
Systémová sklerodermie
Systemic sclerosis
Systemic sclerosis (SSc) is a rare, chronic connective tissue disease affecting the skin, musculoskeletal system and internal organs. SSc occurs predominantly in women, begins usually in middle age, and the overall survival rate is reduced – 70% of patients survive 10 years. Despite the advances in pharmacotherapy of organ complications and new insights into the pathogenesis of SSc, there is no effective treatment for this serious disorder. The aim of this review article is to introduce this rare disease, its main symptoms, basic principles of diagnosis and current treatment, and contribute to early diagnosis and better prognosis of patients with SSc.
Keywords:
systemic sclerosis – rheumatic diseases – connective tissue disease – fibrosis – vasculopathy – autoimmunity
:
M. Tomčík
:
Revmatologický ústav a Revmatologická klinika 1. LF UK, Praha
:
Kardiol Rev Int Med 2014, 16(5): 414-419
:
Internal Medicine
Systémová sklerodermie (SSc) je vzácné, chronické systémové onemocnění pojiva postihující kůži, pohybové ústrojí a vnitřní orgány. SSc postihuje častěji ženy, začíná obvykle ve středním věku a celková doba přežití je zkrácená – 70 % pacientů přežívá 10 let. I přes pokroky ve farmakoterapii orgánových komplikací a nové poznatky v patogenezi SSc dosud neexistuje účinná léčba tohoto závažného onemocnění. Cílem tohoto přehledového článku je seznámit čitatele s touto vzácnou nemocí, hlavními příznaky, základy diagnostiky a principy současné léčby, a přispět tak ke včasné diagnóze a k lepší prognóze pacientů se SSc.
Klíčová slova:
systémová sklerodermie – revmatická onemocnění – systémové onemocnění pojiva – fibróza – vaskulopatie – autoimunita
Úvod
Systémová sklerodermie (SSc) je vzácné, chronické systémové onemocnění pojiva postihující kůži, pohybové ústrojí a vnitřní orgány. Název tohoto onemocnění je odvozen z řeckých slov „scleros“ a „derma“ – tuhá kůže a byl prvně použit Gintracem v roce 1847. První zmínka o tuhé kůži, na které nelze vytvořit řasu, však pochází již od Hippokrata asi 400 let př. n. l. Etiologie SSc dosud nebyla objasněna a v patogenezi tohoto onemocnění se uplatňují tři základní procesy – vaskulopatie, fibróza a porucha imunity. SSc postihuje častěji ženy, začíná obvykle ve středním věku a celková doba přežití je zkrácená – 10 let přežívá 70 % pacientů. I přes pokroky ve farmakoterapii orgánových komplikací a nové poznatky v patogenezi SSc dosud neexistuje účinná léčba tohoto závažného onemocnění [1].
Základní příznaky
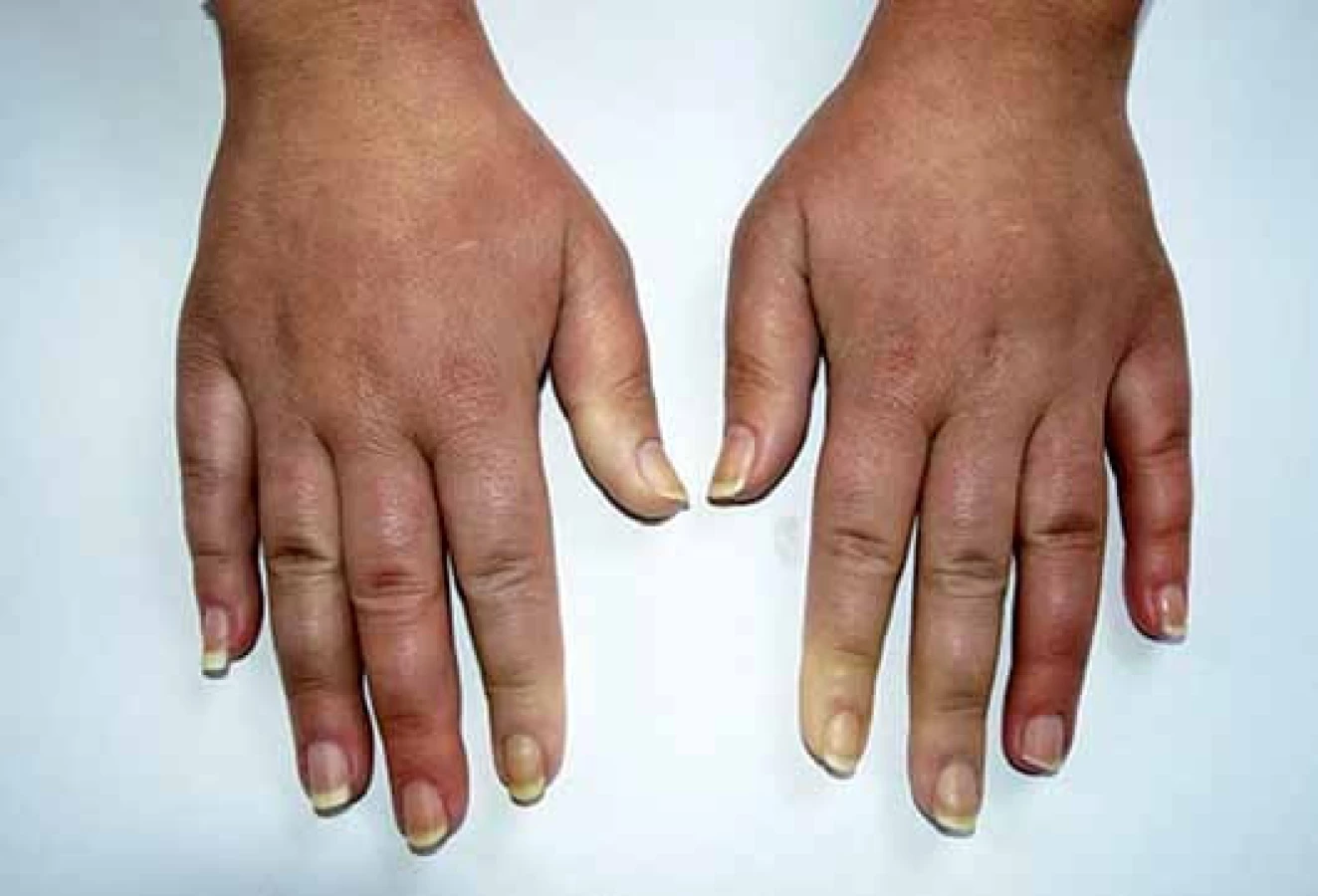
Klinickým projevům dominuje Raynaudův fenomén (epizodické barevné změny aker od bílé přes fialovou až po červenou – vyvolané chladem nebo stresem) (obr. 1), trofické změny, tuhnutí a ztluštění kůže (obr. 2) a postižení pohybového ústrojí a vnitřních orgánů – gastrointestinálního traktu, plic, srdce a ledvin. SSc je značně heterogenní a polymorfní onemocnění a třídí se podle rozsahu postižení kůže a vnitřních orgánů na několik forem (tab. 1) [2]. V laboratorním nálezu je charakteristický průkaz nespecifických antinukleárních protilátek (ANA) a specifických autoprotilátek, které jsou asociovány s různým typem kožního a/ nebo orgánového postižení u SSc: zejména proti DNA topoizomeráze I (Scl70), centromerám (ACA) a RNA polymeráze III (tab. 2) [1,3].
![Třídění systémové sklerodermie [2].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/d3bcab39d63502ea247b8110e0d5e369.png)
![Autoprotilátky a jejich asociace s klinickým obrazem systémové sklerodermie [3].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/9d9b7b0913679e9b32806d84ca3abab7.png)
Epidemiologie
Prevalence SSc je 31 – 350 případů na milion a incidence činí 1,2 – 23 nových případů na milion za rok. SSc postihuje ženy průměrně třikrát častěji než muže (ještě výraznější poměr je u mladších jedinců) a manifestuje se převážně ve středním věku (průměrný věk 45 – 50 let). Častější výskyt SSc byl popsán u Afroameričanů, indiánů kmene Choctaw, Australanů a Japonců [4,5].
Etiologie a patogeneze
Etiologie SSc je neznámá. Při vzniku tohoto onemocnění se mohou uplatnit genetické i vnější faktory a významnou roli zde mají humorální a imunologické děje. Bylo popsáno několik genů, které mohou být asociovány s predispozicí ke vzniku SSc, ale etiologii SSc nelze vysvětlit jen na základě genetických faktorů. Konkordance u dvojčat je jen 5 %, ale prevalence SSc v rodinách (1,6 %) je několikrát vyšší než v běžné populaci (0,003 – 0,03 %) a pozitivní rodinná anamnéza pořád zůstává nejsilnějším rizikovým faktorem pro rozvoj SSc [6]. Asociace mezi vnějšími vlivy (profesní expozice, viry a bakterie) a rozvojem SSc byly dostatečně zkoumány, ale role těchto faktorů není dosud uspokojivě objasněna. Nejsilnější důkazy existují pro sloučeniny křemíku a organická rozpouštědla, avšak jejich podíl na všech případech SSc je jen minimální [7].
V patogenezi SSc dominují tři navzájem se prolínající patologické procesy:
- poškození endotelu a obliterující vaskulopatie cévních lůžek,
- zánět a autoimunita a
- progresivní fibróza [8].
Vaskulární poškození je nejčasnějším a pravděpodobně i primárním procesem v patogenezi SSc a je histopatologicky prokazatelné před rozvojem fibrózy a objevením se prvního klinického příznaku, kterým obvykle bývá Raynaudův fenomén (RAF). K dalším manifestacím vaskulopatie u SSc patří teleangiektazie, typické změny na kapilárách nehtových valů, trofické defekty, plicní arteriální hypertenze, antrální vaskulární ektazie a renální krize s maligní hypertenzí [9]. V patogenezi SSc má význam jak vrozená, tak získaná imunita. V postižených tkáních lze histopatologicky prokázat perivaskulární infiltráty (zejména T a B lymfocyty, makrofágy a žírné buňky), které přispívají k rozvoji SSc sekrecí různých profibrotických cytokinů a chemokinů a k produkci autoprotilátek [10]. Fibróza bývá často dominantním rysem SSc a postihuje jak kůži, tak vnitřní orgány – zejména plíce, srdce a gastrointestinální trakt. Dochází k postupnému nahrazení funkční tkáně postižených orgánů kolagenními vlákny a dalšími složkami extracelulární matrix, což vede ke ztrátě jejich funkce a selhávání [8].
Klinický obraz
SSc je velmi heterogenní nemoc co do průběhu i rozsahu postižení. Podle nedávné studie u téměř 500 pacientů se SSc patří mezi pět nejvýznamnějších symptomů uváděných pacienty únava (89 %), RAF (86 %), ztuhlost rukou (81 %), artralgie (81 %) a poruchy spánku (76 %). K celkovým projevům dále patří nechutenství, hubnutí, febrilie a později reaktivní deprese [1,8,11].
Kožní postižení
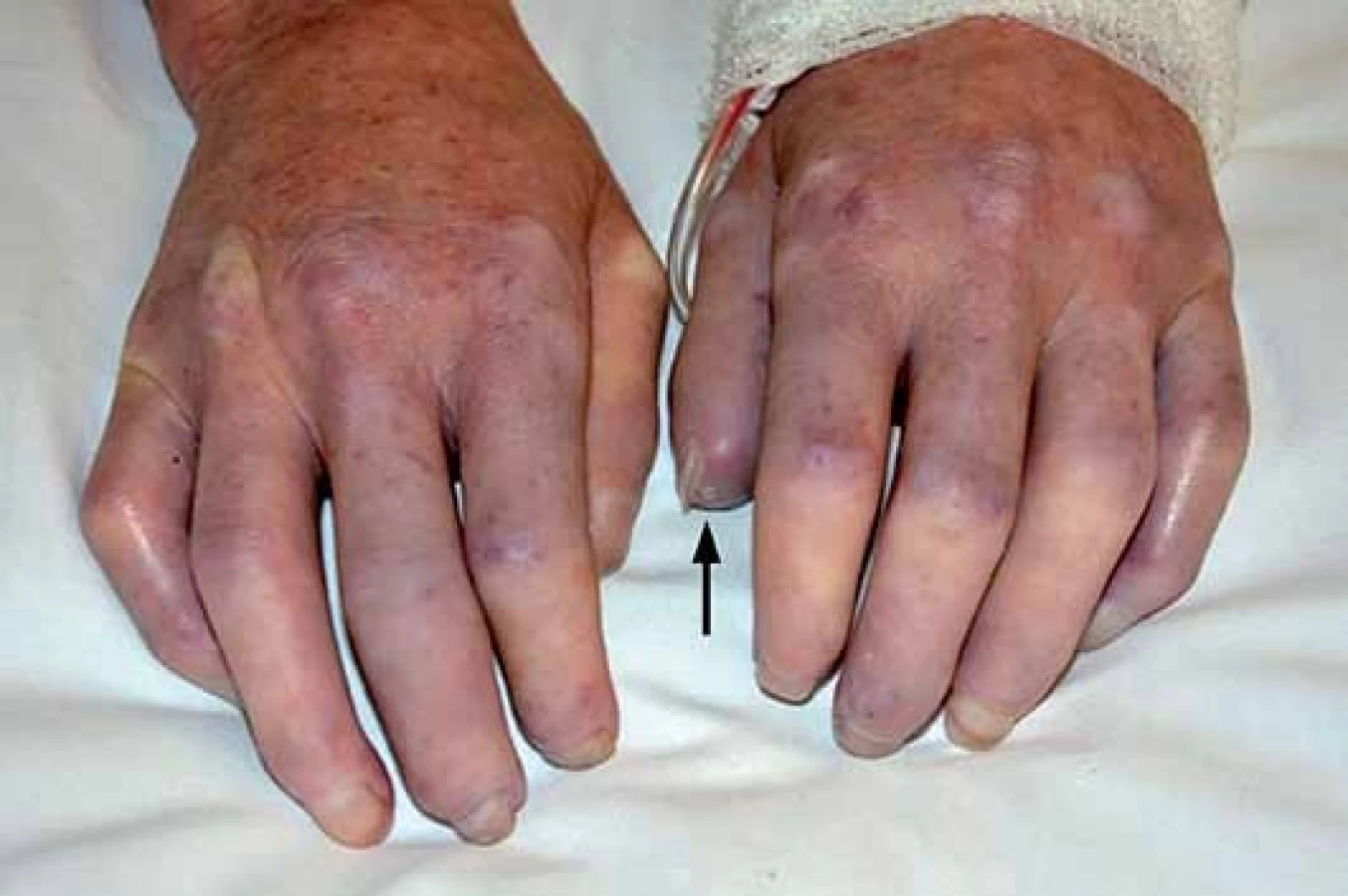
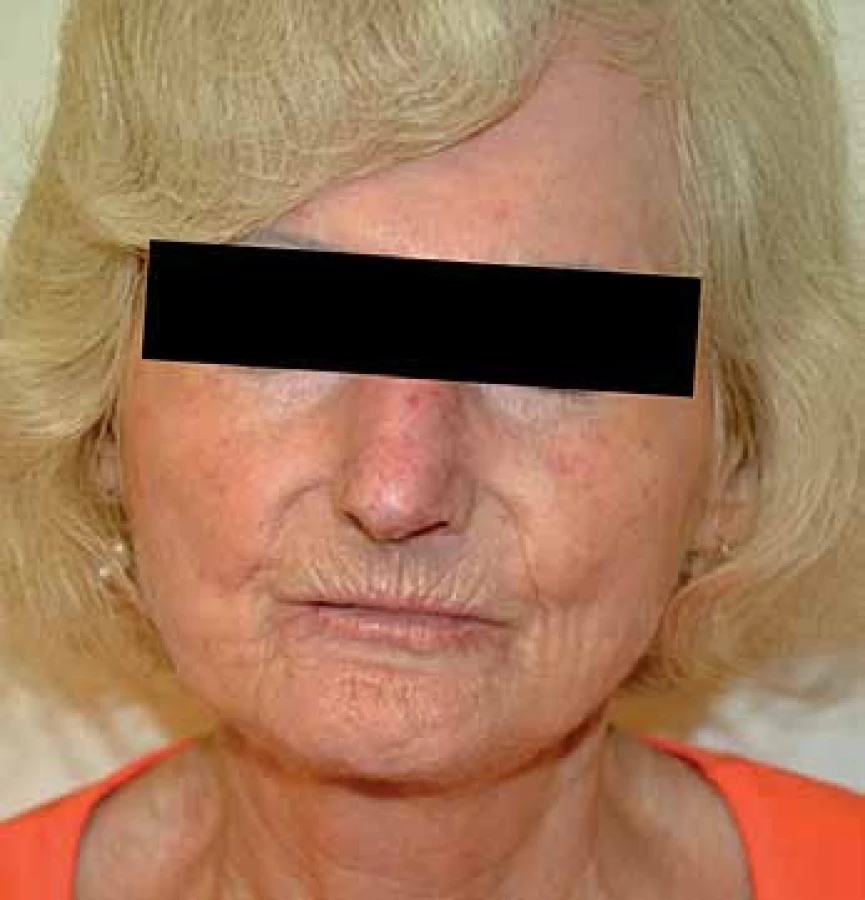
Jedná se o téměř univerzální a nejtypičtější projev SSc. V počátku nemoci dochází k difuzním edematózním změnám na prstech horních končetin, předloktích a obličeji, které mohou být provázené pruritem (obr. 1). Následuje ztuhnutí a ztluštění kůže, jejichž rozsah a závažnost je variabilní (tab. 1). Pokožka je lesklá, tuhá, napnutá, nelze vytvořit kožní řasu, dochází ke ztrátě adnex a depigmentaci anebo hyperpigmentaci (obr. 2). Obličej obvykle mívá maskovitý vzhled, úzký nos a kolem úst se tvoří radiální rýhy (obr. 3). Často se tvoří podkožní kalcifikace a teleangiektazie (obr. 3). Po několika letech dochází k vystřídání zánětlivé a fibrotické fáze atrofickou fází a kůže se pak stává měkčí, dá se řasit a je atrofická [1,8,12].
Postižení cév
U SSc dochází k fibrointimální proliferaci a vazospastickým epizodám malých cév, což se projevuje jako nejčasnější a zároveň nejčastější příznak této nemoci – Raynaudův fenomén (97 %). Je charakterizován epizodickými barevnými změnami aker (nejčastěji prsty horních a/ nebo dolních končetin, vzácněji uši, nos a jazyk) vyvolanými chladem nebo stresem. Typicky má tři fáze – zbělání (palor – způsobený vazospazmem, vnímán jako pocit tuposti až necitlivosti), zmodrání anebo zfialovění (cyanóza – způsobená tkáňovou hypoxií) a zčervenání (rubor – způsoben reperfuzní hyperemií, vnímán jako pálení a brnění) (obr. 1). Progresivní strukturální změny v malých cévách vedou k trvalé poruše toku krve, k prodloužení epizod RAF a k rozvoji ischemických změn (digitální ulcerace, nekrózy a gangrény prstů) (obr. 2). Postižení větších cév u SSc se může manifestovat jako plicní arteriální hypertenze (PAH), sklerodermická renální krize a antrální vaskulární ektazie [1,8,9].
Muskuloskeletální příznaky
K nejčasnějším a častým symptomům SSc patří myalgie a slabost proximálních svalových skupin, artralgie a ranní ztuhlost kloubů. Později se objevují třecí šelesty šlachových pouzder (zejména flexory prstů, nad lokty a rameny) a akroosteolýza (resorpce distálních článků v důsledku protrahované ischemie). Synovitida, podobná té u revmatoidní artritidy (RA), bývá vzácným nálezem, častějším však u překryvných syndromů (SSc/ RA). Obdobně bývá svalová slabost a atrofie častějším nálezem u překryvných syndromů SSc s idiopatickými zánětlivými myopatiemi (polymyozitida (PM) nebo dermatomyozitida (DM)), což může být spojené s pozitivitou anti‑PM – Scl protilátek (tab. 2) [1,8,13].
Gastrointestinální postižení
U SSc dochází k častému postižení trávicího ústrojí, které charakterizuje porucha motility v důsledku abnormální inervace, atrofie hladké svaloviny a tkáňové fibrózy. Mohou být postiženy prakticky všechny části gastrointestinálního traktu (GIT) s různou frekvencí. Nejčastěji bývá postižen jícen (90 %) s nálezem poruchy motility, dilatací dolních dvou třetin a dysfunkcí dolního svěrače. K nejčastějším příznakům postižení GIT u SSc patří dysfagie, pyróza, nadýmání, průjem, zácpa a inkontinence stolice [1,8,14].
Plicní postižení
Postižení respiračního traktu můžeme nalézt až u 70 % pacientů se SSc. Jde hlavně o postižení plicního intersticia (pod obrazem intersticiální plicní fibrózy a/ nebo alveolitidy, nejčastěji subpleurálně při bázích plic; projevuje se progredující dušností a suchým kašlem) a plicních cév (pod obrazem plicní arteriální hypertenze (PAH) – 1. primární, bez postižení intersticia, anebo 2. sekundární, jako následek plicní fibrózy; projevuje se rychle progredující dušností a známkami pravostranného srdečního selhávání) [1,8,15].
Postižení srdce
K postižení srdce dochází u 50 % nemocných se SSc – častěji sekundárně v důsledku PAH než primárně na podkladě fibrózy myokardu (postihující predilekčně vodivý systém) anebo myokarditidy (u překryvných syndromů SSc/ PM/ DM). Častým, ale spíše subklinickým nálezem bývá perikarditida. K nejčastějším manifestacím patří palpitace, dušnost a atypické bolesti na hrudi [1,8,16].
Postižení ledvin
Nejdůležitější manifestací renálního postižení u SSc je sklerodermická renální krize (10 %) pod obrazem rychle progredující renální insuficience s oligurií až anurií a akcelerované arteriální hypertenze. U části pacientů vídáme pouze nízkou proteinurii, pokles renálních funkcí a hypertenzi [1,8,17].
Postižení nervového systému, slinných žláz a pohlavních orgánů
Mezi méně časté manifestace SSc patří neuropatie hlavových nervů (hlavně senzorická neuropatie trigeminu), periferních nervů (zejména senzomotorického typu u nervových vláken v kůži) a syndrom karpálního tunelu. Asi u 25 % pacientů se SSc nalézáme sekundární Sjögrenův syndrom v důsledku fibrotických změn v slinných žlázách. Mezi časté projevy patří i sexuální dysfunkce (poruchy erekce při neurovaskulárním postižení penisu), snížená fertilita pacientek, spontánní aborty a předčasné porody [1,8].
Diagnostika
Diagnózu SSc stanovujeme na základě klinického obrazu (RAF, typické kožní postižení, orgánové manifestace), průkazu specifických autoprotilátek a typických nálezů z cévních a orgánových změn. Jelikož pro SSc chybí specifický diagnostický test, byla vytvořena klasifikační kritéria. V roce 2013 vydala společná pracovní skupina American College of Rheumatology (ACR) a European League Against Rheumatism (EULAR) nová klasifikační kritéria pro SSc s vyšší senzitivitou (0,91) i specificitou (0,92) (tab. 3). Pacienti s celkovým skóre ≥ 9 splňují kritéria pro definitivní SSc [18].
![Klasifikační kritéria pro systémovou sklerodermii podle ACR-EULAR 2013 [18].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/0fad29f3f45ac82795be1e295607e1e3.png)
V laboratorním obraze obvykle detekujeme mírnou normochromní normocytární anémii, trombocytopenii, normální až lehce zvýšené reaktanty akutní fáze, vyšší hodnoty cirkulujících imunokomplexů, revmatoidní faktory a kryoglobuliny (poslední dva parametry až u 40 % pacientů). V imunologickém vyšetření často nalézáme autoprotilátky – nespecifické (ANA) a specifické (nejčastěji Scl70, ACA, RNA polymeráza III, U1 – RNP a PM Scl) (tab. 2) [1,8].
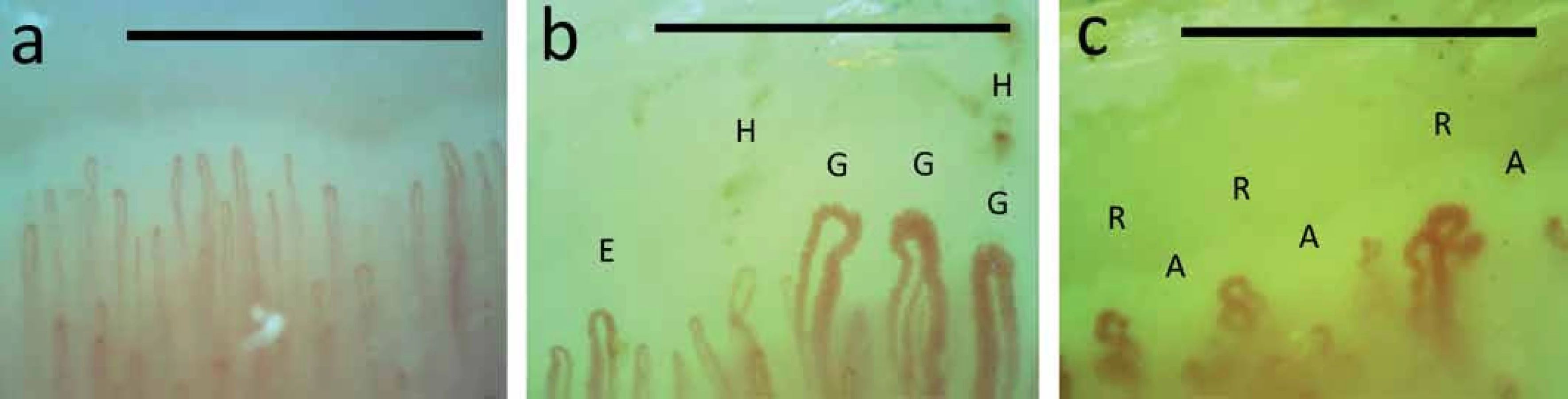
Rozsah a stupeň ztluštění kůže se hodnotí pomocí modifikovaného Rodnanova kožního skóre a při nejasném kožním obraze se provádí kožní excize s histologickým vyšetřením. RAF lze objektivizovat angiologickými metodami (chladové testy, infračervená tomografie a další) a je indikací pro kapilaroskopické vyšetření nehtových valů. Toto nenákladné a pacienta nezatěžující vyšetření odliší, zda se jedná o primární RAF (až 5 % populace – zejména mladé ženy; bez strukturálního postižení kapilár) anebo o sekundární RAF vyskytující se např. u systémových onemocnění pojiva – konkrétně u SSc s charakteristickým obrazem (tvorba megakapilár, přítomnost mikrohemoragií, avaskulárních zón a známek neoangiogeneze) (obr. 4) [1,8,19].
K verifikaci postižení gastrointestinálního traktu se používá nejčastěji jícnová manometrie, polykací akt a pasáž GIT pomocí kontrastní látky, gastroskopie a koloskopie. Muskuloskeletální postižení hodnotíme pomocí RTG postižených kloubů, svalového testu, elektromyografie, případně magnetické rezonance svalů a svalové biopsie. Pro zhodnocení plicního postižení se používá vyšetření plicních funkcí (spirometrie a difuzní kapacita pro CO), RTG a HRCT hrudníku a pro verifikaci aktivní alveolitidy zejména bronchoalveolární laváž s cytologickým vyšetřením. Screening PAH se provádí pomocí echokardiografického vyšetření a k potvrzení této diagnózy je nutná pravostranná srdeční katetrizace. Postižení srdce hodnotíme rutinně pomocí EKG nebo EKG podle Holtera a pomocí ECHO [1,8].
Diferenciální diagnostika
V časných stadiích SSc musíme vyloučit další systémová onemocnění pojiva (RA, systémový lupus erythematodes, PM, DM). Při orgánovém postižení je nutno myslet na nozologické jednotky postihující jednotlivé orgánové systémy (např. primární PAH, primární biliární cirhóza, idiopatická plicní fibróza a další). Při postižení kůže je důležité vyloučit jiné formy sklerodermie (lokalizovaná sklerodermie – morfea a lineární sklerodermie) a syndromy podobné sklerodermii podmíněné endogenně (scleroedema adultorum Buschke, skleromyxedém, lichen sclerosus et atrophicus), metabolicky (hypotyreóza, diabetes mellitus, amyloidóza), geneticky (eozinofilní fascitida, paraneoplastické dermopatie) a exogenně (reakce štěpu vůči hostiteli, kožní syndromy indukované bleomycinem a vinyl chloridem). Zásadní roli v diferenciální diagnóze má přítomnost RAF, nález na kapilaroskopii specifický pro SSc, autoprotilátky typické pro SSc a typ postižení kůže a orgánů charakteristický pro SSc [20].
Terapie
Univerzální, účinný lék pro SSc dosud nebyl nalezen, proto je u pacientů se SSc na místě komplexní přístup zahrnující režimová opatření (zákaz kouření, zákaz práce a pobytu v prašném či chladném prostředí a zákaz kontaktu s toxickými látkami), rehabilitaci (masáž, manuální lymfodrenáž, techniky měkkých tkání, udržování rozsahu pohybu v kloubu, mobilizace, aktivní a pasivní cvičení k prevenci kontraktur, dechová gymnastika, mechanoterapie a cílená cvičení na pohyblivost svalů tváře) a orgánově specifickou léčbu. V terapii RAF a akrálních ulcerací se uplatňují blokátory kalciových kanálů, pentoxifyllin, série infuzí analog prostaglandinů a hyperbarická komora. V prevenci nových defektů a léčbě PAH mají své místo antagonisté receptorů pro endotelin a blokátory 5 - fosfodiesterázy a v léčbě ulcerací dále chirurgické a lokální přístupy. U PAH se navíc podávají warfarin, diuretika, případně prostanoidy. Gastrointestinální projevy se snažíme ovlivnit prokinetiky, antacidy a inhibitory protonové pumpy, v pokročilejších stadiích je nezbytná enterální výživa. K ovlivnění kožních, kloubních a svalových projevů se používá metotrexát a glukokortikoidy v nízkých až středních dávkách s velmi individuální, variabilní účinností. Při aktivní alveolitidě v rámci intersticiálního plicního procesu používáme opakované (po měsíci) intravenózní pulzy cyklofosfamidu. Slibné se zdá být použití mykofenolát mofetilu při postižení kůže a plic. Recentní studie s vysokodávkovou imunosupresí s následnou autologní transplantací kmenových buněk demonstrují účinnost této metody, ale také mortalitu s ní spojenou – zdá se být záchranným a účinným přístupem pro pacienty s rychle progredujícím multiorgánovým postižením, které není ještě v pokročilém stadiu. Z biologických preparátů se jako nejslibnější zatím jeví rituximab – co do ovlivnění plicního a kožního postižení. První volbou při sklerodermické renální krizi jsou ACE inhibitory, nutná bývá dlouhodobá dialýza i transplantace ledvin. Při postižení srdce se uplatňují antiarytmika a trvalá stimulace pacemakerem [21].
Prognóza
Přirozený průběh limitované formy SSc (lcSSc) je příznivější než difuzní SSc (dcSSc). Celková doba přežití je zkrácená – 10 let přežívá průměrně 70 % pacientů (49 % dcSSc/ 82 % lcSSc, 70 % mužů/ 92 % žen) [22,23]. V průběhu posledních 20 let se snížila mortalita v důsledku SSc jen u dcSSc, zatímco u lcSSc zůstává na stejné úrovni [24]. Mezi nejčastější příčiny úmrtí u pacientů se SSc v současné době patří plicní fibróza (35 %), plicní arteriální hypertenze (26 %), srdeční selhání a arytmie (26 %). Sklerodermická renální krize ztratila své dominantní postavení mezi příčinami mortality se začátkem používání ACE inhibitorů [25]. K prognosticky nepříznivým faktorům patří mužské pohlaví, vysoký věk (nad 65 let), difuzní kožní forma a postižení vnitřních orgánů [4].
Závěr
Prevalence SSc může být výrazně podhodnocena v běžné populaci. Jednotlivé případy bývají často nesprávně anebo nedostatečně diagnostikovány – zejména se jedná o pacienty s mírným kožním postižením anebo bez kožních projevů. Kvůli pozdnímu odeslání těchto jedinců do péče revmatologa progreduje mnoho středně závažných případů do konečných stadií nereverzibilního orgánového postižení, kterému by se dalo předejít včasným stanovením správné diagnózy. I přes dosud neexistující univerzální účinnou léčbu SSc došlo v posledních pár dekádách k výraznému pokroku v objasnění patogeneze této nemoci a ve farmakoterapii jednotlivých orgánových manifestací a snížení morbidity a mortality spojené se SSc. Proto je nutné na tuto diagnózu myslet zejména při častých a protrahovaných atakách RAF a difuzních otocích prstů a odeslat pacienta k vyšetření kapilaroskopie a antinukleárních protilátek.
Poděkování
Práce vznikla za podpory MZČR – Institucionální podpora, Koncepční rozvoj výzkumné organizace č. MZO 023728.
Doručeno do redakce: 30. 9. 2014
Přijato po recenzi: 10. 10. 2014
MU Dr. Michal Tomčík, Ph.D.
www.revma.cz
tomcik@revma.cz
Sources
1. Gabrielli A, Avvedimento EV, Krieg T. Mechanisms of Disease: Scleroderma. N Engl J Med 2009; 360 : 1989 – 2003. doi: 10.1056/ NEJMra0806188.
2. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum 1980; 23 : 581 – 590.
3. Boin F, Rosen A. Autoimmunity in systemic sclerosis: current concepts. Curr Rheumatol Rep 2007; 9 : 165 – 172.
4. Ranque B, Mouthon L. Geoepidemiology of systemic sclerosis. Autoimmun Rev 2010; 9: A311 – A318. doi: 10.1016/ j.autrev.2009.11.003.
5. Chifflot H, Fautrel B, Sordet C et al. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 2008; 37 : 223 – 235.
6. Feghali – Bostwick C, Medsger TA Jr, Wright TM. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum 2003; 48 : 1956 – 1963.
7. Mora GF. Systemic sclerosis: environmental factors. J Rheumatol 2009; 36 : 2383 – 2396. doi: 10.3899/ jrheum.090207.
8. Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 2007; 117 : 557 – 567.
9. Kahaleh MB, Sherer GK, LeRoy EC. Endothelial injury in scleroderma. J Exp Med 1979; 149 : 1326 – 1335.
10. Artlett CM. Immunology of systemic sclerosis. Front Biosci 2005; 10 : 1707 – 1719.
11. Bassel M, Hudson M, Taillefer SS et al. Frequency and impact of symptoms experienced by patients with systemic sclerosis: results from a Canadian National Survey. Rheumatology 2011; 50 : 762 – 767. doi: 10.1093/ rheumatology/ keq310.
12. Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology 2009; 48 (3 Suppl): 14 – 18. doi: 10.1093/ rheumatology/ kep108.
13. Avouac J, Walker UA, Hachulla E et al. Joint and tendon involvement predict disease progression in systemic sclerosis: a EUSTAR prospective study. Ann Rheum Dis 2014; pii: annrheumdis – 2014 – 205295. doi: 10.1136/ annrheumdis ‑ 2014 ‑ 205295.
14. Gyger G, Baron M. Gastrointestinal manifestations of scleroderma: recent progress in evaluation, pathogenesis, and management. Curr Rheumatol Rep 2012; 14 : 22 – 29. doi: 10.1007/ s11926 ‑ 011 ‑ 0217 ‑ 3.
15. Hassoun PM. Lung involvement in systemic sclerosis. Presse med 2011; 40 (1 Pt 2): e3 – e17. doi: 10.1016/ j.lpm.2010.08.006.
16. Parks JL, Taylor MH, Parks LP et al. Systemic sclerosis and the heart. Rheum Dis Clin North Am 2014; 40 : 87 – 102. doi: 10.1016/ j.rdc.2013.10.007.
17. Sabir O, Younas H, Tanvir I, Tarif N. Scleroderma renal crises: case report and review of literature. J Pak Med Assoc 2013; 63 : 916 – 918.
18. van den Hoogen F, Khanna D, Fransen J et al.2013 classification criteria for systemic sclerosis: an American college of rheumatology/ European league against rheumatism collaborative initiative. Ann Rheum Dis 2013; 72 : 1747 – 1755. doi: 10.1136/ annrheumdis ‑ 2013 ‑ 204424.
19. Ingegnoli F, Gualtierotti R. A systematic overview on the use and relevance of capillaroscopy in systemic sclerosis. Expert Rev Clin Immunol 2013; 9 : 1091 – 1097. doi: 10.1586/ 1744666X.2013.849198.
20. Nashel J, Steen V. Scleroderma mimics. Curr Rheum Rep 2012; 14 : 39 – 46. doi: 10.1007/ s11926 ‑ 011 ‑ 0220 ‑ 8.
21. Avouac J, Kowal - Bielecka O, Landewe R et al. European League Against Rheumatism (EULAR) Scleroderma Trial and Research group (EUSTAR) recommendations for the treatment of systemic sclerosis: methods of elaboration and results of systematic literature research. Ann Rheum Dis 2009; 68 : 629 – 634. doi: 10.1136/ ard.2008.095299.
22. Czirjak L, Kumanovics G, Varju C et al. Survival and causes of death in 366 Hungarian patients with systemic sclerosis. Ann Rheum Dis 2008; 67 : 59 – 63.
23. Hissaria P, Lester S, Hakendorf P et al. Survival in scleroderma: results from the population‑based South Australian Register. Intern Med J 2011; 41 : 381 – 390. doi: 10.1111/ j.1445 ‑ 5994.2010.02281.x.
24. Nihtyanova SI, Tang EC, Coghlan JG et al. Improved survival in systemic sclerosis is associated with better ascertainment of internal organ disease: a retrospective cohort study. QJM 2010; 103 : 109 – 115. doi: 10.1093/ qjmed/ hcp174.
25. Tyndall AJ, Bannert B, Vonk M et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69 : 1809 – 1815. doi: 10.1136/ ard.2009.114264.
Labels
Paediatric cardiology Internal medicine Cardiac surgery CardiologyArticle was published in
Cardiology Review

2014 Issue 5
Most read in this issue
- Treatment of anemia and iron deficiency from the hematologist’s perspective
- Antiphospholipid syndrome – diagnostics, manifestation and treatment
- Adverse effects of biological therapy in rheumatology
- Systemic sclerosis