
Únikové strategie nádorů pozornosti imunitního systému
Escape Strategies of Tumors from Immune Surveillence
Immune system must be able to protect us from foreign dangerous pathogens, but on the other side, it must be able to recognize our own tissues and organs. Activity of the immune system is affected by many positive (stimulatory) and negative (inhibitory) signals. Some of these negative receptors protect us from damage of our tissues at a place of inflammation as it blocks too intensive or long‑lasting immune reaction. Thereby, they have a physiological protective function against strong inflammatory reaction and possible subsequent autoimmune pathology. However, some of these mechanisms are also utilized by tumors to avoid immune recognition and attention of the immune cells. Other tumor escape mechanisms involve increased production of cytokines and factors which are responsible for immunosuppressive tumor microenvironment where effective immune response is actively blocked. This review summarizes the most frequently used strategies, which are utilized by tumors to avoid immune recognition and/ or killing by the immune cells.
Key words:
immune evasion – tumor escape – immunotherapy – CTLA-4 – PD-1 – immune checkpoint
* I declare that, in connection with this contribution of which I am the co-author, I have a conflict of interest with following company: Bristol-Myers Squibb al. s r. o.
Author is former employee of Institute of Microbiology of the AS CR, v. v. i., Prague.
** The author declares she has no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
4. 8. 2015
Accepted:
1. 10. 2015
Authors:
M. Šťastný 1*; B. Říhová 2**
Authors‘ workplace:
Bristol‑ Myers Squibb spol. s r. o., Praha2 Mikrobiologický ústav AV ČR, v. v. i., Praha
1
Published in:
Klin Onkol 2015; 28(Supplementum 4): 28-37
Category:
Generals
doi:
https://doi.org/10.14735/amko20154S28
Overview
Imunitní systém musí být na jedné straně schopen efektivně zasáhnout proti cizím a nebezpečným patogenům, na straně druhé musí být schopen rozpoznat a tolerovat naše vlastní tkáně a orgány. Aktivita imunitního systému je ovlivňována celou řadou pozitivních (stimulačních) a negativních (inhibičních) signálů. některé z těchto inhibičních receptorů zabraňují poškození našich tkání v místě zánětu tím, že tlumí příliš silnou či dlouhou imunitní reakci. Plní tak fyziologickou ochrannou funkci před silnou zánětlivou reakcí a možnou autoimunitní patologií. některé z těchto mechanizmů jsou ovšem využívány nádory k tomu, aby unikly pozornosti imunitního systému. Další únikové strategie spočívají v produkci cytokinů a faktorů vytvářejících v nádorovém mikroprostředí silnou imunosupresi, která zabraňuje efektivní imunitní odpovědi. Tato práce si klade za cíl popsat nejčastější strategie, které jsou nádory využívány k potlačení imunitní reakce.
Klíčová slova:
imunitní úniky – únikové mechanizmy nádorů – imunoterapie – CTLA ‑ 4 – PD ‑ 1 – kontrolní body imunitní reakce
Úvod
Imunitní systém savců se vyvíjel desítky miliónů let a učil se reagovat na cizorodá (ohrožující) agens. Jedním z hlavních úkolů imunitního systému bylo zabránit úmrtí jedince na infekci a umožnit dosažení reprodukčního období. Imunitní systém se tedy vyvinul primárně pro boj s bakteriálními a virovými infekcemi, ne pro boj s nádory. V průběhu evoluce se totiž imunitní systém neměl příliš šanci s nádory potkat. Přestože jsou v archeologických výzkumech popisovány nálezy kostí neandrtálců deformovaných nádory [1], naprostá většina malignit se v současné době objevuje až po 50. roce života (graf 1), přičemž historická data ukazují, že ještě v polovině 19. století byla očekávaná doba přežití ve Velké Británii jen 25 – 30 let [2]. S jistou formou nadsázky lze tedy tvrdit, že imunitní systém člověka se s nádory „potkává“ teprve posledních 150 let, kdy došlo k významnému prodloužení průměrné doby přežití, a tím k vyššímu výskytu nádorových onemocnění.

Někdy se říkává, že maligní nádory se chovají jako vlastní tkáň a vysvětluje se tím absence jejich rozpoznávání a následného odstranění imunitními mechanizmy. Není to přesná formulace. Spíše se ukazuje, že imunitní systém některé nádory, které mutacemi nevytvořily dostatek antigenně cizích proteinů, glykoproteinů a lipoproteinů, prostě nerozpoznává jako potenciální nebezpečí. Nádory jako buňky tělu „vlastní“ představují mnohem slabší imunogeny pro imunitní systém a navíc mají – stejně jako normální netransformované buňky – řadu mechanizmů, jak se chránit před napadením buňkami imunitního systému.
Myšlenka „imunitního dozoru“ byla u nádorů poprvé vyslovena Paulem Ehrlichem a později byla reformulována Burnetem a Thomasem v roce 1957. Tato teorie hovoří o tom, že imunitní systém je u imunokompetentního jedince (spolu)zodpovědný za prevenci nádorového bujení. Tato teorie budila řadu kontroverzí, protože se původně nepodařilo prokázat, že by athymické myši měly vyšší výskyt nádorů než imunokompetentní zvířata s funkčním thymem [3]. Později se však ukázalo, že tyto nude myši nejsou zcela imunodeficitní, a teprve s rozvojem geneticky modifikovaných myších modelů imunodeficience v 90. letech minulého století se prokázalo, že imunitní systém hraje důležitou roli v kontrole nádorů. Skupina kolem prof. Schreibera zdokumentovala, že zvířata deficitní v genech pro IFN ‑ γ či pro všechny IFN receptory mají vyšší výskyt chemicky indukovaných nebo spontánně se objevujících nádorů v porovnání s imunokompetentními zvířaty [4,5]. Navíc se zjistilo, že imunitní systém nejen kontroluje počet nádorových buněk, ale i jejich imunogenicitu. Nádory rostoucí v imunodeficitních zvířatech byly totiž více imunogenní („needitované“) v porovnání s nádory rostoucími v imunokompetentních zvířatech („editované“ nádory) [6]. Tato teorie byla nazvána „nádorovou imunoeditací“. V dalších experimentech se pak ukázalo, že vztah mezi imunitním systémem a nádory prochází třemi fázemi, a proto se také někdy hovoří o teorii 3E, z anglického elimination – equilibrium – escape [7]:
1. fáze eliminace
Tato fáze odpovídá v podstatě původní teorii imunitního dohledu, ve které dochází ke spolupráci vrozené a adaptivní složky imunitního systému při detekci rozvíjejícího se nádoru – v ideálním případě k jeho likvidaci – ještě předtím, než je nádor klinicky zjistitelný. Mechanizmy, kterými je imunitní systém „varován“ o potenciálním nebezpečí, jsou komplexní, ne zcela jasné, ale mohou se na nich podílet tzv. DAMPs molekuly (damage‑associated molecular patterns), které jsou uvolňovány z hynoucích nádorových buněk či ze stromálních buněk nádoru. Jedná se o molekulární vzory spojené s poškozením/ nebezpečím, mezi než patří např. nukleární či cytosolické proteiny (DNA, HSP, HMGB1, ATP a další) [7].
2. fáze rovnováhy
Část nádorových buněk může přežít fázi eliminace, které se tím se dostanou do fáze rovnováhy mezi imunitním systémem a nádorovými buňkami. V této fázi se adaptivní imunitní systém podílí na kontrole nádorového bujení a preferenčně dochází k likvidaci více imunogenních variant nádorových buněk. Na druhé straně může zcela dle Darwinových principů docházet k tomu, že se mohou objevovat méně imunogenní varianty nádorových buněk (ztráta povrchového antigenu/ markeru nebo schopnost přežít útok imunitního systému), které nejsou kompletně eliminovány. V této fázi, která může být různě dlouhá (někdy může trvat roky či po celý život pacienta), je imunitní systém schopen držet reziduální nádorové buňky pod určitou kontrolou ve stavu funkční dormance. Nedochází sice ke kompletní eliminaci nádorových buněk, ale může být blokována jejich schopnost nekontrolovaného růstu či metastáz [7].
3. fáze úniku
V této fázi už dochází k tomu, že nádorové buňky získávají schopnost vyhnout se kontrole buňkami imunitního systému a rozpoznání či destrukci. Tato schopnost úniku imunitnímu systému dokonce patří k základním charakteristikám nádorů [7,8]. Proto v okamžiku, kdy je nádor klinicky detekovatelný, má už obvykle celou řadu mechanizmů, které mu umožňují uniknout imunitní odpovědi hostitele (exprese cytokinů, chemokinů či dalších faktorů, jako je IL‑10, TGF‑β, VEGF, IDO).
Tyto fáze nádorové imunoeditace jsou velmi pěkně dokumentovány na preklinických modelech. Objevuje se vyšší výskyt chemicky indukovaných nádorů u imunodeficitních myší v porovnání s imunokompetentními jedinci. Naopak likvidací T buněčných subpopulací (CD4+, CD8+ IFN ‑ γ+) dojde u 40 – 50 % myší k objevu nádoru, přestože u nich nízké dávky karcinogenu nádor neindukovaly [6,7,9]. Podrobná vyšetření pak ukázala, že u těchto zvířat jsou sice sarkomové buňky přítomny, ale jsou drženy pod kontrolou imunitním systémem zvířete. V klinickém prostředí je přesvědčujících dat z logických důvodů výrazně méně, nicméně se ví, že pacienti s výraznou imunosupresí (transplantace) či oslabeným/ nefunkčním imunitním systémem (AIDS) mají výrazně vyšší výskyt některých nádorů [10,11]. Velmi zajímavé jsou kazuistiky, kdy se u imunosuprimovaného pacienta s transplantací ledviny objeví melanom a zjistí se, že nejde o jeho vlastní nádor, ale o nádor pocházející z dárce [12]. Zpětným dohledáním se pak zjistilo, že dárci byl před desítkami let odstraněn melanom a on sám byl až do svého úmrtí zcela v pořádku. Zřejmě došlo k tomu, že v transplantovaném orgánu přežívaly desítky let dormantní melanomové buňky, které držel pod kontrolou imunitní systém dárce, a teprve poté, kdy byla ledvina přenesena do imunosuprimovaného jedince, došlo k růstu melanomu. Podobně se objevují kazuistiky, kdy došlo k metastázování renálního karcinomu do plic osm let po nefrektomii, přičemž v té době měl pacient silnou imunosupresi kvůli transplantované ledvině [13]. U karcinomu prsu se ví, že u 30 – 40 % žen, které podstoupily mastektomii, jsou v krvi detekovány cirkulující nádorové buňky, a přesto u všech těchto žen nedochází 8 – 22 let po zákroku k relapsu karcinomu prsu [14]. Naopak existují i zajímavé případy, kdy došlo ke spontánní regresi plicních metastáz renálního karcinomu u pacienta s psoriázou v období, kdy došlo k exacerbaci choroby, a naopak remise psoriázy byla spojena s progresí nádoru [15].
Nádory unikají vrozené a získané imunitní odpovědi buď pasivně, nebo aktivně. Pasivní mechanizmy se týkají přímo vlastností nádorových buněk. Při aktivním úniku jsou využívány i další buněčné systémy. některé z těchto mechanizmů ukazuje obr. 1.
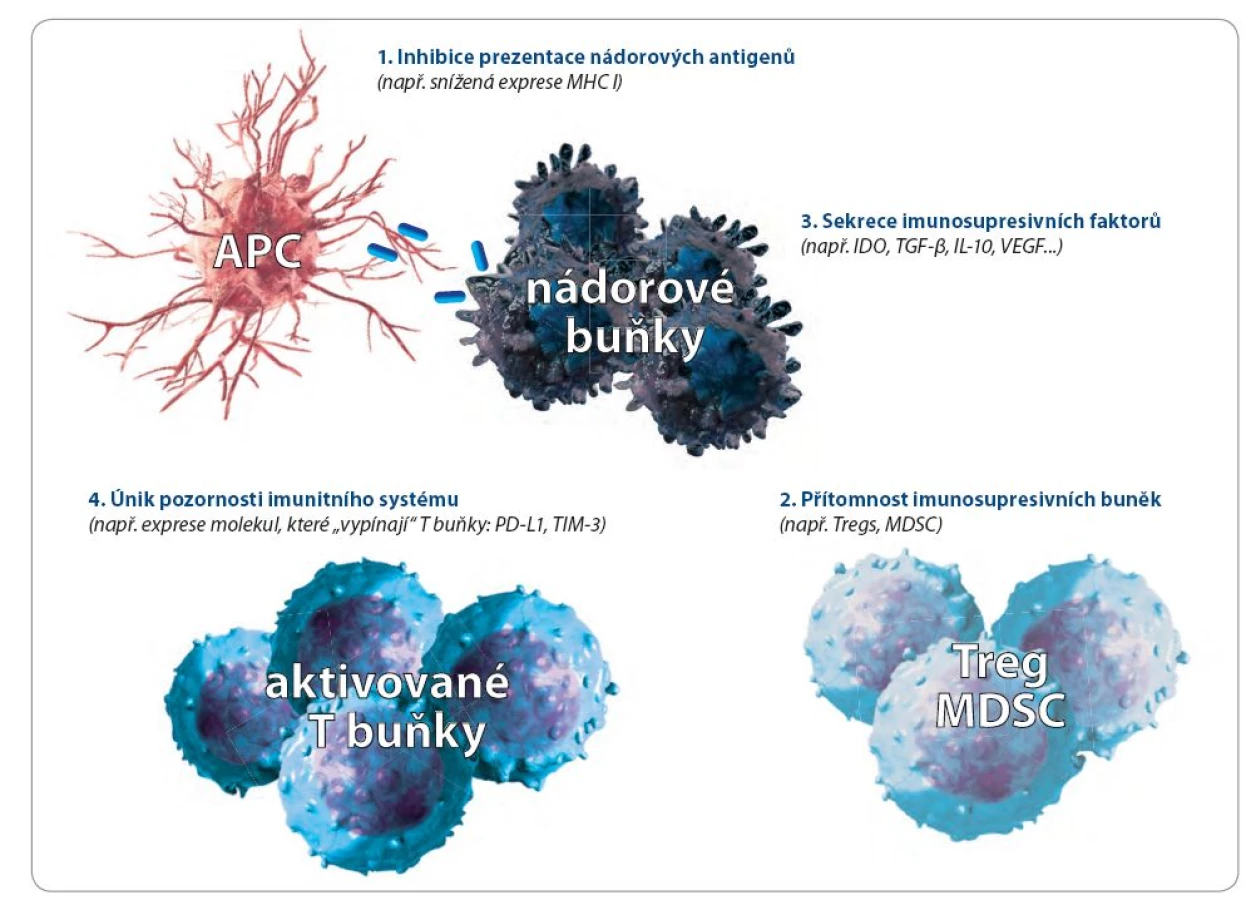
Pasivní obrana (imunoselekce, imunoeditace)
Nádory mutacemi ztrácejí antigeny, které by mohly imunitní reakci vyvolat
Princip imunoselekce spočívá v tom, že populace nádorových buněk je po určité době expanzivního růstu nádoru mimořádně heterogenní. Je geneticky nestabilní nejenom z imunologického, ale i z metabolického hlediska. A tato různorodost se neustále zvyšuje. Jsou nádory, u kterých se v průběhu několika let prokazují až tisíce mutací nebo delecí genů kódujících nádorové antigeny. Pokud nejde o antigeny vyžadované pro růst nádorových buněk nebo udržování transformovaného fenotypu, jsou imunologicky rozpoznatelné nádorové buňky během fáze ekvilibria imunitní editace odstraňovány a přežívají jen imunitním systémem nerozpoznatelné nádorové varianty. Proto je dnes imunoeditace považována za jeden z hlavních důvodů, proč nádory imunitnímu dohledu unikají. Experimentálně to bylo potvrzeno porovnáním růstu nádorů u konvenčních a imunodeficitních myší a jejich následnou transplantací do dalších imunokompetentních myší. Nádory původně rostoucí na imunodefektních myších imunokompetentní myši častěji odhojovaly. Je to důkaz toho, že nádory, které se vyvíjejí v prostředí normálního imunitního systému, se časem stávají méně imunogenní, protože přerůstají méně imunogenní varianty nádorových buněk [7].
Exprese MHC glykoproteinů I. třídy může být na nádorových buňkách snížena tak, že nejsou následně rozpoznávány cytotoxickými T lymfocyty
Nádory, podobně jako některé viry infikované buňky, snižují, nebo dokonce ztrácejí povrchovou expresi molekul MHC (major histocompability complex) glykoproteinů I. třídy, které jsou pro rozpoznávání cytotoxickými T lymfocyty (CTL) zásadní. Obě hlavní T buněčné subpopulace, CD4+ (pomocné) a CD8+ (cytotoxické) T buňky, rozpoznají cílovou, tj. pro organizmus nebezpečnou buňku jedině tehdy, vystavuje‑li své antigeny na pozadí výše zmíněných MHC molekul. CD4+ T lymfocyty rozpoznávají antigenní peptidy vystavené na pozadí MHC glykoproteinů II. třídy, CD8+ T lymfocyty pak antigenní peptidy vystavené na pozadí MHC glykoproteinů I. třídy. Pokud takové molekuly na povrchu nejsou, antigeny na nich nemohou být vystavovány a pro T buňky imunitního systému se taková buňka teoreticky stává neviditelnou. Nejsou ale bohužel zcela exaktní důkazy o korelaci mezi hladinou MHC exprese a růstu nádorů, nicméně jsou známy případy tzv. smíšené odpovědi nádoru na imunoterapii, kdy část nádorových lézí regreduje a část se zvětšuje [16]. V neodpovídající lézi pak byla prokázána snížená exprese MHC molekul I. třídy. Nádorové snížení MHC exprese ale není jediný mechanizmus, který umožňuje nádorovým buňkám pasivně uniknout imunitnímu dozoru. Nádorové buňky snižují nejenom syntézu celých MHC molekul, ale také jen β2 mikroglobulinu nebo buněčných komponent typu transportérů zajišťujících převod zpracovaných peptidů z proteazomů do endoplazmatického retikula anebo jen některých podjednotek proteazomů. Jedná se nepochybně o reakci nádorů na selekční tlak ze strany imunitního systému pacienta a umožňuje to nádorovým buňkám uniknout T buněčné odpovědi.
Nádorové Ag jsou exprimovány tak,že nejsou imunitním systémem rozpoznatelné
Tento mechanizmus bývá také někdy nazýván „maskování antigenu“ a je způsoben tím, že povrchové antigeny nádoru jsou překryty molekulami glykokalyxu. Jako příklad může sloužit sialová kyselina v mukopolysacharidech. Nádory často exprimují více glykokalyxových molekul než buňky normální.
Aktivní obrana
Nádory mohou samy aktivně využívat další buněčné systémy, aby zabránily imunitnímu systému v jejich rozpoznání. Za normálních okolností musí být imunitní systém schopen na jedné straně rozpoznávat a likvidovat nebezpečné patogeny (viry, bakterie, parazity), na straně druhé musí být zachována jeho schopnost tolerovat vlastní tkáně a orgány [17 – 21]. Protože klíčovou roli v rozpoznávání a likvidaci patogenů a nádorových buněk hrají T buňky, jejich aktivita musí být velmi striktně regulována. Akti-vita T buněk je regulována pozitivními, kostimulačními signály a zároveň existuje velký počet negativních, inhibičních signálů (tzv. immune check-points nebo kontrolní body imunitní reakce – KBIR) (obr. 2). Tyto inhibiční receptory modulují trvání a intenzitu imunitní odpovědi a jejich význam je diskutován níže.
![T buňky jsou ovlivňovány řadou pozitivních a negativních signálů (upraveno dle [21]).](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/997f0107ad2f5d447e7d01b7675a423a.jpg)
Aktivace T buněk a kostimulační molekuly
Prvním krokem v aktivaci „naivních“ T buněk je vazba T buněčného receptoru (T - cell receptor – TCR) na MHC molekuly s navázaným antigenním peptidem na povrchu antigen prezentujících buněk (antigen presenting cells – APC; dendritické buňky, aktivované makrofágy či B buňky). APC pohlcují z okolního prostředí antigeny, zpracují je a v ideálním případě vystaví peptidový fragment z nádoru na svém povrchovém MHC. Po vazbě specifického TCR na MHC s příslušným antigenním peptidem dostane T buňka 1. signál [22]. Tento signál není dostatečný pro optimální aktivaci a T buňka musí dostat 2., kostimulační signál. Ten dostává vazbou receptoru CD28 na svém povrchu na molekuly B7 (B7.1/ CD80 či B7.2/ CD86) na povrchu aktivované APC. Tím dojde k plné aktivaci T buněčné odpovědi, sekreci příslušných cytokinů včetně IL‑2, který indukuje proliferaci T buněk a vznik mnoha „kopií“ efektorových, aktivovaných T buněk [22 – 24]. T buňky, které byly aktivovány ve spádové uzlině, migrují zpět do místa primárního nádoru. Jakmile jsou tyto buňky aktivovány, už nevyžadují 2. kostimulační signál, ale stačí jim rozpoznání MHC antigenu s navázaným peptidovým fregmentem, což vede k napadení nádorové buňky a v ideálním případě k její likvidaci. Část aktivovaných T buněk se na konci imunitní odpovědi mění v tzv. paměťové T buňky, které jsou schopny rychlejší a silnější reakce v případě opětovného setkání s antigenním peptidem [22]. Molekuly, které dávají T buňkám 2. signál, aby došlo k jejich plné aktivaci a proliferaci, se nazývají kostimulační molekuly a část těchto molekul ukazuje obr. 1. Kromě již výše zmiňované molekuly CD28 existuje celá řada dalších kostimulačních molekul, které jsou důležité pro optimální efektorové funkce T buněk a pro tvorbu paměťových T buněk, které v organizmu přežívají roky [25]. Vazba těchto molekul na své ligandy zesiluje aktivaci T buněk, zvyšuje proliferaci či sekreci cytokinů a obecně lze říci, že tyto molekuly představují potenciální cíl pro imunomodulační léčbu [26].
Inhibiční molekuly („immune checkpoints“ neboli kontrolní body imunitní reakce)
Kromě pozitivní kostimulace, kdy se z naivních T buněk stávají imunologicky aktivní cytotoxické T buňky, existuje také celá řada inhibičních receptorů a molekul, které přispívají k imunologické homeostáze a zabraňují nechtěným projevům autoimunity či nadměrnému poškození vlastních tkání v místě intenzivního zánětu. Aktivní blokáda imunitního dozoru patří také od roku 2011 k jedné z nových charakteristik nádorů [8]. Mezi nejdůležitější molekuly, které patří mezi tzv. immune checkpoints (kontrolní body imunitní reakce), patří CTLA ‑ 4 (cytotoxic T‑lymphocyte antigen ‑ 4), receptor PD ‑ 1 (programmed death ‑ 1), LAG ‑ 3 (lymphocyte activation gene ‑ 3), TIM ‑ 3 (T ‑ cell immunoglobulin and mucin‑domain containing ‑ 3) a další [26].
CTLA ‑ 4
CTLA ‑ 4 je klíčový negativní regulátor imunitní odpovědi. Je exprimován exkluzivně na T buňkách, kde se objevuje 2 – 3 dny po jejich aktivaci, a jeho hlavní funkcí je regulovat aktivitu T buněk. Váže se na stejné molekuly jako kostimulační molekula CD28 (tedy na ligandy CD80 a CD86 na povrchu APC), a protože má k těmto molekulám vyšší afinitu než CD28, vytěsňuje tuto kostimulační molekulu z vazby a tím tlumí aktivitu T buněk [27 – 28]. Tato interakce je extrémně důležitá pro udržení imunologické rovnováhy a indukci periferní tolerance k vlastním antigenům. Je to „bezpečnostní pojistka“ či „brzda“, která zabraňuje nadměrné imunitní odpovědi a nechtěné autoimunitě. Studie na knockout myších ukazují, jak extrémně důležitou molekulou CTLA ‑ 4 je. Zvířata bez genu pro CTLA ‑ 4 se sice narodí, ale hynou 3 – 4 týdny po narození na fatální periferní lymfoproliferaci s infiltrací orgánů T buňkami [29].
V počátku imunitní odpovědi je CTLA ‑ 4 na povrchu T buněk téměř nedetekovatelný a na povrchu se objevuje během 48 – 72 hod po jejich aktivaci [30]. CTLA ‑ 4 je tedy rozhodujícím „imunitním checkpointem“, který kontroluje trvání a intenzitu imunitní odpovědi [18,31]. Kromě toho CTLA ‑ 4 přispívá k imunomodulační aktivitě regulačních T buněk (Tregs), kde je (na rozdíl od efektorových T buněk) experimován konstitutivně a přispívá k lokální imunosupresi vyvolané právě Tregs v nádorovém mikroprostředí [32,33]. Paradoxně se ukazuje, že zatímco CTLA ‑ 4 inhibuje efektorové T buňky, u regulačních T buněk dochází k opačné situaci – CTLA ‑ 4 zesiluje aktivitu Tregs a jejich proliferaci. Zdá se, že část protinádorového působení anti‑CTLA ‑ 4 protilátek (ipilimumab, tremelimumab) je zprostředkována právě tím, že blokují imunosupresivní aktivitu Tregs [21,34].
PD ‑ 1 receptor
PD ‑ 1 je transmembránový imunomodulační receptor, který patří k nejintenzivněji studovaným molekulám v oblasti protinádorového působení. Byl původně objeven jako gen indukovaný u T buněčných hybridomů při průchodu apoptózou [35]. Na klidových buňkách imunitního systému je minimální exprese PD ‑ 1, ale po aktivaci se PD ‑ 1 objevuje jak na T buňkách, tak na B buňkách, NK buňkách, NKT buňkách, ale i na dendritických buňkách (dendritic cells – DC) a makrofázích. Velký zájem pak vzbudila data, která ukazovala, že PD ‑ 1 je silně exprimován vyčerpanými nefunkčními T buňkami při chronické virové infekci a že blokáda PD ‑ L1 obnovila funkci T buněk a kontrolu virové replikace [36,37]. Ovšem u PD ‑ L1 deficitních myší vyvolala chronická virová infekce fatální autoimunitní reakci, což ukázalo na důležitou roli PD ‑ 1 dráhy v ochraně organizmu před imunitně mediovanou destrukcí vlastních tkání při chronické antigenní stimulaci [38].
Fenotyp myší deficitních v genu pro PD ‑ 1 ukazuje, že role PD ‑ 1 je především v inhibici nadměrné aktivace T buněk v periferních tkáních. Na rozdíl od rychlého a fatálního nástupu autoimunity u zvířat deficitních v genu pro CTLA ‑ 4 zvířata bez genu pro PD ‑ 1 mají opožděný výskyt orgánově specifických autoimunit, jejichž spektrum se ještě liší podle použitého inbredního kmene. U PD ‑ 1 deficitních myší kmene C57BL/ 6 dochází po šesti měsících k rozvoji lupus‑like syndromu, zatímco u BALB/ c myší dochází především k rozvoji autoimunitní kardiomyopatie [39,40]. Tyto a další důkazy ukazují, že PD ‑ 1 slouží jako inhibitor T buněčné odpovědi v periferii.
Exprese PD ‑ 1 a jeho funkce
PD ‑ 1 se váže na dva různé ligandy, PD ‑ L1 a PD ‑ L2, jejichž exprese se výrazně liší. PD ‑ L1 je exprimován na relativně širokém spektru buněk, od klidových i aktivovaných T buněk (včetně Tregs) přes B buňky, DC až po nehematopoietické buňky (např. endoteliální či epitelální buňky), což ukazuje na důležitou roli PD ‑ L1 v regulaci periferní tolerance a v prevenci autoimunit. Oproti tomu PD ‑ L2 exprese je spíše omezená na hematopoietické buňky (hlavně DC a makrofágy). Prozánětlivé signály včetně INF ‑ γ indukují expresi PD ‑ L1 a ukazuje se, že celá řada nádorů exprimuje PD ‑ L1 molekulu jako „ochranu“ před protinádorovou odpovědí T buněk. Po vazbě PD ‑ 1 na své ligandy tedy dochází k „vypnutí“ imunitní odpovědi, ke snížené produkci cytokinů a inhibici proliferace [41,42]. Zajímavé je však zjištění, že blokáda PD ‑ 1 monoklonálními protilátkami nejen zvrátí anergii na úrovni T buněk, ale je schopna i vyvolat zesílenou T buněčnou odpověď proti dalším „chronickým“ chorobám, jako jsou např. nádory [21,43]. Preklinická data ukázala, že protilátky blokující osu PD ‑ 1/ PD ‑ L1 zvyšují protinádorovou aktivitu na zvířecích modelech, a ukazuje se, že se možná jedná o relativně univerzální mechanizmus, jak se nádory brání napadení T buňkami [21].
PD ‑ 1 a protinádorová terapie
PD ‑ L1 ligand je exprimován poměrně širokým spektrem nádorů. Kromě toho se ukazuje, že tumor infiltrující lymfocyty (TILs) velmi často exprimují PD ‑ 1 molekulu, což nahrává domněnkám, že nádor jako „tělu vlastní“ tkáň je schopen využívat podobné mechanizmy, které využívají normální tkáně v ochraně před nadměrnou imunitní reakcí. Myší modely ukazují, že zvýšená exprese PD ‑ L1 na nádorových buňkách snižuje schopnost T buněk zabíjet tyto nádorové buňky in vitro, a naopak in vivo tyto nádorové linie rostou rychleji. Blokáda PD ‑ 1 však tento fenomén inhibuje a ukazuje se, že tato protinádorová terapie funguje napříč nádorovými modely: model karcinomu prsu 4T1, myší model karcinomu pankreatu, melanom B16 či model kolorektálního karcinomu CT26 [44 – 47]. Podobná data se objevují i u lidských malignit. Exprese PD ‑ 1 na povrchu tumor ‑ infiltrujících lymfocytů spolu s expresí PD ‑ L1 mnoha různými nádory ukazují, že by se mohlo jednat o univerzální mechanizmus využívaný nádory k úniku pozornosti imnitního systému. Ukazuje se, že exprese PD ‑ L1 na nádorové tkáni bývá často spojena s horší prognózou u hepatocelulárního karcinomu, melanomu, renálního karcinomu, nemalobuněčného karcinomu plic, ovariálního karcinomu, glioblastomu, Hodgkinova lymfomu a dalších [48 – 55].
Na základě těchto dat se v posledních pěti letech velmi intenzivně testují protilátky blokující osu PD-1/ PD-L1 v klinických studiích [56 – 67]. V roce 2014 byly první anti‑PD-1 molekuly registrovány pro léčbu metastatického melanomu a v roce 2015 pro skvamózní nemalobuněčný karcinom plic. S ohledem na počet studií, které se objevují (k 17. 6. 2015 více než 250 studií s anti‑PD-1/ /anti‑PD ‑ L1 molekulami; www.clinicaltrials.gov), bude velmi zajímavé sledovat vývoj v této oblasti.
LAG ‑ 3
LAG ‑ 3 je další inhibiční molekula, která je ve zvýšené míře exprimována aktivovanými pomocnými (CD4+) nebo cytotoxickými (CD8+) T buňkami [68]. Ačkoliv byl LAG ‑ 3 klonován před více než 20 lety, jeho funkce „imunitního check-pointu“ byla definovaná teprve v roce 2005, kdy se ukázalo, že zesiluje funkci Tregs [69]. Kromě toho LAG ‑ 3 také přímo inhibuje funkci efektorových CD8+ buněk [70]. Jediným známým ligandem LAG ‑ 3 jsou MHC antigeny II. třídy, ovšem funkce této interakce a způsob ovlivnění efektorových a regulačních T buněk není zcela objasněn. Ukazuje se však, že duální blokáda LAG ‑ 3 a PD ‑ 1 synergistickým způsobem blokuje anergii u nádorově či virově specifických CD8+ T buněk při chronické, perzistentní infekci. Studie na dvojitých knock out myších (Pd1– / – Lag3 – / – ) naznačily, že tato zvířata kompletně odhojovala i slabě imunogenní nádory, a naopak se u nich rychleji objevovaly autoimunitní syndromy než u myší s knock out genem buď pro PD ‑ 1, nebo pro LAG ‑ 3 [71].
TIM ‑ 3
TIM ‑ 3 (T ‑ cell immunoglobulin and mucin protein‑3), jehož ligandem je galectin 9, inhibuje funkci pomocných TH1 buněk [72] a protilátky proti TIM ‑ 3 zesilují protinádorovou odpověď [73]. V několika studiích byla pozorována společná exprese molekul TIM ‑ 3 a PD ‑ 1 na nádorově specifických CD8+ T buňkách. Na zvířecích modelech se ukázalo, že simultánní blokáda PD ‑ 1 a TIM ‑ 3 zesiluje protinádorovou imunitní odpověď a vyvolává odhojení nádoru v situacích, kdy je blokáda jednotlivých receptorů TIM ‑ 3 nebo PD ‑ 1 jen slabě účinná [74]. Zajímavostí, která se ukázala u nádorů, je fakt, že dysfunkční T buňky exprimující jak Tim ‑ 3, tak PD ‑ 1 byly nalezeny pouze v nádorové tkáni, ale ne v periferní krvi. Navíc se ukázalo, že TIM ‑ 3 exprese charakterizuje regulační T buňky a byla spojena s progresí karcinomu plic [75]. Podobná data publikovala nedávno česká skupina kolem Špíška et al [76], která na úrovni mRNA u karcinomu hlavy a krku prokázala vysoké procento Tim ‑ 3+ PD ‑ 1+ buněk pouze v nádorové tkáni. Pomocí průtokové cytometrie prokázali, že cytotoxické CD8+ buňky s dvojitě pozitivním fenotypem (Tim ‑ 3+/ PD ‑ 1+) mají výrazně nižší procento buněk produkujících IFN‑γ v porovnání s fenotypem Tim ‑ 3– / PD ‑ 1+ či Tim ‑ 3+/ PD ‑ 1– . Právě exprese Tim ‑ 3 spolu s PD ‑ 1 může být považována u karcinomu hlavy a krku za lepší marker vyčerpanosti T buněk než jen samotný PD ‑ 1, a proto by tyto dvě molekuly mohly představovat zajímavý cíl pro budoucí imunoterapeutické přístupy v léčbě karcinomu hlavy a krku [76].
Produkty nádorů mohou imunitní reakci potlačovat
Deregulacemi získávají některé nádorové buňky schopnost produkovat imunosupresivní cytokiny a molekuly, které ovlivňují imunitní odpověď a utlumují či zcela inhibují funkci T buněk, případně způsobují, že jsou v nádorech přítomny takové subpopulace buněk, které nejenže nádorový růst neblokují, ale ještě mu pomáhají růst. Mezi nimi jsou nejdůležitější IDO, TGF‑β a IL‑10. Jsou to buněčné hormony, které výrazně inhibují proliferaci a efektorové funkce lymfocytů a makrofágů [24].
Indoleamin 2,3 - dioxygenáza
Indoleamin 2,3 - dioxygenáza (IDO) je enzym, který katabolizuje rozklad esenciální aminokyseliny L ‑ tryptofanu na N ‑ formylkynurenin [77]. Tryptofan je nezbytný pro správné fungování T buněčné odpovědi. IDO je produkován některými subpopulacemi makrofágů, regulačními T buňkami (což přispívá k vytvoření lokálního imunosupresivního prostředí), ale je to i strategie mnoha nádorů, jak se vyhnout pozornosti imunitního systému [78]. U některých nádorů se ví (ovariální karcinom), že zvýšená exprese IDO se podílí na progresi choroby a horším přežívání pacientů, pravděpodobně prostřednictvím útlumu lokální imunitní odpovědi [79]. Protože se jedná o mechanizmus přispívající k imunosupresi v nádorovém mikroprostředí, objevují se první klinické studie s inhibitory IDO v monoterapii či v kombinaci s blokátory kontrolních bodů imunitní reakce [80].
Interleukin 10
Interleukin 10 (IL‑10) je cytokin s protizánětlivými vlastnostmi a má centrální úlohu v tom, že inhibuje nadměrnou imunitní odpověď proti patogenům, čímž zabraňuje zánětlivé a autoimunitní patologii. IL‑10 je produkován monocyty a v menší míře Th2 lymfocyty, mastocyty a regulačními T buňkami [81]. Myši deficitní v genu pro IL‑10 vykazují zánětlivé procesy ve střevě po kolonizaci zažívacího traktu určitými mikroorganizmy. Původně byl IL‑10 popsán jako cytokin, který inhibuje diferenciaci T buněk na Th1 fenotyp, ale podílí se i na diferenciaci tolerizujících nebo Treg, které mají obvykle imunosupresivní efekt na protinádorovou imunitu [82]. Navíc se ukazuje, že zatímco v počátcích tumorigeneze má IL‑10 spíše stimulační efekt na NK buňky a cytotoxické T buňky, v pokročilých fázích růstu mohou mít nádorové buňky na svém povrchu receptor pro IL‑10 a IL‑10 pak funguje jako silný promotor nádorového bujení [82].
Transforming growth factor β
Transforming growth factor β (TGF‑β) představuje pleiotropní cytokin, který reguluje různé biologické procesy včetně vývoje orgánů a tkání, karcionogeneze a imunitní odpovědi. U normálních a premaligních buněk (časný rozvoj nádoru) hraje TGF‑β důležitou roli v homeostáze a má tumor supresivní vlastnosti (blokáda růstu a vyvolání apoptózy). Ukazuje se však, že nádorové buňky mohou přestat reagovat na inhibiční působení TGF‑β a naopak mohou získat schopnost využít TGF‑β ke svému prospěchu, např. blokádou imunitní reakce a zvýšenou schopností tvořit metastatické kolonie [83]. TGF‑β má také schopnost vyvolávat tzv. epiteliálně‑mezenchymální tranzici (EMT), což je proces charakterizovaný ztrátou E ‑ cadherinu, což vede následně k diferenciaci na invazivnější fenotyp [84]. TGF‑β byl totiž původně objeven jako imunosupresivní cytokin, jehož přidání do buněčných kultur inhibovalo proliferaci T buněk [85]. Následně se ukázalo, že myši, které nemají TGF‑β1 nebo receptory pro něj, hynou velmi časně na systémové autoimunitní onemocnění vyvolané hyperaktivací a zvýšenou proliferací T buněk [86]. Při normálních fyziologických podmínkách tak TGF‑β aktivně udržuje T buněčnou homeostázu a reguluje funkci T buněk. Na druhou stranu hraje TGF‑β klíčovou roli při diferenciaci a expanzi Treg na periferii. To může spolu s expresí dalších imunosupresivních cytokinů a s následnou převahou Tregs v nádorovém mikroprostředí vyvolávat silnou lokální inhibici protinádorové imunity, kdy infiltrující CD8+ T buňky nejsou schopny se dělit a zabíjet nádorové buňky [87].
Regulační T buňky
Důležitou, i když ne zatím zcela pochopenou funkci má podskupina T lymfocytů, tzv. Treg. Je to populace T buněk, která tvoří 5 – 10 % všech T buněk, inhibuje aktivaci jiných T lymfocytů a je nezbytná k udržení periferní tolerance vůči vlastním antigenům [88]. Většina regulačních buněk jsou typu CD4+ a exprimují α řetězec IL‑2 receptoru (také označovaný jako CD25), CTLA ‑ 4 a transkripční faktor FoxP3. Deplece Treg u myších nádorových modelů většinou vede k zlepšení protinádorové imunity a k redukci nádorového růstu. Naopakna myších modelech může přidání Treg zmírnit autoimunitní projevy [24]. Výsledky z experimentálních modelových systémů a od pacientů ukazují, že u některých typů nádorů je jejich hladina jak v periferní krvi, tak v buněčných infiltrátech významně zvýšena [88]. Není ale absolutním pravidlem, že jejich vyšší počet by byl vždy korelován s horší prognózou (graf 2). Oproti tomu přítomnost paměťových (CD45RO+), cytotoxických buněk (CD8+) či Th1 buněk (produkují IFN‑γ a IL‑2) v nádoru je téměř vždy spojena s příznivým vlivem na prognózu pacientů [89].
![Asociace mezi infiltrací nádoru různými buňkami imunitního systému a prognózou pacienta: analýza 124 publikací (upraveno dle [88]).](https://pl-master.mdcdn.cz/media/image/9b321bba294fd487915476c3a9d78383.jpg?version=1537795499)
Tumor‑ asociované makrofágy, M1 a M2
Další z buněčných populací, které zřejmě přímo podporují růst nádoru tím, že mění nádorové mikroprostředí a potlačují T buněčnou odpověď, jsou nádorově asociované makrofágy typu M2 [24]. Ty jsou, na rozdíl od klasických makrofágů označovaných jako M1, aktivovány cytokiny IL‑4 a IL‑13. Mají nejenom tkáňově‑reparační, ale hlavně protizánětlivý charakter. Sekretují mediátory, jako je IL‑10 a prostaglandin E2. Ty potlačují T buněčnou aktivaci a efektorové funkce [90]. M2 kromě toho produkují i velká množství TFG ‑ β a VEGF, které podporují neoangiogenezi a tím urychlují růst nádoru [91].
MDSC
MDSC populace (myeloid ‑ derived suppressor cells) představuje nezralé myeloidní prekurzory, které vznikají v kostní dřeni a akumulují se v lymfoidních tkáních, krvi a v nádorech jak experimentálních zvířat, tak nádorových pacientů, kde potlačují protinádorovou reakci jak vrozeného, tak získaného (specifického) charakteru [92]. Je to heterogenní populace několika buněčných typů, mezi které se počítají i prekurzory DC, monocyty a neutrofily. Vyznačují se některými společnými povrchovými znaky včetně Ly6C a Ly6G a CD11b u myší a CD33, CD11b a CD15 u lidí. Vyplavování z kostní dřeně do lymfatických uzlin a dalších tkání vyvolává řada prozánětlivých mediátorů, z nichž některé jsou také produkovány nádory. Patří mezi ně prostaglandin E2, IL‑6, VEGF a komplementový fragment C5a. Nejsou pro ně ale unikátní. MDSC se akumulují v místech chronického zánětu, který primárně není nádorem, i když se může během doby na vzniku nádoru rozhodujícím způsobem podílet. MDSC potlačují vrozenou imunitu tím, že syntetizují a uvolňují IL‑10, který inhibuje různé zánětlivé procesy, na kterých se podílejí aktivované makrofágy M1 typu a DC. MDSC potlačují také T buněčnou odpověď, a to různými mechanizmy. Mezi nimi je třeba poukázat na produkci volných radikálů, jako je peroxynitril a IDO, která katabolizuje tryptofan nezbytný pro T buněčnou proliferaci. MDSC kromě toho poškozují protinádorovou T buněčnou odpověď tím, že podporují vývoj T regulačních lymfocytů a ovlivňují T buněčnou diferenciaci směrem k Th2 populaci, která, na rozdíl od Th1, má jen omezenou protinádorovou aktivitu [26].
Exprese FasL, aktivní protiútok
Probíhající imunitní reakce bývá po několika týdnech zastavena fyziologickou likvidací až 90 % specifických aktivovaných buněk. Je k tomu využívána řada mechanizmů. Patří k nim aktivací indukovaná buněčná smrt (activation‑induced cell death – AICD), což je apoptóza aktivovaných T buněk využívající inhibiční mechanizmy, jako je vazba Fas ligandy (FasL) s membránovým Fas receptorem. Fas (CD95) je povrchový protein, který po vazbě na svůj ligand FasL (CD95L) indukuje apoptózu. Fas je relativně hojně exprimován napříč tkáněmi, nicméně silná exprese je v thymu, játrech, srdci či ledvinách. CD95L je exprimován hlavně aktivovanými T buňkami a NK buňkami a vyskytuje se též konstitutivně v tzv. imunoprivilegovaných tkáních, jako jsou varlata nebo oči. Genové experimenty se zvířaty, která nemají CD95, ukázala na důležitost této molekuly v udržení buněčné homeostázy a v ochraně organizmu před autoreaktivními T buňkami [26]. Myši kmene gld (generalized lymphoproliferative disease) či lpr (lymphoproliferation) trpí autoimunitními projevy z důvodu nedostatečné eliminace potenciálně autoreaktivních lymfocytů [93].
Zastavení nepotřebné antiinfekční imunity je po vyřešení akutní infekční hrozby nutné z hlediska homeostázy. Interakce Fas (CD95) a Fas ligandu sice byla původně popsána jako součást regulačních mechanizmů přispívajících k regulaci periferní tolerance, nicméně čím dál více důkazů ukazuje na důležitou roli v karcinogenezi, nádorovém růstu a metastazování [94]. Ukazuje se, že nádory jsou relativně často rezistentní k apoptóze indukované vazbou CD95L na povrchu T lymfocytů na CD95 na povrchu nádorových buněk. Naopak nádory nebo endotelie nádorové tkáně mohou exprimovat CD95L, který přispívá k lokální supresi imunitní reakce tím, že indukuje apoptózu v infiltrujících T buňkách exprimujících CD95 tzv. nádorovým protiútokem [95,96]. Charakter, stejně tak jako dynamika protinádorové reakce se od reakce protiinfekční významně liší a v tomto ohledu je předčasné ukončení imunitní odpovědi možné považovat za jednu ze strategií, které nádory využívají k úniku [26].
Závěr
Posledních několik let se ukazuje, že imuno ‑ onkologické přístupy v léčbě nádorů představují novou, nadějnou modalitu v léčbě pacientů s nádorovým onemocněním. Přestože byl protinádorový výzkum v oblasti imunoterapie a vakcín spojen velmi často se zklamáním, poslední roky ukazují, že koncept imnoterapie a imuno ‑ onkologických přístupů se velmi intenzivně rozvíjí. Spolu se zvyšujícími se znalostmi o tom, jak je imunitní systém regulován, se objevují další a další nadějné molekuly a přístupy využívající sílu imunitního systému. Kromě toho se zdá, že imuno ‑ onkologické přístupy představují modalitu s potenciálem dostat se do možných kombinací nejen s chemoterapií, ale i s radioterapií, cílenou terapií, vakcínami, DC či tumor infiltrujícími lymfocyty. Imunitní systém je dynamický a má schopnost reagovat na měnící se nádor, který je schopen potlačovat imunitní systém a blokovat aktivitu T buněk. Navíc je imunitní systém vybaven něčím, co se může ukázat jako zásadní – tedy imunologickou pamětí. Možná i to je jeden z důvodů, proč je délka trvání odpovědí po imunoterapii v porovnání s chemoterapií výrazně delší. Ne bezdůvodně nazval časopis Science protinádorovou imunoterapii průlomem roku 2013 [97].
* Prohlašuji, že v souvislosti s výše uvedeným příspěvkem, jehož jsem spoluautorem, mám střet zájmů se společností Bristol-Myers Squibb spol. s r. o.
Autor je bývalý pracovník Mikrobiologického ústavu AV ČR, v.v.i., Praha.
** Autorka deklaruje, že v souvislosti s předmětem studie nemá žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Dr. Marek Šťastný, Ph.D.
Bristol-Myers Squibb spol. s r. o.
Budějovická 778/3
140 00 Praha 4
e-mail: marek.stastny@bms.com
Obdrženo: 4. 8. 2015
Přijato: 1. 10. 2015
Sources
1. Monge J, Kricun M, Radovčić J et al. Fibrous dysplasia in a 120,000+ year old Neandertal from Krapina, Croatia. PLoS One 2013; 8(6): e64539. doi:10.1371/ journal.pone.0064539.
2. Casanova JL, Abel L. The genetic theory of infectious diseases: a brief history and selected illustrations. Annu Rev Genomics Hum Genet 2013; 14 : 215 – 243. doi: 10.1146/ annurev genom ‑ 091212 ‑ 153448.
3. Stutman O. Tumor development after 3 - methylcholanthrene in immunologically deficient athymic - nude mice. Science 1974; 183(4124): 534 – 536.
4. Kaplan DH, Shankaran V, Dighe AS et al. Demonstration of an interferon γ ‑ dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A 1998; 95(13): 7556 – 7561.
5. Shankaran VH, Ikeda A, Bruce T et al. IFNγ, and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001; 410(6832): 1107 – 1111.
6. Dunn GP, Bruce AT, Sheehan KC et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol 2005; 6(7): 722 – 729.
7. Schreiber RD, Old LJ, Smyth MJ. Suppression and promotion cancer immunoediting: integrating immunity‘s roles in cancer. Science 2011; 331(6024): 1565 – 1570. doi: 10.1126/ science.1203486.
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144(5): 646 – 674. doi: 10.1016/ j.cell.2011.02.013.
9. Koebel CM, Vermi W, Swann JB et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007; 450(7171): 903 – 907.
10. Kasiske BL, Snyder JJ, Gilbertson DT et al. Cancer after kidney transplantation in the United States. Am J Transplant 2004; 4(6): 905 – 913.
11. Robbins HA, Pfeiffer RM, Shiels MS et al. Excess cancers among HIV ‑ infected people in the United States. J Natl Cancer Inst 2015; 107(4). pii: dju503. doi: 10.1093/ jnci/ dju503.
12. MacKie RM, Reid R, Junor B. Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N Engl J Med 2003; 348(6): 567 – 568.
13. Cozar JM, Aptsiauri N, Tallada M et al. Late pulmonary metastases of rena cell carcinoma immediately after post‑transplantation immunosuppressive treatment: a case report. J Med Case Rep 2008; 2 : 111. doi: 10.1186/ 1752 ‑ 1947 ‑ 2 ‑ 111.
14. Meng S, Tripathy D, Frenkel EP et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res 2004; 10(24): 8152 – 8162.
15. Melichar B, Vanecková J, Morávek P et al. Spontaneous regression of renal cell carcinoma lung metastases in a patient with psoriasis. Acta Oncol 2009; 48(6): 925 – 927. doi: 10.1080/ 02841860902882451.
16. Jäger E, Ringhoffer M, Altmannsberger M et al. Immunoselection in vivo: independent loss of MHC class I and melanocyte differentiation antigen expression in metastatic melanoma. Int J Cancer 1997; 71(2): 142 – 147.
17. Driessens G, Kline J, Gajewski TF. Costimulatory and coinhibitory receptors in anti‑tumor immunity. Immunol Rev 2009; 229(1): 126 – 144. doi: 10.1111/ j.1600 ‑ 065X.2009.00771.x.
18. Peggs KS, Quezada SA, Korman AJ et al. Principles and use of anti‑CTLA4 antibody in human cancer. Curr Opin Immunol 2006; 18(2): 206 – 213.
19. Nurieva RI, Liu X, Dong C. Yin‑Yang of costimulation: crucial controls of immune tolerance and function. Immunol Rev 2009; 229(1): 88 – 100. doi: 10.1111/ j.1600 ‑ 065X.2009.00769.x.
20. Peggs KS, Quezada SA, Allison JP. Cancer immunotherapy: co ‑ stimulatory agonists and co‑inhibitory antagonists. Clin Exp Immunol 2009; 157(1): 9 – 19. doi: 10.1111/ j.1365 ‑ 2249.2009.03912.x.
21. Pardoll DM. Immunology beats cancer: a blueprint for successful translation. Nat Immunol 2012; 13(12): 1129 – 1132. doi: 10.1038/ ni.2392.
22. Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol 1993; 11 : 191 – 212.
23. Jenkins MK, Johnson JG. Molecules involved in T ‑ cell costimulation. Curr Opin Immunol 1993; 5(3): 361 – 367.
24. Abbas AK, Lichtman AH, Pillai S (eds). Cellular and molecular immunology. 6th ed. Philadelphia, PA: Elsevier Saunders 2010.
25. Moran AE, Kovacsovics ‑ Bankowski M, Weinberg AD. The TNFRs OX40, 4 - 1BB, and CD40 as targets for cancer immunotherapy. Curr Opin Immunol 2013; 25(2): 230 – 237. doi: 10.1016/ j.coi.2013.01.004.
26. Perez ‑ Gracia JL, Labiano S, Rodriguez ‑ Ruiz ME et al. Orchestrating immune check ‑ point blockade for cancer immunotherapy in combinations. Curr Opin Immunol 2014; 27 : 89 – 97. doi: 10.1016/ j.coi.2014.01.002.
27. Krummel MF, CTLA ‑ 4 have opposingAllison JP. CD28 and effects on the response of T cells to stimulation. J Exp Med 1995; 182(2): 459 – 465.
28. Sharma P, Allison JP. The future of immune check-point therapy. Science 2015; 348 (6230): 56 – 61. doi: 10.1126/ science.aaa8172.
29. Tivol EA, Borriello F, Schweitzer AN et al. Loss of CTLA ‑ 4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA ‑ 4. Immunity 1995; 3(5): 541 – 547.
30. Alegre ML, Frauwirth KA, Thompson CB. T ‑ cell regulation by CD28 and CTLA ‑ 4. Nat Rev Immunol 2001; 1(3): 220 – 228.
31. Greenfield EA, Nguyen KA, Kuchroo VK. CD28/ B7 costimulation: a review. Crit Rev Immunol 1998; 18(5): 389 – 418.
32. Petrausch U, Poehlein CH, Jensen SM et al. Cancer immunotherapy: the role regulatory T cells play and what can be done to overcome their inhibitory effects. Curr Mol Med 2009; 9(6): 673 – 682.
33. Liu VC, Wong LY, Jang T et al. Tumor evasion of the immune system by converting CD4+CD25 – T cells into CD4+CD25+ T regulatory cells: role of tumor ‑ derived TGF‑beta. J Immunol 2007; 178(5): 2883 – 2892.
34. Wing K, Onishi Y, Prieto ‑ Martin P et al. CTLA ‑ 4 control over Foxp3+ regulatory T cell function. Science 2008; 322(5899): 271 – 275. doi: 10.1126/ science.1160062.
35. Ishida Y, Agata Y, Shibahara K et al. Induced expression of PD ‑ 1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992; 11(11): 3887 – 3895.
36. Barber DL, Wherry EJ, Masopust D et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006; 439(7077): 682 – 687.
37. Day CL, Kaufmann DE, Kiepiela P et al. PD ‑ 1 expression on HIV ‑ specific T cells is associated with T ‑ cell exhaustion and disease progression. Nature 2006; 443(7109): 350 – 354.
38. Intlekofer AM, Thompson CB. At the bench: preclinical rationale for CTLA ‑ 4 and PD ‑ 1 blockade as cancer immunotherapy. J Leukoc Biol 2013; 94(1): 25 – 39. doi: 10.1189/ jlb.1212621.
39. Nishimura H, Okazaki T, Tanaka Y et al. Autoimmune dilated cardiomyopathy in PD ‑ 1 receptor ‑ deficient mice. Science 2001; 291(5502): 319 – 322.
40. Okazaki T, Tanaka Y, Nishio R et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD ‑ 1-deficient mice. Nat Med 2003; 9(12): 1477 – 1483.
41. Keir ME, Butte MJ, Freeman GJ et al. PD ‑ 1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008; 26 : 677 – 704. doi: 10.1146/ annurev.immunol.26.021607.090331.
42. Blank C, Mackensen A. Contribution of the PD ‑ L1/ PD ‑ 1 pathway to T ‑ cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother 2007; 56(5): 739 – 745.
43. Weber J. Immune checkpoint proteins: a new therapeutic paradigm for cancer – preclinical background: CTLA ‑ 4 and PD ‑ 1 blockade. Semin Oncol 2010; 37(5): 430 – 439. doi: 10.1053/ j.seminoncol.2010.09.005.
44. Hirano F, Kaneko K, Tamura H et al. Blockade of B7 - H1 and PD ‑ 1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 2005; 65(3): 1089 – 1096.
45. Nomi T, Sho M, Akahori T et al. Clinical significance and therapeutic potential of the programmed death ‑ 1 ligand/ programmed death ‑ 1 pathway in human pancreatic cancer. Clin Cancer Res 2007; 13(7): 2151 – 2157.
46. Peng W, Liu C, Xu C et al. PD ‑ 1 blockade enhances T ‑ cell migration to tumors by elevating IFN ‑ gamma inducible chemokines. Cancer Res 2012; 72(20): 5209 – 5218. doi: 10.1158/ 0008 ‑ 5472.CAN ‑ 12 ‑ 1187.
47. Iwai Y, Terawaki S, Honjo T. PD ‑ 1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol 2005; 17(2): 133 – 144.
48. Shi F, Shi M, Zeng Z et al. PD ‑ 1 and PD ‑ L1 upregulation promotes CD8(+) T ‑ cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer 2011; 128(4): 887 – 896. doi: 10.1002/ ijc.25397.
49. Hino R, Kabashima K, Kato Y et al. Tumor cell expression of programmed cell death ‑ 1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010; 116(7): 1757 – 1766. doi: 10.1002/ cncr.24899.
50. Richendollar BG, Pohlman B, Elson P et al. Follicular programmed death 1 - positive lymphocytes in the tumor microenvironment are an independent prognostic factor in follicular lymphoma. Human Pathol 2011; 42(4): 552 – 527. doi: 10.1016/ j.humpath.2010.08.015.
51. Dorfman DM, Brown JA, Shahsafaei A et al. Programmed death ‑ 1 (PD ‑ 1) is a marker of germinal center‑associated T-cells and angioimmunoblastic T ‑ cell lymphoma. Am J Surg Pathol 2006; 30(7): 802 – 810.
52. Liu J, Hamrouni A, Wolowiec D et al. Plasma cells from multiple myeloma patients express B7 - H1 (PD ‑ L1) and increase expression after stimulation with IFN ‑ {gamma} and TLR ligands via a MyD88 - , TRAF6 - , and MEK ‑ dependent pathway. Blood 2007; 110(1): 296 – 304.
53. Iwai Y, Ishida M, Tanaka Y et al. Involvement of PD ‑ L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD ‑ L1 blockade. Proc Natl Acad Sci USA 2002; 99(19): 12293 – 12297.
54. Hamanishi J, Mandai M, Iwasaki M et al. Programmed cell death 1 ligand 1 and tumor ‑ infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A 2007; 104(9): 3360 – 3365.
55. Ishikawa T, Fujita T, Suzuki Y et al. Tumor ‑ specific immunological recognition of frameshift ‑ mutated peptides in colon cancer with microsatellite instability. Cancer Res 2003; 63(7): 5564 – 5572.
56. Wolchok JD, Kluger H, Callahan MK et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013; 369(2): 122 – 133. doi: 10.1056/ NEJMoa1302369.
57. Herbst RS, Soria JC, Kowanetz M et al. Predictive correlates of response to the anti‑PD ‑ L1 antibody MPDL3280A in cancer patients. Nature 2014; 515(7528): 563 – 567. doi: 10.1038/ nature14011.
58. Robert C, Ribas A, Wolchok JD et al. Anti‑programmed ‑ death ‑ receptor ‑ 1 treatment with pembrolizumab in ipilimumab ‑ refractory advanced melanoma: a randomized dose‑comparison cohort of a phase 1 trial. Lancet 2014; 384(9948): 1109 – 1117. doi: 10.1016/ S0140 ‑ 6736(14)60958 ‑ 2.
59. Weber JS, D‘Angelo SP, Minor D et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti‑CTLA ‑ 4 treatment (CheckMate 037): a randomized, controlled, open ‑ label, phase 3 trial. Lancet Oncol 2015; 16(4): 375 – 384. doi: 10.1016/ S1470 ‑ 2045(15)70076 ‑ 8.
60. Robert C, Long GV, Brady B et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372(4): 320 – 330. doi: 10.1056/ NEJMoa1412082.
61. Larkin J, Chiarion ‑ Sileni V, Gonzalez R et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373(1): 23 – 34. doi: 10.1056/ NEJMoa1504030.
62. Postow MA, Chesney J, Pavlick AC et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015; 372(21): 2006 – 2017. doi: 10.1056/ NEJMoa1414428.
63. Rizvi NA, Hellmann MD, Snyder A et al. Cancer immunology. Mutational landscape determines sensitivity to PD ‑ 1 blockade in non‑small cell lung cancer. Science 2015; 348(6230): 124 – 128. doi: 10.1126/ science.aaa1348.
64. Rizvi NA, Mazières J, Planchard D et al. Activity and safety of nivolumab, an anti‑PD ‑ 1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non‑small‑cell lung cancer (CheckMate 063): a phase 2, single‑arm trial. Lancet Oncol 2015; 16(3): 257 – 265. doi: 10.1016/ S1470 ‑ 2045(15)70054 ‑ 9.
65. Brahmer J, Reckamp KL, Baas P et al. Nivolumab versus docetaxel in advanced squamous ‑ cell non‑small‑cell lung cancer. N Engl J Med 2015; 373(2): 123 – 135. doi: 10.1056/ NEJMoa1504627.
66. Gettinger SN, Horn L, Gandhi L et al. Overall survival and long‑term safety of nivolumab (anti‑programmed death 1 antibody, BMS ‑ 936558, ONO ‑ 4538) in patients with previously treated advanced non‑small‑cell lung cancer. J Clin Oncol 2015; 33(18): 2004 – 2012. doi: 10.1200/ JCO.2014.58.3708.
67. Garon EB, Rizvi NA, Hui R et al. Pembrolizumab for the treatment of non‑small‑cell lung cancer. N Engl J Med 2015; 372(21): 2018 – 2028. doi: 10.1056/ NEJMoa1501824.
68. Sierro S, Romero P, Speiser DE. The CD4‑like molecule LAG ‑ 3, biology and therapeutic applications. Expert Opin Ther Targets 2011; 15(1): 91 – 101. doi: 10.1517/ 14712598.2011.540563.
69. Goldberg MV, Drake CG. LAG ‑ 3 in cancer immunotherapy. Curr Top Microbiol Immunol 2011; 344 : 269 – 278. doi: 10.1007/ 82_2010_114.
70. Grosso JF, Kelleher CC, Harris TJ et al. LAG ‑ 3 regulates CD8+ T cell accumulation and effector function in murine self ‑ and tumor ‑ tolerance systems. J Clin Invest 2007; 117(11): 3383 – 3392.
71. Woo SR, Turnis ME, Goldberg MV et al. Immune inhibitory molecules LAG ‑ 3 and PD ‑ 1 synergistically regulate T ‑ cell function to promote tumoral immune escape. Cancer Res 2012; 72(4): 917 – 927. doi: 10.1158/ 0008 ‑ 5472.CAN ‑ 11 ‑ 1620.
72. Zhu C, Anderson AC, Schubart A et al. The Tim 3 ligand galectin 9 negatively regulates T helper type 1 immunity. Nature Immunol 2005; 6(12): 1245 – 1252.
73. Ngiow SF, von Scheidt B, Akiba H et al. Anti TIM3 antibody promotes T cell IFN γ ‑ mediated antitumor immunity and suppresses established tumors. Cancer Res 2011; 71(10): 3540 – 3551. doi: 10.1158/ 0008 ‑ 5472.CAN ‑ 11 ‑ 0096.
74. Sakuishi K, Apetoh L, Sullivan JM et al. Targeting Tim 3and PD 1 pathways to reverse T cell exhaustion and restore anti‑tumor immunity. J Exp Med 2010; 207(10): 2187 – 2194. doi: 10.1084/ jem.20100643.
75. Gao X, Zhu Y, Li G et al. TIM ‑ 3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One 2012; 7(2): e30676. doi: 10.1371/ journal.pone.0030676.
76. Partlová S, Bouček J, Kloudová K et al. Distinct patterns of intratumoral immune cell infiltrates in patients with HPV‑associated compared to non‑virally induced head and neck squamous cell carcinoma. Oncoimmunology 2015; 4(1): e965570.
77. Uyttenhove L, Pilotte I, Théate I et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 2003; 9(10): 1269 – 1274.
78. Pilotte L, Larrieu P, Stroobant V et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3 - dioxygenase. Proct Natl Acad Sci U S A 2012; 109(7): 2497 – 2502. doi: 10.1073/ pnas.1113873109.
79. Tanizaki Y, Kobayashi A, Toujima S et al. Indoleamine 2,3 - dioxygenase promotes peritoneal metastasis of ovarian cancer by inducing an immunosuppressive environment. Cancer Sci 2014; 105(8): 966 – 973. doi: 10.1111/ cas.12445.
80. Jiang T, Sun Y, Yin Z et al. Research progress of indoleamine 2,3 - dioxygenase inhibitors. Future Med Chem 2015; 7(2): 185 – 201. doi: 10.4155/ fmc.14.151.
81. Saraiva M, O‘Garra A. The regulation of IL‑10 production by immune cells. Nat Rev Immunol 2010; 10(3): 170 – 181. doi: 10.1038/ nri2711.
82. Mannino MH, Zhu Z, Xiao H et al. The paradoxical role of IL‑10 in immunity and cancer. Cancer Lett 2015; 367(2): 103 – 107. doi: 10.1016/ j.canlet.2015.07.009.
83. Massague J. TGFbeta in Cancer. Cell 2008; 134(2): 215 – 230. doi: 10.1016/ j.cell.2008.07.001.
84. Elliott RL, Blobe GC. Role of transforming growth factor beta in human cancer. J Clin Oncol 2005; 23(9): 2078 – 2093.
85. Kehrl JH, Wakefield LM, Roberts AB et al. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med 1986; 163(5): 1037 – 1050.
86. Shull MM, Ormsby I, Kier AB et al. Targeted disruption of the mouse transforming growth factor‑beta 1 gene results in multifocal inflammatory disease. Nature 1992; 359(6397): 693 – 699.
87. Tu E, Chia PZ, Chen W. TGFβ in T cell biology and tumor imunity: angel or devil? Cytokine Growth Factor Rev 2014; 25(4): 423 – 435. doi: 10.1016/ j.cytogfr.2014.07.014.
88. Kretschmer K, Apostolou I, Jaeckel E et al. Making regulatory T cells with defined antigen specificity: role in autoimmunity and cancer. Immunol Rev 2006; 212 : 163 – 169.
89. Fridman WH, Pagès F, Sautès ‑ Fridman C et al. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012; 12(4): 298 – 306. doi: 10.1038/ nrc3245.
90. Italiani P, Boraschi D. From monocytes to M1/ M2 macrophages: phenotypical vs. functional differentiation. Front Immunol 2014; 5 : 514. doi: 10.3389/ fimmu.2014.00514.
91. Galdiero MR, Garlanda C, Jaillon S et al. Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol 2013; 228(7): 1404 – 1412. doi: 10.1002/ jcp.24260.
92. Parker KH, Beury DW, Ostrand ‑ Rosenberg S. Myeloid ‑ derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res 2015; 128 : 95 – 139. doi: 10.1016/ bs.acr.2015.04.002.
93. Cohen PL, Eisenberg RA. The lpr and gld genes in systemic autoimmunity: life and death in the Fas lane. Immunol Today 1992; 13(11): 427 – 428.
94. Owen ‑ Schaub L, Chan H, Cusack JC et al. Fas and Fas ligand interactions in malignant disease. Int J Oncol 2000; 17(1): 5 – 12.
95. Igney FH, Behrens CK, Krammer PH. Tumor counterattack ‑ concept and reality. Eur J Immunol 2000; 30(3): 725 – 731.
96. Motz GT, Santoro SP, Wang LP et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 2014; 20(6): 607 – 615. doi: 10.1038/ nm.3541.
97. Couzin‑Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013; 342(6165): 1432 – 1433. doi: 10.1126/science.342.6165.1432.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2015 Issue Supplementum 4
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Nežádoucí účinky moderní imunoterapie a jejich řešení v klinické praxi
- Imunoterapie uroteliálního karcinomu močového měchýře – od BCG vakcín k cílené imunoterapii
- Únikové strategie nádorů pozornosti imunitního systému
- Význam imunogenní buněčné smrti v protinádorové imunitě