
Ferroptóza jako nový typ buněčné smrti a její role v léčbě rakoviny
Ferroptosis as a New Type of Cell Death and its Role in Cancer Treatment
Background:
Ferroptosis is a recently discovered type of cell death. It is genetically, morphologically, and biochemically distinct from other types of programmed cell death, such as necrosis, apoptosis, and autophagy. The level of intracellular free iron and reactive oxygen species formation are important for ferroptosis activation, which can occur through either of two key inhibitory processes. The first one involves inhibition of cystine transfer into cells by the cystine/glutamate antiporter system (Xc–). Cystine serves as a precursor for the synthesis of glutathione, a major cellular antioxidant. The second one involves the inhibition of glutathione peroxidase 4, which protects cells from lipid peroxidation. Ferroptosis is associated with many metabolic disorders, including neurological diseases and cancer. Molecules involved in the activation of ferroptotic pathways are involved in protecting cells against stress conditions, and in the maintenance of nicotinamide adenine dinucleotide phosphate and glutathione levels, as well as iron homeostasis. Also important is the connection with autophagy, so called ferritinophagy, in which iron is released from lysosomes into the cytosol. Cascade reactions of free unstable iron atoms with other molecules result in the production of reactive oxygen species that initiate the cellular stress that triggers ferroptosis. In diseases such as cancer where cell death inducing mechanisms, including apoptosis, are usually suppressed by genetic changes, the induction of alternative pathways leading to cell death could provide an attractive treatment strategy.
Conclusion:
In recent years, research into new antimetastatic drugs has focused on the activation of alternative cell death pathways that might overcome disturbed metabolic processes inside cancer cells or the chemotherapy resistance acquired in the course of routine treatment. A number of molecules have been found to induce ferroptosis in tumor cells, suggesting that they may offer new alternatives for anticancer treatment.
Key words:
cell death – cancer – autophagy – ferroptosis – ferritinophagy – cellular stress – ROS
This work was supported by the projects GAČR 17-05838S, MEYS – NPS I – LO1413 and MH CZ - -DRO (MMCI, 00209805).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Accepted: 31. 8. 2018
:
Skoupilová Hana; Michalová Eva; Hrstka Roman
:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
:
Klin Onkol 2018; 31(Supplementum 2): 21-26
:
Review
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amko20182S21
Východiska:
Ferroptóza je jedním z nově objevených typů buněčné smrti. Je geneticky, morfologicky i biochemicky odlišitelná od dalších typů programované buněčné smrti, jako je nekróza, apoptóza i autofagie. Pro indukci ferroptózy je důležitá hladina volného železa uvnitř buněk a tvorba reaktivních kyslíkových radikálů (reactive oxygen species – ROS), které aktivují mechanizmy vedoucí k buněčné smrti. K indukci ferroptózy může dojít prostřednictvím inhibice jednoho ze dvou klíčových procesů. Prvním je zablokování přenosu cystinu do buněk pomocí cystin/glutamátového transportního systému (Xc–), neboť cystin slouží jako prekurzor k syntéze glutathionu, hlavního buněčného antioxidantu. Druhým je inhibice glutathion peroxidázy 4, která chrání buňky před peroxidací lipidů. Ferroptóza bývá asociovaná s mnoha metabolickými poruchami, vč. neurologických chorob nebo rakoviny. Molekuly zapojené do drah aktivujících ferroptózu se podílejí na obraně buněk před stresovými podmínkami, napomáhají udržovat hladinu nikotinamidadenindinukleotidfosfátu, glutathionu a homeostázu železa. Důležitá je také spojitost s autofagií. Pomocí tzv. ferritinofagie dochází k uvolňování železa z lysozomů do cytosolu buněk. Následnou kaskádou reakcí uvolněných nestabilních atomů železa s jinými molekulami dochází k produkci ROS a iniciaci buněčného stresu, jež je spouštěčem ferroptózy. U onemocnění, jako je rakovina, při které jsou mechanizmy indukující buněčnou smrt, vč. apoptózy potlačeny obvykle z důvodu genetických změn, může indukce alternativního způsobu buněčné smrti představovat atraktivní léčebnou strategii.
Závěr:
V posledních letech je výzkum nových protinádorových léků směřován na aktivaci alternativních drah buněčné smrti, které mohou překonat narušené metabolické procesy uvnitř rakovinných buněk nebo rezistenci buněk k běžně podávané chemoterapii. Byla nalezena celá řada molekul, které mohou ferroptózu v nádorových buňkách indukovat, což dodává jejímu výzkumu na významu, neboť by mohl vést k objevu nových alternativ při léčbě nádorových onemocnění.
Klíčová slova:
buněčná smrt – rakovina – autofagie – ferroptóza – ferritinofagie – buněčný stres – ROS
Úvod
Buněčná smrt je klíčovým fyziologickým mechanizmem, na kterém je závislý vývoj organizmu prakticky již od počátku embryogeneze, vč. plnohodnotné funkce imunitního systému nebo udržování správné funkce tkání a orgánů u dospělých mnohobuněčných organizmů [1–5]. Původně se předpokládalo, že existují pouze 2 typy buněčné smrti, programovaná – apoptóza – nebo neprogramovaná – nekróza. Následkem vnějších faktorů, jako jsou zranění nebo infekce, buňka obvykle reaguje na nepříznivé podmínky nevratnými procesy a bývá likvidována prostřednictvím nekrózy. Spolu s nekrózou existují i další typy buněčné smrti, které jsou geneticky programované a bývají buňkou záměrně vyvolávány. Slouží převážně jako obranný mechanizmus před poškozením buněk následkem patogenů, buněčného stresu nebo např. kumulací mutací v genech důležitých pro zásadní procesy uvnitř buňky [6]. Jedním z nejlépe prostudovaných typů programované buněčné smrti (programmed cell death – PCD) je apoptóza. Důsledkem apoptózy je např. diferenciace prstů v rozvíjejících se embryích [7]. Její deregulace může vést ke vzniku vrozených vývojových vad nebo k rozvoji rakoviny [8–11]. Během posledních desetiletí se však ukázalo, že apoptóza není zdaleka jediným typem PCD. Zkoumání jejích nových typů se stalo předmětem intenzivního výzkumu. Mezi nově objevené typy PCD patří autofagie, nekroptóza, pyroptóza, pyronekróza a také ferroptóza [6,12–15].
Ferroptóza je relativně nově objeveným typem PCD, která je morfologicky, biochemicky i geneticky odlišitelná od dalších typů buněčné smrti. Morfologické odlišnosti byly prokázány transmisní elektronovou mikroskopií, která potvrdila, že ferroptotické buňky mají jasné substrukturní změny [14]. Pro buňky podléhající ferroptóze jsou charakteristické menší mitochondrie s nižší hustotou mitochondriálního obsahu a naopak zvýšeným mitochondriálním membránovým potenciálem [14,16]. Naopak klasické znaky objevující se u ostatních typů buněčné smrti, jako je kondenzace chromatinu u apoptózy nebo ruptury buněčné stěny u nekrózy, nebyly u ferroptotických buněk pozorovány [14]. Z biochemického hlediska dochází při ferroptóze k produkci cytosolických a lipidových reaktivních kyslíkových radikálů (reactive oxygen species – ROS), jejichž tvorba ústí k vyčerpání antioxidantů v buňkách a následné buněčné smrti [14].
Historie objevu ferroptózy
Pojem ferroptóza byl poprvé zaveden v roce 2012 americkými vědci pod vedením profesora Brenta R. Stockwella [14] během výzkumu malých molekul erastinu, RSL3 a RSL5. Jedná se o sloučeniny selektivně letální pro nádorové buňky nesoucí mutace v onkogenu RAS. Z tohoto důvodu byly pojmenovány jako RAS-selektivně letální molekuly (RSL) [17,18]. Jejich význam podtrhuje skutečnost, že geny rodiny malých GTPáz RAS (HRAS, NRAS a KRAS) jsou mutovány přibližně u 30 % všech nádorů [19], vč. nemalobuněčných nádorů plic [20], nádorů tlustého střeva [21], slinivky [22] nebo štítné žlázy [21,23]. Současně jsou pak tyto mutace u řady nádorů spojeny i se špatnou prognózou [23].
U nových RSL molekul však bylo třeba objasnit mechanizmus účinku, neboť nebyly pozorovány klasické rysy apoptózy, jako je aktivace kaspáz, fragmentace chromatinu nebo uvolňování cytochromu c z mitochondrií [17,18,24]. Všechny tři sloučeniny naopak indukovaly RAS-RAF-MEK-závislou oxidativní buněčnou smrt [17,24]. Dále bylo zjištěno, že erastin spolu s RSL5 pro svoji funkci vyžadují přítomnost mitochondriálního anion-selektivního kanálu 3 (voltage-dependent anion channel – VDAC3), kdežto RSL3 funguje nezávisle na VDAC kanálech [24]. U všech tří molekul pak dochází k potlačení účinku po použití různých chelátorů železa. To úzce souvisí se skutečností, že mutantní Ras zvyšuje hladinu transferinových receptorů (TfR1) [24], které převádějí komplexy železa do cytoplazmy, kde následně dochází k jeho uvolnění (schéma 1) [25,26]. Stejně tak mutantní Ras snižuje množství ferritinu, jehož molekula slouží ke skladování železa v buňkách [27]. Zesíleným transportem a současně snížením množství molekul, které udržují železo v navázaném stavu, pak dochází ke zvyšování volného železa v buňce [24]. Volné reaktivní železo Fe2+ uvolněné z endozomů nebo lysozomů je zapojeno do produkce ROS prostřednictvím Fentonovy reakce [28]. Smrt indukovaná prostřednictvím RSL je tedy spojena se zvýšením produkce ROS díky zabránění chelatace železa, popř. genetickou inhibicí hladiny železa v cytoplazmě [17,24]. I další proteiny zapojené v podobně působících drahách mohou rovněž aktivovat ferroptózu [17]. Dokonce bylo prokázáno, že jeden z nejdůležitějších nádorových supresorových proteinů, protein p53, často označovaný jako strážce genomu [29], může být zapojen do některých mechanizmů iniciujících ferroptotickou buněčnou smrt [30].

Metabolizmus železa při ferroptóze
Buněčné železo se zdá být jedním z nejdůležitějších faktorů při tvorbě lipidových peroxidových radikálů během ferroptózy [14,17]. Volné atomy železa Fe2+ se mohou za přítomnosti peroxidu vodíku pomocí tzv. Fentonovy reakce oxidovat na Fe3+, z peroxidu je současně odebrán elektron a dochází k produkci hydroxylových radikálů přímo uvnitř buněk [31]. To vede k indukci buněčného stresu, poškození buňky volnými radikály a spuštění ferroptózy [14]. Z tohoto důvodu se buňky snaží chránit před vlivy volného železa hned několika způsoby. Jedním z nejdůležitějších proteinů, který plní v buňce tuto ochrannou funkci, je ferritin [32]. Jedná se o protein tvořený 24 podjednotkami lehkých a těžkých řetězců [32], přičemž těžké řetězce vykazují navíc ferroxidázovou aktivitu. Přijímané železo Fe2+ je pomocí těchto řetězců oxidováno na Fe3+ a skladováno uvnitř ferritinu v netoxických a biologicky dostupných formách [33]. Při zvýšené potřebě reaktivní formy železa je ferritin v lysozomech degradován a železo je uvolněno do cytosolu [34,35].
Transferin je dalším důležitým proteinem, který se uplatňuje především při transportu železa. Tento extracelulární protein je schopen vázat 1–2 ionty železa Fe3+, které přenáší krevním řečištěm, dokud není zachycen TfR1 lokalizovaným na povrchu buněk [25,36]. Komplex receptoru s navázaným transferinem je posléze pohlcován endocytózou dovnitř buněk. Z endozomů jsou následně za účasti transmembránového transportéru DMT1 (divalent metal transporter-1), známého také jako Nramp2 (natural resistance-associated macrophage) uvolňovány do cytoplazmy nestabilní atomy železa Fe2+ spolu s volným transferinem a TfR1, který je připraven k dalšímu transportu [36,37].
Pro udržování správné hladiny železa v buňce je kromě transportu dovnitř důležitý také transport železa ven z buňky. Jediným známým transportérem, který převádí železo ven přes buněčnou membránu do krve, je ferroportin 1. Je kódován genem SLC40A1 [38]. Jeho správná regulace je rozhodující pro udržování optimální hladiny železa. Změny v hladině ferroportinu mohou vést k nedostatku, nebo naopak k nadbytku železa uvnitř buněk. Bylo prokázáno, že nedostatečná funkce ferroportinu může mít za následek ztrátu homeostázy a způsobit např. anémii u myší [39,40].
Mechanizmus ferroptózy
Ferroptóza, tj. na intracelulárním železe závislá regulovaná forma buněčné smrti, může být indukována dvěma různými mechanizmy. Prvním z nich je inhibice cystin/glutamátového transportního systému Xc– (na sodíku nezávislém), který je vyžadován při transportu cystinu do buněk, kde dochází k syntéze glutathionu, hlavního buněčného antioxidantu. Narušením tohoto systému dochází k vyčerpání glutathionu a vzniku oxidativního stresu [41,42], což může být mimo jiné spouštěno i malými molekulami, tzv. induktory ferroptózy třídy I (FINs I) [42]. Druhým podnětem pro vznik ferroptotické buněčné smrti je inhibice glutathion peroxidázy 4 (GPX4), která používá glutathion k detoxikaci lipidových peroxidů [41,42]. GPX4 může být přímo inhibována tzv. induktory ferroptózy třídy II (FINs II) [42].
Cystin/glutamátový transportní systém Xc–
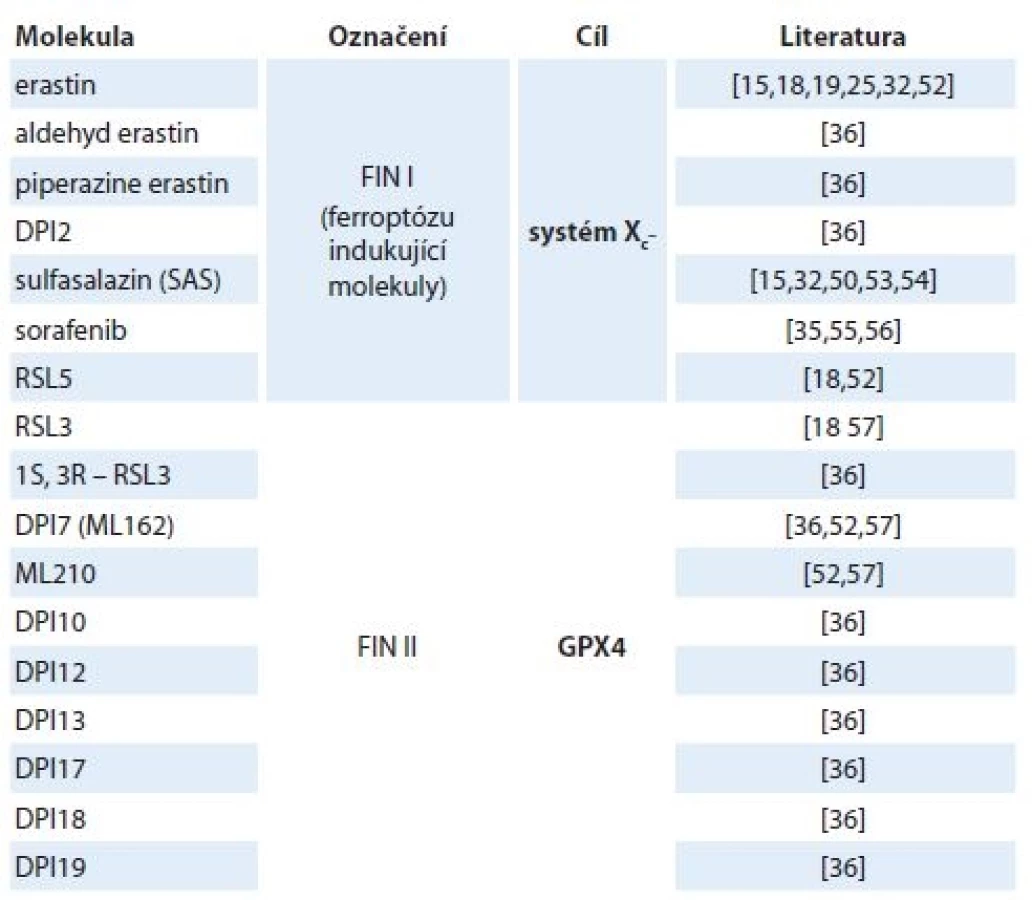
V roce 1980 Bannai a Kitamura poprvé zdokumentovali Xc– transportní systém v lidských embryonálních plicních fibroblastech [43]. O 2 roky později pak v podstatě totéž ukázali Makowske a Christensen v transformovaných krysích buněčných liniích hepatomu [44]. Systém Xc– působí jako obousměrný přenašeč aniontových forem cystinu a glutamátu a je závislý na Cl– a nezávislý na Na+ [45]. I když může transportovat obě aminokyseliny v obou směrech, jeho hlavním úkolem je zásobovat buňku cystinem a odvádět glutamát ven z buňky. Intracelulární zásoba cystinu je poměrně malá, zatímco koncentrace intracelulárního glutamátu je v buňce obecně vyšší než v extracelulárním prostoru, proto jsou molekuly přenášeny především po koncentračním spádu [46]. Extracelulární glutamát pak funguje jako kompetitivní inhibitor vychytávající cystin prostřednictvím transportního systému Xc– [44]. Bylo prokázáno, že nejen radikály, ale i různá elektrofilní činidla [47], bakteriální lipopolysacharidy a zánětlivý cytokin (faktor nádorové nekrózy α (tumor necrosis factor – TNFα)) [48] nebo např. i pěstování buněčných kultur neutrofilů [49] nebo makrofágů [50] v podmínkách 21% hladiny kyslíku může systém Xc– aktivovat. V roce 2012 Dixon et al prokázali souvislost mezi aktivací ferroptózy a cystin/glutamátovým transportním systémem, a to díky výzkumu zaměřenému na studium mechanizmu účinku ferrostatinu 1, který je selektivním inhibitorem ferroptózy indukované erastinem [14]. Současně byla objevena celá řada molekul, které jsou naopak schopny ferroptotickou buněčnou smrt navodit (tab. 1).
Značný vliv na hladinu transportéru systému Xc– může mít také protein p53. Bylo prokázáno, že p53 indukuje ferroptózu snížením exprese genu SLC7A11, který kóduje cystin/glutamátový transportní systém [51–53]. V důsledku toho dochází ke snížení příjmu cysteinu, vyčerpání hladiny antioxidantu glutathionu, což vede k neschopnosti buňky bojovat s oxidativním stresem [41,42].
Glutathion peroxidáza 4
Glutathion peroxidáza 4 (GPX4) je jednou z osmi glutathion peroxidáz známých u savců. S výjimkou GPX5, -7 a -8 (a GPX6 u hlodavců) obsahují všechny glutathion peroxidázy místo cysteinu selenocystein, který má jeden atom síry nahrazen selenem [62]. Uvádí se, že selenocystein zde pravděpodobně zvyšuje účinnost v redoxních reakcích [63].
GPX4 funguje jako monomerní peroxidáza [64]. Kromě jednoduchých peroxidů a malých hydroperoxidů je schopná redukovat také hydroperoxidy v komplexních lipidech, např. fosfolipidové, cholesterolové a cholesterolesterové hydroperoxidy začleněné v biomembránách a lipoproteinech [64,65]. Vyčerpáním GPX4 tedy dochází ke snížení ochrany buněk před negativním účinkem reaktivních radikálů, hromadění lipidových peroxidů a k vyčerpání polynenasycených mastných kyselin v lipidových dvojvrstvách, což může vést k indukci ferroptózy [14,42,56,66–68].
Ferroptóza a její spojení s autofagií
Autofagie (z řečtiny „vlastní stravování“) je evolučně konzervovaný homeostatický mechanizmus, který je zapojen do mnoha procesů udržujících správný chod buňky, jako je odstraňování špatně složených proteinů, poškozených organel a buněk napadených patogeny nebo také patologických stavů, vč. infekce nebo procesů souvisejících s rakovinou [69,70]. Nedávno bylo zjištěno, že aktivace autofagie je také jedním z důležitých induktorů ferroptózy v nádorových i normálních lidských buňkách [71–73]. Inhibitory autofagie jsou schopny ochránit buňky před vstupem do ferroptózy pravděpodobně prostřednictvím procesů zabraňujících tvorbě cytosolických a lipidových ROS [73]. K degradaci ferritinu jako zásobárny železa v buňkách dochází prostřednictvím lysozomů během autofagického procesu. V roce 2014 byl tento proces pojmenován jako ferritinofagie a současně s tím byl identifikován jaderný receptorový koaktivátor 4 (nuclear receptor coactivator 4 – NCOA4) přispívající k degradaci ferritinu během ferritinofagie [34]. Snížení aktivity receptoru NCOA4 stejně tak jako inhibice Atg5 a Atg7 omezuje degradaci ferritinu, snižuje hladinu reaktivního železa v cytosolu a potlačuje ferroptózu v normálních fibroblastech i nádorových buňkách [72]. Oproti tomu zvýšení hladiny NCOA4 urychluje ferroptózu, přičemž degradací ferritinu dochází k uvolnění železa do cytosolu [72]. Tato zjištění naznačují, že selektivní autofagie může hrát významnou úlohu při zprostředkování ferroptózy [72].
Ferroptóza a nádorová onemocnění
Nádorová buňka nese ve své genetické výbavě celou škálu mutací narušujících základní obranné mechanizmy. Mimo mutace v genech, jako jsou např. TP53, RB1 nebo RAS, které dovolují buňce překonat kontrolní mechanizmy zastavující buněčný cyklus či indukující buněčnou smrt, se nádorové buňky často setkávají také s nárůstem oxidativního stresu následkem změn v metabolických procesech a okolním mikroprostředí [74]. V reakci na zvýšenou hladinu ROS nádorová buňka indukuje zvýšení antioxidační aktivity s cílem udržet redoxní homeostázu [75]. To přímo souvisí také s hladinami volného železa v cytoplazmě. Bylo prokázáno, že některé onkogeny, vč. RAS a MYC, mění metabolizmus železa a jeho množství v buňkách tím, že ovlivňují expresi ferritinu nebo TfR1 [24,76–79].
Porozumění těmto procesům může mít velký vliv při hledání nových způsobů léčby rakoviny díky možnosti přesného zacílení na konkrétní části metabolických drah, které úzce souvisejí s redoxním stresem. Jednou z variant může být právě navození ferroptózy, která byla pozorována v ledvinových buňkách, neuronech a dále také v širokém spektru nádorových buněk, např. prsních, plicních, ovariálních, melanomech, gliomech a dokonce i u nádorů kostí [14,41,42,51–54,66,67,80–82].
Závěr
Ferroptóza jako jeden z nových typů programované buněčné smrti představuje proces, který je zcela závislý na množství reaktivního železa uvnitř cytoplazmy buněk. K aktivaci dochází narušením systémů, které v buňkách odbourávají ROS a lipidové peroxidy vznikající při chemických reakcích nebo nadměrném vystavení buněk oxidativnímu stresu. Na této skutečnosti je založen i současný výzkum, který se cíleně věnuje možnosti aktivovat alternativní dráhy buněčné smrti, vč. ferroptózy, s cílem obejít narušené metabolické procesy uvnitř nádorových buněk, se kterými je často spojena i rezistence ke standardně podávané terapii. Bylo prokázáno, že velké množství nádorových buněčných linií vykazuje citlivost k induktorům vyvolávajícím ferroptózu, proto i hledání nových sloučenin spouštějících ferroptózu představuje značně atraktivní alternativu, která může vést k objevu nových, účinnějších léků.
Práce byla podpořena projekty GAČR 17 - 05838S, MŠMT – NPU I – LO1413 a MZ ČR – RVO (MOÚ, 00209805).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Hana Skoupilová
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7 656 53 Brno
e-mail: hana.skoupilova@mou.cz
Přijato: 31. 8. 2018
Sources
1. Akera T, Brody TM, Wiest SA. Saturable adenosine 5‘-triphosphate-independent binding of [3H]-ouabain to brain and cardiac tissue in vitro. Br J Pharmacol 1979; 65 (3): 403–409.
2. Gunawardena AH. Programmed cell death and tissue remodelling in plants. J Exp Bot 2008; 59 (3): 445–451. doi: 10.1093/jxb/erm189.
3. Gaussand GM, Jia Q, van der Graaff E et al. Programmed cell death in the leaves of the arabidopsis spontaneous necrotic spots (sns-D) mutant correlates with increased expression of the eukaryotic translation initiation factor eIF4B2. Front Plant Sci 2011; 2 : 9. doi: 10.3389/fpls.2011.00009.
4. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004; 116 (2): 205–219.
5. Alberts B, Johnson A, Lewis J et al. Programmed cell death (apoptosis), in molecular biology of the cell. 4th edition. New York: Garland Science 2002.
6. Galluzzi L, Vitale I, Abrams JM et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ 2012; 19 (1): 107–120. doi: 10.1038/cdd.2011.96.
7. Wood W, Turmaine M, Weber R et al. Mesenchymal cells engulf and clear apoptotic footplate cells in macrophageless PU.1 null mouse embryos. Development 2000; 127 (24): 5245–5252.
8. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature 2001; 411 (6835): 342–348. doi: 10.1038/35077213.
9. Rock KL, Kono H. The inflammatory response to cell death. Annu Rev Pathol 2008; 3 : 99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456.
10. Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer 2009; 9 (7): 501–507. doi: 10.1038/nrc2663.
11. Ranger AM, Malynn BA, Korsmeyer SJ. Mouse models of cell death. Nat Genet 2001; 28 (2): 113–118. doi: 10.1038/88815.
12. Tait SW, Ichim G, Green DR. Die another way-nonapoptotic mechanisms of cell death. J Cell Sci 2014; 127 (Pt 10): 2135–2144. doi: 10.1242/jcs.093575.
13. McKenzie BA, Mamik MK, Saito LB et al. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc Natl Acad Sci U S A 2018; 115 (26): E6065–E6074. doi: 10.1073/pnas.1722041115.
14. Dixon SJ, Lemberg KM, Lamprecht MR et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149 (5): 1060–1072. doi: 10.1016/j.cell.2012.03.042.
15. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S et al. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014; 15 (2): 135–147. doi: 10.1038/nrm3737.
16. Kang R, Tang D. Autophagy andferroptosis – what‘s the connection? Curr Pathobiol Rep 2017; 5 (2): 153–159. doi: 10.1007/s40139-017-0139-5.
17. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 2008; 15 (3): 234–245. doi: 10.1016/j.chembiol.2008.02.010.
18. Dolma S, Lessnick SL, Hahn WC et al. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003; 3 (3): 285–296.
19. Vigil D, Cherfils J, Rossman KL et al. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 2010; 10 (12): 842–857. doi: 10.1038/nrc2960.
20. Mitsuuchi Y, Testa JR. Cytogenetics and molecular genetics of lung cancer. Am J Med Genet 2002; 115 (3): 183–188. doi: 10.1002/ajmg.10692.
21. Ofir Dovrat T, Sokol E, Frampton G et al. Unusually long-term responses to vemurafenib in BRAF V600E mutated colon and thyroid cancers followed by the development of rare RAS activating mutations. Cancer Biol Ther 2018 : 1–4. doi: 10.1080/15384047.2018.1480289.
22. Sekita-Hatakeyama Y, Nishikawa T, Takeuchi M et al. K-ras mutation analysis of residual liquid-based cytology specimens from endoscopic ultrasound-guided fine needle aspiration improves cell block diagnosis of pancreatic ductal adenocarcinoma. PLoS One 2018; 13 (3): e0193692. doi: 10.1371/journal.pone.0193692.
23. Garcia-Rostan G, Zhao H, Camp RL et al. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J Clin Oncol 2003; 21 (17): 3226–3235. doi: 10.1200/JCO.2003.10.130.
24. Yagoda N, von Rechenberg M, Zaganjor E et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007; 447 (7146): 864–868. doi: 10.1038/nature05859.
25. Cheng Y, Zak O, Aisen P et al. Structure of the human transferrin receptor-transferrin complex. Cell 2004; 116 (4): 565–576.
26. Chan RY, Ponka P, Schulman HM. Transferrin-receptor-independent but iron-dependent proliferation of variant Chinese hamster ovary cells. Exp Cell Res 1992; 202 (2): 326–336.
27. Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1996; 1275 (3): 161–203.
28. Terman A, Kurz T. Lysosomal iron, iron chelation, and cell death. Antioxid Redox Signal 2013; 18 (8): 888–898. doi: 10.1089/ars.2012.4885.
29. Guo G, Cui Y. New perspective on targeting the tumor suppressor p53 pathway in the tumor microenvironment to enhance the efficacy of immunotherapy. J Immunother Cancer 2015; 3 : 9. doi: 10.1186/s40425-015-0053-5.
30. Wang SJ, Li D, Ou Y et al. Acetylation is crucial for p53-mediated Ferroptosis and tumor suppression. Cell Rep 2016; 17 (2): 366–373. doi: 10.1016/j.celrep.2016.09.022.
31. Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett 1995; 82–83 : 969–974.
32. Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol 2001; 33 (10): 940–959.
33. Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell 2004; 117 (3): 285–297.
34. Mancias JD, Wang X, Gygi SP et al. Quantitative proteomics identifies NCOA4 as the cargo receptor media-ting ferritinophagy. Nature 2014; 509 (7498): 105–109. doi: 10.1038/nature13148.
35. Asano T, Komatsu M, Yamaguchi-Iwai Y et al. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol 2011; 31 (10): 2040–2052. doi: 10.1128/MCB.01437-10.
36. Gunshin H, Mackenzie B, Berger UV et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997; 388 (6641): 482–488. doi: 10.1038/41343.
37. Qian ZM, Li H, Sun H et al. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev 2002; 54 (4): 561–587.
38. Donovan A, Lima CA, Pinkus JL et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1 (3): 191–200. doi: 10.1016/j.cmet.2005.01.003.
39. Zohn IE, De Domenico I, Pollock A et al. The flatiron mutation in mouse ferroportin acts as a dominant negative to cause ferroportin disease. Blood 2007; 109 (10): 4174–4180. doi: 10.1182/blood-2007-01-066 068.
40. Zhang Z, Zhang F, An P et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 2011; 118 (7): 1912–1922. doi: 10.1182/blood-2011-01-330324.
41. Dixon SJ, Patel DN, Welsch M et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014; 3e02523. doi: 10.7554/eLife.02523.
42. Yang WS, SriRamaratnam R, Welsch ME et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014; 156 (1–2): 317–331. doi: 10.1016/j.cell.2013.12.010.
43. Bannai S, Kitamura E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem 1980; 255 (6): 2372–2376.
44. Makowske M, Christensen HN. Contrasts in transport systems for anionic amino acids in hepatocytes and a hepatoma cell line HTC. J Biol Chem 1982; 257 (10): 5663–5670.
45. Patel SA, Warren BA, Rhoderick JF et al. Differentiation of substrate and non-substrate inhibitors of transport system xc (-): an obligate exchanger of L-glutamate and L-cystine. Neuropharmacology 2004; 46 (2): 273–284.
46. Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem 1986; 261 (5): 2256-2263.
47. Bannai S. Induction of cystine and glutamate transport activity in human fibroblasts by diethyl maleate and other electrophilic agents. J Biol Chem 1984; 259 (4): 2435–2440.
48. Sato H, Fujiwara K, Sagara J et al. Induction of cystine transport activity in mouse peritoneal macrophages by bacterial lipopolysaccharide. Biochem J 1995; 310 (Pt 2): 547–551.
49. Sakakura Y, Sato H, Shiiya A et al. Expression and function of cystine/glutamate transporter in neutrophils. J Leukoc Biol 2007; 81 (4): 974–982. doi: 10.1189/jlb.0606385.
50. Watanabe H, Bannai S. Induction of cystine transport activity in mouse peritoneal macrophages. J Exp Med 1987; 165 (3): 628–640.
51. Jiang L, Kon N, Li T et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015; 520 (7545): 57–62. doi: 10.1038/nature14344.
52. Liu XX, Li XJ, Zhang B et al. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targeting SLC7A11. FEBS Lett 2011; 585 (9): 1363–1367. doi: 10.1016/j.febslet.2011.04.018.
53. Robert SM, Buckingham SC, Campbell SL et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci Transl Med 2015; 7 (289): 289ra286. doi: 10.1126/scitranslmed.aaa8103.
54. Linkermann A, Skouta R, Himmerkus N et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A 2014; 111 (47): 16836–16841. doi: 10.1073/pnas.1415518111.
55. Weiwer M, Bittker JA, Lewis TA et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett 2012; 22 (4): 1822–1826. doi: 10.1016/j.bmcl.2011.09.047.
56. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci 2016; 73 (11–12): 2195–2209. doi: 10.1007/s00018-016-2194-1.
57. Gout PW, Buckley AR, Simms CR et al. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x (c) – cystine transporter: a new action for an old drug. Leukemia 2001; 15 (10): 1633–1640.
58. Eling N, Reuter L, Hazin J et al. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015; 2 (5): 517–532. doi: 10.18632/oncoscience.160.
59. Lachaier E, Louandre C, Godin C et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res 2014; 34 (11): 6417–6422.
60. Louandre C, Ezzoukhry Z, Godin C et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer 2013; 133 (7): 1732–1742. doi: 10.1002/ijc.28159.
61. Viswanathan VS, Ryan MJ, Dhruv HD et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017; 547 (7664): 453–457. doi: 10.1038/nature23007.
62. Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta 2013; 1830 (5): 3289–3303. doi: 10.1016/j.bbagen.2012.11.020.
63. Conrad M, Friedmann Angeli JP. Glutathione peroxidase 4 (Gpx4) and ferroptosis: what‘s so special about it? Mol Cell Oncol 2015; 2 (3): e995047. doi: 10.4161/23723556.2014.995047.
64. Takebe G, Yarimizu J, Saito Y et al. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J Biol Chem 2002; 277 (43): 41254–41258. doi: 10.1074/jbc.M202773200.
65. Thomas JP, Geiger PG, Maiorino M et al. Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins. Biochim Biophys Acta 1990; 1045 (3): 252–260.
66. Friedmann Angeli JP, Schneider M, Proneth B et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014; 16 (12): 1180–1191. doi: 10.1038/ncb3064.
67. Skouta R, Dixon SJ, Wang J et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 2014; 136 (12): 4551–4556. doi: 10.1021/ja411006a.
68. Magtanong L, Ko PJ, Dixon SJ. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ 2016; 23 (7): 1099–1109. doi: 10.1038/cdd.2016.25.
69. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132 (1): 27–42. doi: 10.1016/j.cell.2007.12.018.
70. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147 (4): 728–741. doi: 10.1016/j.cell.2011.10.026.
71. Gao M, Monian P, Pan Q et al. Ferroptosis is an autophagic cell death process. Cell Res 2016; 26 (9): 1021–1032. doi: 10.1038/cr.2016.95.
72. Hou W, Xie Y, Song X et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016; 12 (8): 1425–1428. doi: 10.1080/15548627.2016.1187366.
73. Torii S, Shintoku R, Kubota C et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J 2016; 473 (6): 769–777. doi: 10.1042/BJ20150658.
74. Cross CE, Halliwell B, Borish ET et al. Oxygen radicals and human disease. Ann Intern Med 1987; 107 (4): 526–545.
75. Chio IIC, Tuveson DA. ROS in Cancer: The Burning Question. Trends Mol Med 2017; 23 (5): 411–429. doi: 10.1016/j.molmed.2017.03.004.
76. Kakhlon O, Gruenbaum Y, Cabantchik ZI. Ferritin expression modulates cell cycle dynamics and cell responsiveness to H-ras-induced growth via expansion of the labile iron pool. Biochem J 2002; 363 (Pt 3): 431–436.
77. Kakhlon O, Gruenbaum Y, Cabantchik ZI. Repression of ferritin expression modulates cell responsiveness to H-ras-induced growth. Biochem Soc Trans 2002; 30 (4): 777–780. doi: 10.1042.
78. O‘Donnell KA, Yu D, Zeller KI et al. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol Cell Biol 2006; 26 (6): 2373–2386. doi: 10.1128/MCB.26.6.2373-2386.2006.
79. Wu KJ, Polack A, Dalla-Favera R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science 1999; 283 (5402): 676–679.
80. Hangauer MJ, Viswanathan VS, Ryan MJ et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017; 551 (7679): 247–250. doi: 10.1038/nature24297.
81. Drayton RM, Dudziec E, Peter S et al. Reduced expression of miRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin Cancer Res 2014; 20 (7): 1990–2000. doi: 10.1158/1078-0432.CCR-13-2805.
82. Miess H, Dankworth B, Gouw AM et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene. In press 2018. doi: 10.1038/s41388-018-0315-z.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2018 Issue Supplementum 2
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Effect of DNA Methylation on the Development of Cancer
- Ferroptosis as a New Type of Cell Death and its Role in Cancer Treatment
- Possible Usage of p63 in Bioptic Diagnostics
- Current Methods of microRNA Analysis