijenC, IriarteM, et al. (1998) The virulence plasmid of Yersinia, an antihost genome. Microbiol Mol Biol Rev 62: 1315–1352.
4. CornelisGR (2006) The type III secretion injectisome. Nat Rev Microbiol 4: 811–825.
5. KudryashevM, StentaM, SchmelzS, AmstutzM, WiesandU, et al. (2013) In situ structural analysis of the Yersinia enterocolitica injectisome. eLIFE 2: e00792.
6. BliskaJB (2000) Yop effectors of Yersinia spp. and actin rearrangements. Trends Microbiol 8: 205–208.
7. BlackDS, BliskaJB (1997) Identification of p130Cas as a substrate of Yersinia YopH (Yop51), a bacterial protein tyrosine phosphatase that translocates into mammalian cells and targets focal adhesions. Embo J 16: 2730–2744.
8. BliskaJB, GuanKL, DixonJE, FalkowS (1991) Tyrosine phosphate hydrolysis of host proteins by an essential Yersinia virulence determinant. Proc Natl Acad Sci U S A 88: 1187–1191.
9. PerssonC, CarballeiraN, Wolf-WatzH, FallmanM (1997) The PTPase YopH inhibits uptake of Yersinia, tyrosine phosphorylation of p130Cas and FAK, and the associated accumulation of these proteins in peripheral focal adhesions. EMBO J 16: 2307–2318.
10. GuanKL, DixonJE (1990) Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science 249: 553–556.
11. WongKW, IsbergRR (2005) Yersinia pseudotuberculosis spatially controls activation and misregulation of host cell Rac1. PLoS Pathog 1: e16.
12. LemaitreN, SebbaneF, LongD, HinnebuschBJ (2006) Yersinia pestis YopJ suppresses tumor necrosis factor alpha induction and contributes to apoptosis of immune cells in the lymph node but is not required for virulence in a rat model of bubonic plague. Infect Immun 74: 5126–5131.
13. MonackDM, MecsasJ, GhoriN, FalkowS (1997) Yersinia signals macrophages to undergo apoptosis and YopJ is necessary for this cell death. Proc Natl Acad Sci U S A 94: 10385–10390.
14. MukherjeeS, KeitanyG, LiY, WangY, BallHL, et al. (2006) Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312: 1211–1214.
15. ZhengY, LiloS, BrodskyIE, ZhangY, MedzhitovR, et al. (2011) A Yersinia effector with enhanced inhibitory activity on the NF-kappaB pathway activates the NLRP3/ASC/caspase-1 inflammasome in macrophages. PLoS Pathog 7: e1002026.
16. ZhangY, BliskaJB (2010) YopJ-promoted cytotoxicity and systemic colonization are associated with high levels of murine interleukin-18, gamma interferon, and neutrophils in a live vaccine model of Yersinia pseudotuberculosis infection. Infect Immun 78: 2329–2341.
17. SkrzypekE, CowanC, StraleySC (1998) Targeting of the Yersinia pestis YopM protein into HeLa cells and intracellular trafficking to the nucleus. Mol Microbiol 30: 1051–1065.
18. HentschkeM, BernekingL, Belmar CamposC, BuckF, RuckdeschelK, et al. (2010) Yersinia virulence factor YopM induces sustained RSK activation by interfering with dephosphorylation. PLoS One 5: e13165.
19. McDonaldC, VacratsisPO, BliskaJB, DixonJE (2003) The Yersinia virulence factor YopM forms a novel protein complex with two cellular kinases. J Biol Chem 278: 18514–18523.
20. YeZ, KerschenEJ, CohenDA, KaplanAM, van RooijenN, et al. (2009) Gr1+ cells control growth of YopM-negative Yersinia pestis during systemic plague. Infect Immun 77: 3791–3806.
21. KerschenEJ, CohenDA, KaplanAM, StraleySC (2004) The plague virulence protein YopM targets the innate immune response by causing a global depletion of NK cells. Infect Immun 72: 4589–4602.
22. ThorslundSE, ErmertD, FahlgrenA, ErttmannSF, NilssonK, et al. (2012) Role of YopK in Yersinia pseudotuberculosis resistance against polymorphonuclear leukocyte defense. Infect Immun 81: 11–22.
23. ThorslundSE, EdgrenT, PetterssonJ, NordfelthR, SellinME, et al. (2011) The RACK1 signaling scaffold protein selectively interacts with Yersinia pseudotuberculosis virulence function. PLoS One 6: e16784.
24. DewoodyR, MerrittPM, HouppertAS, MarketonMM (2011) YopK regulates the Yersinia pestis type III secretion system from within host cells. Mol Microbiol 79: 1445–1461.
25. ViboudGI, BliskaJB (2001) A bacterial type III secretion system inhibits actin polymerization to prevent pore formation in host cell membranes. EMBO J 20: 5373–5382.
26. MejiaE, BliskaJB, ViboudGI (2008) Yersinia controls type III effector delivery into host cells by modulating Rho activity. PLoS Pathog 4: e3.
27. LockmanHA, GillespieRA, BakerBD, ShakhnovichE (2002) Yersinia pseudotuberculosis produces a cytotoxic necrotizing factor. Infect Immun 70: 2708–2714.
28. HoffmannC, PopM, LeemhuisJ, SchirmerJ, AktoriesK, et al. (2004) The Yersinia pseudotuberculosis cytotoxic necrotizing factor (CNFY) selectively activates RhoA. J Biol Chem 279: 16026–16032.
29. LemonnierM, LandraudL, LemichezE (2007) Rho GTPase-activating bacterial toxins: from bacterial virulence regulation to eukaryotic cell biology. FEMS Microbiol Rev 31: 515–534.
30. KnustZ, SchmidtG (2010) Cytotoxic Necrotizing Factors (CNFs)-A Growing Toxin Family. Toxins (Basel) 2: 116–127.
31. CaprioliA, FalboV, RodaLG, RuggeriFM, ZonaC (1983) Partial purification and characterization of an Escherichia coli toxic factor that induces morphological cell alterations. Infect Immun 39: 1300–1306.
32. BlancoJ, GonzalezEA, GarciaS, BlancoM, RegueiroB, et al. (1988) Production of toxins by Escherichia coli strains isolated from calves with diarrhoea in galicia (north-western Spain). Vet Microbiol 18: 297–311.
33. KouokamJC, WaiSN, FallmanM, DobrindtU, HackerJ, et al. (2006) Active cytotoxic necrotizing factor 1 associated with outer membrane vesicles from uropathogenic Escherichia coli. Infect Immun 74: 2022–2030.
34. DavisJM, CarvalhoHM, RasmussenSB, O'BrienAD (2006) Cytotoxic necrotizing factor type 1 delivered by outer membrane vesicles of uropathogenic Escherichia coli attenuates polymorphonuclear leukocyte antimicrobial activity and chemotaxis. Infect Immun 74: 4401–4408.
35. BuetowL, FlatauG, ChiuK, BoquetP, GhoshP (2001) Structure of the Rho-activating domain of Escherichia coli cytotoxic necrotizing factor 1. Nat Struct Biol 8: 584–588.
36. LemichezE, FlatauG, BruzzoneM, BoquetP, GauthierM (1997) Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol Microbiol 24: 1061–1070.
37. ContaminS, GalmicheA, DoyeA, FlatauG, BenmerahA, et al. (2000) The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endocytosed by a clathrin-independent mechanism and enters the cytosol by an acidic-dependent membrane translocation step. Mol Biol Cell 11: 1775–1787.
38. BlumenthalB, HoffmannC, AktoriesK, BackertS, SchmidtG (2007) The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect Immun 75: 3344–3353.
39. KnustZ, BlumenthalB, AktoriesK, SchmidtG (2009) Cleavage of Escherichia coli cytotoxic necrotizing factor 1 is required for full biologic activity. Infect Immun 77: 1835–1841.
40. GerhardR, SchmidtG, HofmannF, AktoriesK (1998) Activation of Rho GTPases by Escherichia coli cytotoxic necrotizing factor 1 increases intestinal permeability in Caco-2 cells. Infect Immun 66: 5125–5131.
41. SchmidtG, SehrP, WilmM, SelzerJ, MannM, et al. (1997) Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 387: 725–729.
42. LermM, SelzerJ, HoffmeyerA, RappUR, AktoriesK, et al. (1999) Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: activation of c-Jun N-terminal kinase in HeLa cells. Infect Immun 67: 496–503.
43. SchmidtG, SelzerJ, LermM, AktoriesK (1998) The Rho-deamidating cytotoxic necrotizing factor 1 from Escherichia coli possesses transglutaminase activity. Cysteine 866 and histidine 881 are essential for enzyme activity. J Biol Chem 273: 13669–13674.
44. FlatauG, LemichezE, GauthierM, ChardinP, ParisS, et al. (1997) Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 387: 729–733.
45. MayM, KolbeT, WangT, SchmidtG, GenthH (2012) Increased cell-matrix adhesion upon constitutive activation of Rho proteins by cytotoxic necrotizing factors from E. coli and Y. pseudotuberculosis. J Signal Transduct 2012: 570183.
46. HuelsenbeckSC, MayM, SchmidtG, GenthH (2009) Inhibition of cytokinesis by Clostridium difficile toxin B and cytotoxic necrotizing factors–reinforcing the critical role of RhoA in cytokinesis. Cell Motil Cytoskeleton 66: 967–975.
47. FalzanoL, FiorentiniC, DonelliG, MichelE, KocksC, et al. (1993) Induction of phagocytic behaviour in human epithelial cells by Escherichia coli cytotoxic necrotizing factor type 1. Mol Microbiol 9: 1247–1254.
48. HofmanP, Le NegrateG, MograbiB, HofmanV, BrestP, et al. (2000) Escherichia coli cytotoxic necrotizing factor-1 (CNF-1) increases the adherence to epithelia and the oxidative burst of human polymorphonuclear leukocytes but decreases bacteria phagocytosis. J Leukoc Biol 68: 522–528.
49. VisvikisO, BoyerL, TorrinoS, DoyeA, LemonnierM, et al. (2011) Escherichia coli producing CNF1 toxin hijacks Tollip to trigger Rac1-dependent cell invasion. Traffic 12: 579–590.
50. HopkinsAM, WalshSV, VerkadeP, BoquetP, NusratA (2003) Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. J Cell Sci 116: 725–742.
51. HofmanP, FlatauG, SelvaE, GauthierM, Le NegrateG, et al. (1998) Escherichia coli cytotoxic necrotizing factor 1 effaces microvilli and decreases transmigration of polymorphonuclear leukocytes in intestinal T84 epithelial cell monolayers. Infect Immun 66: 2494–2500.
52. MillsM, MeysickKC, O'BrienAD (2000) Cytotoxic necrotizing factor type 1 of uropathogenic Escherichia coli kills cultured human uroepithelial 5637 cells by an apoptotic mechanism. Infect Immun 68: 5869–5880.
53. GrabensteinJP, MarceauM, PujolC, SimonetM, BliskaJB (2004) The response regulator PhoP of Yersinia pseudotuberculosis is important for replication in macrophages and for virulence. Infect Immun 72: 4973–4984.
54. BöhmeK, SteinmannR, KortmannJ, SeekircherS, HerovenAK, et al. (2012) Concerted actions of a thermo-labile regulator and a unique intergenic RNA thermosensor control Yersinia virulence. PLoS Pathog 8: e1002518.
55. LogsdonLK, MecsasJ (2003) Requirement of the Yersinia pseudotuberculosis effectors YopH and YopE in colonization and persistence in intestinal and lymph tissues. Infect Immun 71: 4595–4607.
56. HarmonDE, DavisAJ, CastilloC, MecsasJ (2010) Identification and characterization of small-molecule inhibitors of Yop translocation in Yersinia pseudotuberculosis. Antimicrob Agents Chemother 54: 3241–3254.
57. GaoW, XingB, TsienRY, RaoJ (2003) Novel fluorogenic substrates for imaging beta-lactamase gene expression. J Am Chem Soc 125: 11146–11147.
58. ZlokarnikG, NegulescuPA, KnappTE, MereL, BurresN, et al. (1998) Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science 279: 84–88.
59. DurandEA, Maldonado-ArochoFJ, CastilloC, WalshRL, MecsasJ (2010) The presence of professional phagocytes dictates the number of host cells targeted for Yop translocation during infection. Cell Microbiol 12: 1064–1082.
60. HuelsenbeckJ, DregerS, GerhardR, BarthH, JustI, et al. (2007) Difference in the cytotoxic effects of toxin B from Clostridium difficile strain VPI 10463 and toxin B from variant Clostridium difficile strain 1470. Infect Immun 75: 801–809.
61. JustI, HuelsenbeckSC, GenthH (2010) Clostridium botulinum C3 exoenzyme: Rho-inactivating tool in cell biology and a neurotrophic agent. The Open Toxicology Journal 3: 19–23.
62. BlackDS, BliskaJB (2000) The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol Microbiol 37: 515–527.
63. AiliM, IsakssonEL, HallbergB, Wolf-WatzH, RosqvistR (2006) Functional analysis of the YopE GTPase-activating protein (GAP) activity of Yersinia pseudotuberculosis. Cell Microbiol 8: 1020–1033.
64. AiliM, HallbergB, Wolf-WatzH, RosqvistR (2002) GAP activity of Yersinia YopE. Methods Enzymol 358: 359–370.
65. SongsungthongW, HigginsMC, RolanHG, MurphyJL, MecsasJ (2010) ROS-inhibitory activity of YopE is required for full virulence of Yersinia in mice. Cell Microbiol 12: 988–1001.
66. BergsbakenT, CooksonBT (2007) Macrophage activation redirects Yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog 3: e161.
67. Etienne-MannevilleS, HallA (2002) Rho GTPases in cell biology. Nature 420: 629–635.
68. Rippere-LampeKE, LangM, CeriH, OlsonM, LockmanHA, et al. (2001) Cytotoxic necrotizing factor type 1-positive Escherichia coli causes increased inflammation and tissue damage to the prostate in a rat prostatitis model. Infect Immun 69: 6515–6519.
69. Rippere-LampeKE, O'BrienAD, ConranR, LockmanHA (2001) Mutation of the gene encoding cytotoxic necrotizing factor type 1 (cnf(1)) attenuates the virulence of uropathogenic Escherichia coli. Infect Immun 69: 3954–3964.
70. BergsbakenT, CooksonBT (2009) Innate immune response during Yersinia infection: critical modulation of cell death mechanisms through phagocyte activation. J Leukoc Biol 86: 1153–1158.
71. PhilipNH, BrodskyIE (2012) Cell death programs in Yersinia immunity and pathogenesis. Front Cell Infect Microbiol 2: 149.
72. FinkSL, CooksonBT (2005) Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73: 1907–1916.
73. SavillJ, DransfieldI, GregoryC, HaslettC (2002) A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol 2: 965–975.
74. BrodskyIE, PalmNW, SadanandS, RyndakMB, SutterwalaFS, et al. (2010) A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7: 376–387.
75. MonackDM, MecsasJ, BouleyD, FalkowS (1998) Yersinia-induced apoptosis in vivo aids in the establishment of a systemic infection of mice. J Exp Med 188: 2127–2137.
76. MeinzerU, BarreauF, Esmiol-WelterlinS, JungC, VillardC, et al. (2012) Yersinia pseudotuberculosis effector YopJ subverts the Nod2/RICK/TAK1 pathway and activates caspase-1 to induce intestinal barrier dysfunction. Cell Host Microbe 11: 337–351.
77. WoltersM, BoyleEC, LardongK, TruelzschK, SteffenA, et al. (2013) Cytotoxic necrotizing factor-Y boosts Yersinia effector translocation by activating Rac. J Biol Chem 288(32): 23543–53.
78. AiliM, IsakssonEL, CarlssonSE, Wolf-WatzH, RosqvistR, et al. (2008) Regulation of Yersinia Yop-effector delivery by translocated YopE. Int J Med Microbiol 298: 183–192.
79. KeestraAM, WinterMG, AuburgerJJ, FrassleSP, XavierMN, et al. (2013) Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature 496: 233–237.
80. BoyerL, TravaglioneS, FalzanoL, GauthierNC, PopoffMR, et al. (2004) Rac GTPase instructs nuclear factor-kappaB activation by conveying the SCF complex and IkBalpha to the ruffling membranes. Mol Biol Cell 15: 1124–1133.
81. Sambrook J (2001) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratories,.
82. Miller JH (1992) A short course in bacterial genetic: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Habor, New York: Laboratories CSH.
83. NagelG, LahrzA, DerschP (2001) Environmental control of invasin expression in Yersinia pseudotuberculosis is mediated by regulation of RovA, a transcriptional activator of the SlyA/Hor family. Mol Microbiol 41: 1249–1269.
84. HerovenAK, DerschP (2006) RovM, a novel LysR-type regulator of the virulence activator gene rovA, controls cell invasion, virulence and motility of Yersinia pseudotuberculosis. Mol Microbiol 62: 1469–1483.
85. MarketonMM, DePaoloRW, DeBordKL, JabriB, SchneewindO (2005) Plague bacteria target immune cells during infection. Science 309: 1739–1741.
86. StudierFW, MoffattBA (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression. J Mol Biol 189: 113–130.
87. ManoilC, BeckwithJ (1986) A genetic approach to analyzing membrane protein topology. Science 233: 1403–1408.
88. SimonR, PrieferU, PuehlerA (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Biotechnology 1: 784–791.
89. BolinI, ForsbergA, NorlanderL, SkurnikM, Wolf-WatzH (1988) Identification and mapping of the temperature-inducible, plasmid-encoded proteins of spp. Infect Immun 56: 343–348.
90. ChainPS, CarnielE, LarimerFW, LamerdinJ, StoutlandPO, et al. (2004) Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A 101: 13826–13831.
91. ChangAC, CohenSN (1978) Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol 134: 1141–1156.
92. MiltonDL, O'TooleR, HorstedtP, Wolf-WatzH (1996) Flagellin A is essential for the virulence of Vibrio anguillarum. J Bacteriol 178: 1310–1319.
93. UliczkaF, PisanoF, KochutA, OpitzW, HerbstK, et al. (2011) Monitoring of gene expression in bacteria during infections using an adaptable set of bioluminescent, fluorescent and colorigenic fusion vectors. PLoS One 6: e20425.
94. PisanoF, KochutA, UliczkaF, GeyerR, StolzT, et al. (2012) In vivo-induced InvA-like autotransporters Ifp and InvC of Yersinia pseudotuberculosis promote interactions with intestinal epithelial cells and contribute to virulence. Infect Immun 80: 1050–1064.
95. DerbiseA, LesicB, DacheuxD, GhigoJM, CarnielE (2003) A rapid and simple method for inactivating chromosomal genes in Yersinia. FEMS Immunol Med Microbiol 38: 113–116.
Štítky
Hygiena a epidemiológia Infekčné lekárstvo LaboratóriumČlánok vyšiel v časopise
PLOS Pathogens
2013 Číslo 11
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Očkování proti virové hemoragické horečce Ebola experimentální vakcínou rVSVDG-ZEBOV-GP
- Koronavirus hýbe světem: Víte jak se chránit a jak postupovat v případě podezření?
Najčítanejšie v tomto čísle
- Baculoviruses: Sophisticated Pathogens of Insects
- Identification of the Adenovirus E4orf4 Protein Binding Site on the B55α and Cdc55 Regulatory Subunits of PP2A: Implications for PP2A Function, Tumor Cell Killing and Viral Replication
- A Unique SUMO-2-Interacting Motif within LANA Is Essential for KSHV Latency
- Turning Defense into Offense: Defensin Mimetics as Novel Antibiotics Targeting Lipid II