
Toxické účinky pesticidů
Toxic Effects of Pesticides
Although intoxication with organophosphates have occurred rather sporadically in the Czech Republic, self-poisoning with organophosphates may represent a serious clinical issue in rural regions of the developing world. According to an estimation from the World Health Organization, up to two million people are poisoned every year. Medical management is usually difficult, associated with the mortality rate of above 15%. Based on chemical-physical properties, the central nervous system is one of the most important targets for organophosphates. Brain damage is defined as a progressive damage resulting from cholinergic neuronal excitotoxicity and dysfunction of the brain cholinergic regions. Loss of neurons, damage to cholinergic and non-cholinergic pathways and degeneration of axons is usually observed in the central nervous system. This delayed secondary neuronal damage might be largely responsible for persistent neuropsychiatric and neurological impairments, such as cognitive, motor and sensory deficits.
Key words:
acetylcholinesterase – pesticides – central nervous system – neuron degeneration – NMDA receptor – cholinergic system
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
:
J. Žďárová Karasová
:
Centrum biomedicínského výzkumu, FN v Hradci Králové
; Katedra toxikologie a vojenské farmacie, Fakulta vojenského zdravotnictví v Hradci Králové, Univerzita obrany, Brno
:
Cesk Slov Neurol N 2017; 80/113(2): 164-171
:
Review Article
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amcsnn2017164
Práce vznikla za podpory Projektu rozvoje organizace 1011 – DZRO ZHN (Ministerstvo obrany, Česká republika) a RVO – FNHK 00179906 (Ministerstvo zdravotnictví, Česká republika).
I přesto, že v České republice jsou intoxikace organofosforovými inhibitory acetylcholinesterázy zaznamenány jen ojediněle, dle World Health Organization roční odhad celosvětově odpovídá 2 milionům. Přibližně 30 % z celkového počtu těchto otrav má vážný průběh, odhadováno je přibližně 300 tisíc úmrtí, zejména v rozvojových zemích. Vzhledem k fyzikálně-chemickým vlastnostem většiny inhibitorů acetylcholinesterázy je za jedno z nejdůležitějších cílových míst jejich toxického působení považován centrální nervový systém. V posledních desetiletích byl vliv organofosforových inhibitorů acetylcholinesterázy na tento systém intenzivně studován. Bylo potvrzeno, že poškození mozku v případě otrav organofosforovými inhibitory bývá progresivního charakteru. Na úrovni centrálního nervového systému je to zejména zánik neuronů, poškození cholinergních i necholinergních drah a degenerace axonů. Pokud dojde k inicializaci změn v mozkové tkáni, rozvíjí se kaskáda patologických dějů, které probíhají i ve velmi vzdálených časových obdobích, tedy řádově po dobu měsíců.
Klíčová slova:
acetylcholinesteráza – pesticidy – centrální nervový systém – degenerace neuronů – NMDA receptor – cholinergní systém
Úvod
Inhibitory cholinesteráz, konkrétně skupina ovlivňující funkci acetylcholinesterázy (AChE; EC 3.1.1.7), je vzhledem k jasně definované a důležité funkci tohoto enzymu v organizmu předmětem intenzivního vědeckého výzkumu. Z hlediska struktury jde o širokou skupinu látek zahrnující přírodní toxiny, látky s léčebným účinkem, pesticidy a také bojové chemické látky.
Charakteristické období pro syntézu a vývoj této skupiny toxických látek je datováno v období mezi dvěma světovými válkami, tedy do let 30. a první poloviny 40. let 20. století. Německý chemik Gerhard Schräder, který se specializoval na vývoj nových insekticidů, definoval základní chemickou strukturu látek s vysokým toxickým účinkem vůči hmyzu. Na základě těchto zjištění byl připraven první komerčně užívaný organofosforový pesticid. Následovaly studie zabývající se thiostrukturami; paraoxon a jeho thioanalog parathion byly připraveny v roce 1944 a jsou i v dnešní době považovány za jedny z nejvýznamnějších pesticidů [1].
Pesticidy jsou celosvětově široce využívány. Zejména v rozvojových zemích jsou zaznamenávány časté otravy těmito látkami, dle World Health Organization se celkový roční odhad blíží 2 milionům. Část těchto otrav má těžký průběh a přibližně 15 % končí úmrtím intoxikovaného jedince [2]. V České republice, stejně jako v jiných rozvinutých zemích, dochází k otravám ojediněle. Jsou popsány otravy dětí při neúmyslném požití roztoků obsahujících pesticidy, popřípadě suicidální pokusy. Není možné v současné době pominout ani určité riziko vojenského zneužití a narůstající nebezpečí teroristických útoků [3]. Všechny tyto skutečnosti jsou důvodem k podrobnějšímu studiu jejich biologických vlastností, zejména na úrovni centrálního nervového systému (CNS), kde se jedná o poškození progresivního charakteru.
Inhibitory AChE mohou být využity i v léčbě, vzhledem k jejich působení na centrální nebo periferní cholinergní systém. V poslední době jsou v centru lékařského zájmu především v souvislosti s Alzheimerovou chorobou a myastenia gravis. Cholinergní nervový systém zastává významnou úlohu v procesech učení a paměti. Cholinergní hypofunkce definovaná nedostatkem neurotransmiteru acetylcholinu (ACh) na neuronálních spojích v CNS zhoršuje kognitivní funkce, a představuje tak jeden z hlavních patologických podkladů vzniku demence [4]. Myastenia gravis je pak spojována s cholinergní hypofunkcí na nervosvalové ploténce [5].
Periferní cholinergní příznaky intoxikace inhibitory AChE
Příznaky intoxikace inhibitory AChE lze definovat na základě ovlivnění (hyperstimulace) příslušných cholinergních receptorů, které vznikne následkem nedostatečného odbourávání ACh v synaptické štěrbině. Cholinergní receptory se rozdělují na dvě základní skupiny, na receptory muskarinového a nikotinového typu, event. na jejich subtypy [6].
Klinické projevy akumulace ACh v parasympatických synapsích inervujících duhovku, bronchiální kmen, močový měchýř, cévy, sekreční žlázy v dýchacím a gastrointestinálním systému a také v sympatických zakončeních inervujících potní žlázy jsou obecně označovány za příznaky muskarinového typu, zatímco nahromadění Ach na zakončeních motorických nervů příčně pruhovaného svalstva a ve vegetativních gangliích za příznaky nikotinového typu. Akumulace ACh v jednotlivých strukturách mozku a míchy je příčinou centrálních příznaků otravy [7].
Periferní muskarinové příznaky intoxikace se projevují zvýšenou sekrecí exokrinních žláz – zvýšená sekrece z nosu a v bronších, pocení, slzení a slinění. Zvýšená hladina ACh v hladkém svalstvu způsobuje miózu, poruchy akomodace, gastrointestinální obtíže se manifestují jako abdominální křeče či průjem, typické je také častější močení a bradykardie [8].
Mezi periferní nikotinové příznaky patří bledost, tachykardie a hypertenze zprostředkovaná hyperstimulací receptorů v autonomních gangliích. S tíží intoxikace výrazně souvisí míra ovlivnění příčně pruhovaného svalstva, které se při mírnějších formách intoxikace manifestuje fascikulacemi, při těžších otravách tonicko-klonickými křečemi přecházejícími v postupnou paralýzu [9]. Rozhodující je paralýza svalů podílejících se na dýchání (mezižeberní svaly a bránice).
Centrální cholinergní a necholinergní příznaky intoxikace organofosforových inhibitorů AChE
Vzhledem k fyzikálně-chemickým vlastnostem většiny inhibitorů AChE je za jedno z nejdůležitějších cílových míst jejich toxického působení považována CNS, která je chráněna hematoencefalickou bariérou (HEB). HEB zároveň omezuje prostup léčiv/ antidot do centrálního kompartmentu. Z tohoto důvodu je v posledním desetiletí zaměřena pozornost na studium prostupu léčiv přes tuto bariéru a možné ovlivnění tohoto procesu.
Ačkoli cholinergní systém zahrnuje malé procento neuronů, má velký funkční význam (obr. 1). Těla buněk, která jsou hlavním zdrojem cholinergní inervace pro oblast kůry mozkové, hipokampu a limbických struktur, jsou lokalizována v bazální části předního mozku (vč. nucleus basalis, diagonálních oblastí a septa) a v mozkovém kmeni [10]. Propojení mezi cholinergními jádry v bazální části předního mozku a mozkovém kmeni ovlivňují přímo i nepřímo aktivitu těchto vyšších center [11]. Striatum také obsahuje lokální okruh cholinergních interneuronů. Největší hustota cholinergní inervace je v komplexu nucleus caudatus-putamen [12].

Postsynaptické účinky ACh v předním mozku jsou zřejmě zprostředkovány hlavně muskarinovými receptory [12]. V mozkové kůře a limbické oblasti mají cholinergní spojení základní úlohu v učení, paměti a intelektuální aktivitě; v corpus striatum způsobuje příliš velká aktivace cholinergních interneuronů nebo nerovnováha mezi těmito neurony a dopaminergním systémem neurologické symptomy (tremor, katalepsie, stereotypní pohyby); poruchy cholinergních prvků v limbickém systému mají úlohu např. v agresivním chování apod. [11].
Expozice organofosforovými inhibitory AChE indukuje akumulaci ACh v synapsích. Ta přispívá k relativně rychlému rozvoji excitotoxicity a dysfunkci cholinergních neuronů z důvodu vysoké stimulace muskarinových receptorů. Tato hyperstimulace souvisí s narušením rovnováhy jiných neurotransmiterových systémů – glutamatergního a GABAergního – a následným rozvojem neuronálních excitotoxických lézí [13]. Dysfunkce cholinergních neuronů snižuje funkčnost specifických cholinergních drah v mozku s různorodými negativními důsledky: charakteristická je ztráta neuronů v cholinergních regionech, jejichž poškození se promítá do jimi inervovaných částí mozku, jako jsou bazální ganglia (piriformní kůra a entorhinální kůra) a limbický systém (hipokampus, amygdala a thalamus) [12].
Mezi akutní centrální příznaky intoxikace jsou řazeny křeče a deprese dýchání. Vznik křečí je vysvětlován hyperstimulací muskarinových receptorů. Poté je vyvolána výrazná aktivace NMDA receptorů s následným nadměrným uvolňováním glutamátu z glutamaergních neuronů vyvolávající intenzivní uvolňování vápníku v postsynaptických neuronech. Je obecně uznáváno, že křeče, které vznikají na základě dysbalance cholinergního systému, mají synergický vliv na následný rozvoj nevratných změn v mozkové tkáni s neurologickými a behaviorálními poruchami [11]. V některých studiích však bylo prokázáno, že k nevratnému poškození mozku dochází i v případech, kdy křeče nebyly zaznamenány, tedy i při velmi nízkých dávkách těchto látek, jež nevyvolávají typické příznaky otravy [14]. Tyto změny mohou být vysvětleny necholinergními účinky organofosforových inhibitorů na mozkovou tkáň.
Deprese dýchání bývá nejčastější příčinou úmrtí intoxikovaného jedince v akutní fázi otravy. Dechové centrum je uloženo v prodloužené míše. Útlum dýchání bývá potencován periferními příznaky otravy, a to zvláště paralýzou dýchacích svalů a sekrecí bronchiálních žláz [8].
Poškození mozku v případě otrav organofosforovými inhibitory bývá progresivního charakteru. Typický je zánik neuronů, poškození cholinergních i necholinergních drah a degenerace axonů [15]. Pokud jsou inicializovány změny v mozkové tkáni, rozvíjí se kaskáda patologických dějů, které probíhají i ve velmi vzdálených časových obdobích, tedy řádově po dobu měsíců [16].
Mezi sekundární procesy, nastávající řádově v hodinách po expozici organofosforovými inhibitory, které mohou indukovat rozvoj patologických změn mozkové tkáně, jsou řazeny zejména: edém mozku [17], zvýšení propustnosti HEB [18], mozkové mikrohemoragie [19], zvýšení množství intracelulárního vápníku [20], oxidativní stres [21] a zánět, popřípadě stresová odpověď [22].
Edém mozku
Edém mozku je sekundární poškození mozku, které vzniká na základě rozvoje kaskády biochemických a patologických procesů typických pro intoxikaci organofosforovými inhibitory AChE. Jeho výskyt ve specifických částech mozku (amygdala, piriformní část mozkové kůry, hipokampus a thalamus) velmi úzce souvisí s rozvojem dalších patologických změn a je považován za první signál, že je indukován rozvoj následného progresivního poškození mozkové tkáně a ztráta neuronů v daných oblastech CNS [23].
Zánět a změny propustnosti HEB
HEB je nutno chápat jako komplexní strukturu tvořenou několika typy buněk (obr. 2), která je charakterizována odlišnou biologickou stavbou, transportními systémy a také přítomností specifických adhezních proteinů. Kombinace všech těchto parametrů výrazně snižuje mimobuněčný (mezibuněčný) transport a výrazně zvyšuje důležitost aktivního buněčného transportu [24].
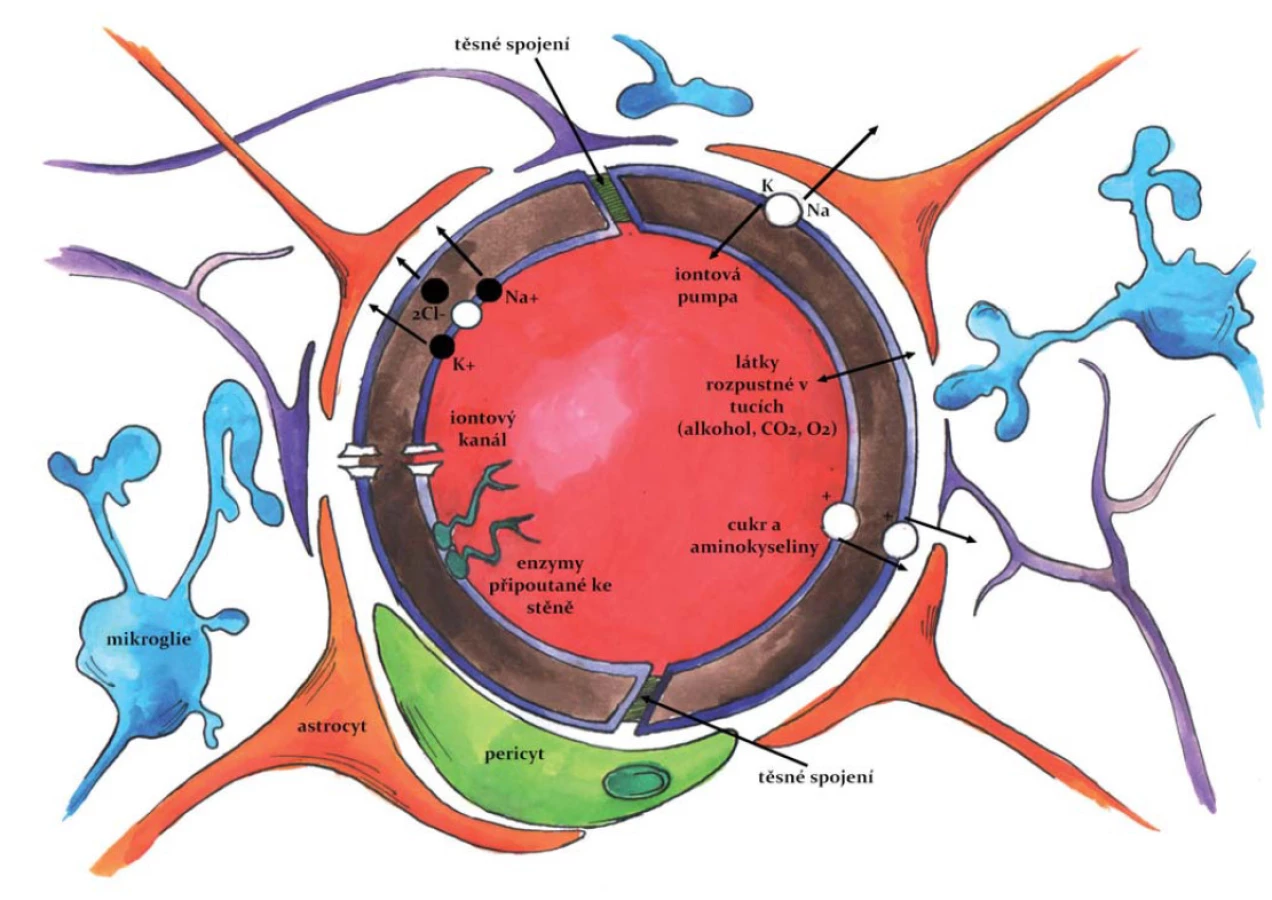
HEB se začíná tvořit v průběhu embryonálního vývoje jako důsledek prostorového vztahu mezi krevními cévami a neuroektodermálními buňkami. Tato neurovaskulární jednotka může být považována za základní funkční jednotku HEB; je definována třemi základními buněčnými subtypy: vaskulární (endotelové buňky, pericyty, buňky hladké svaloviny cév); gliální (astrocyty, mikroglie a oligodendroglie) a neuronální [25].
Zánět je přirozenou reakcí imunitního systému na poškození, typicky na zranění nebo infekci. Zánětlivá odpověď je zprostředkována aktivací makrofágů na periferii, v CNS jsou aktivovány mikroglie a/ nebo astrocyty, které se podílejí na stavbě HEB jakožto multicelulární bariéry [26,27]. Perivaskulární mikroglie jsou imunitní buňky CNS s velmi rychlou schopností odpovědi na poškození mozkové tkáně. Uvolňují působky, které se zapojují do kaskády dalších patologických změn v mozkové tkáni, např. do tvorby amyloidních plaků [28]. Cytokiny uvolňované mikrogliemi aktivují astrocyty, uvolňují se další prozánětlivé mediátory a tato reakce se šíří mozkovou tkání. Cytokiny také výrazně ovlivňují propustnost HEB, vyvolávají dilataci cév a zvýšení adheze leukocytů (neutrofilů) k cerebrálnímu endotelu a jejich migrace do místa poškození. Tím je indukována následná funkční porucha HEB [27]. Současně se lokálně zvyšuje produkce proteáz, které mohou poškozovat proteinové složky myelinu. Neutrofily indukují apoptózu buněk uvolňováním oxidu dusnatého a reaktivních kyslíkových radikálů (ROS) [26].
Po intoxikaci organofosforovými inhibitory AChE se v mozkové tkáni rozvíjí zánět. Tento proces je možné sledovat na základě změny hladin gliálního fibrilárního kyselého proteinu (GFAP), který je produkován aktivovanými astrocyty. Zvýšení jeho hladin bylo zaznamenáno v mnoha částech mozku, mimo jiné i v hipokampu, amygdale, kůře mozkové, mozečku, prodloužené míše, piriformní a entorhinální části mozkové kůry [29,30].
V případě akutní intoxikace bylo prokázáno, že úroveň zánětlivé odpovědi koreluje nejen s hladinou exprese markerů zánětu, ale také s mírou inhibice AChE v CNS a délkou trvání centrálně vyvolaných křečí [31]. V případě chronické expozice byly prokázány zánětlivé změny mozkové tkáně i v případech, kdy nebyly zaznamenány křeče a inhibice AChE, tedy typické příznaky intoxikace. Vysvětlení této aktivace je různé, jedná se o vysoce komplexní proces. Bylo např. prokázáno zvýšené uvolňování ROS a katepsinu D u makrofágů [32], zvýšení fagocytózy u makrofágů [33] nebo zvýšené uvolňování histaminu z bazofilů a žírných buněk [34].
Dále bylo prokázáno, že cholinergní systém zasahuje do regulace zánětlivé odpovědi člověka, která může být tlumena prostřednictvím stimulace bloudivého nervu (nervus vagus) a uvolňování ACh [35]. Diagram tohoto děje je zaznamenán ve schématu 1.
![Schéma 1. Schematický diagram regulace zánětlivé odpovědi cholinergním systémem, diagram zpracován na základě článku [35].](https://pl-master.mdcdn.cz/media/image/6fee7d65c8782e44c1ecb61c70ce8003.jpg?version=1537794552)
V případě akutních intoxikací bylo zaznamenáno také zvýšení produkce prostaglandinů a izoprostanoidů v CNS. Prostaglandiny jsou syntetizovány cyoklooxygenázami (COX) z kyseliny arachidonové uvolněné z membrán fosfolipázami [36]. Prostaglandin E2 (PGE-2) je spojován nejen se zánětem, ale i se změnami propustnosti cévních stěn, regulací krevního toku a vyvoláním hyperalgezie [36]. Izoprostanoidy, jako jsou F2 izoprostanoid a neuronálně specifický F4 neuroprostanoid, jsou odvozeny od prostaglandinů a jsou chápány jako biomarkery oxidačního stresu [37]. Izoprostanoidy usnadňují adhezi neutrofilů k buňkám endotelu, aktivované neutrofily potencují zánětlivou odpověď.
V případě akutní intoxikace organofosforovými inhibitory AChE bylo zaznamenáno zvýšení produkce PGE-2 v kůře mozkové a hipokampu [31]. Opětovné zvýšení hladin PGE-2 bylo zaznamenáno v období 1 – 6 měsíců po akutní intoxikaci, kdy byla zvýšena aktivita COX-2 v neuronech hipokampu, piriformní části mozkové kůry a amygdaly [30].
Zvýšení hladiny intracelulárního vápníku
Extrémní influx vápníku vyvolaný hyperstimulací muskarinových a NMDA receptorů v mozku je považován za jeden z nejdůležitějších faktorů iniciujících odumírání neuronů. Zvýšení hladin intracelulárního vápníku bylo zaznamenáno v synaptozomech izolovaných z mozkové kůry po expozici somanem [38]. Při nadbytku iontů vápníku jsou v buňce aktivovány lipázy, proteázy, kinázy, fosfatázy a endonukleázy v metabolické kaskádě, vedoucí ke snížení syntézy proteinů buňky a deprivaci enzymových systémů důležitých k jejímu přežití [39].
Tento proces je také často výsledkem poškození volnými radikály, vlivem nadměrného oxidačního stresu nebo zánětlivé odpovědi na poškození mozkové tkáně.
Apoptóza, zánik neuronů a nervových spojů a degenerace axonů
Apoptóza je programovaný proces buněčné smrti, který velmi úzce souvisí s imunitní odpovědí organizmu na poškození tkáně. Apoptóza buněk některých oblastí mozkové tkáně je typický patologický nález progredující v časovém období odpovídajícím dvěma a více měsícům po intoxikaci organofosforovými inhibitory AChE. Charakterizována je zpravidla fragmentací jaderné DNA, buněčným scvrkáváním, tvorbou bublin na površích buněk, degradací DNA a proteinového chromatinu v jádře buňky a poškozením mitochondrií [40]. Apoptický program buňky může být aktivován také působením kyslíkových radikálů, aktivací TNF nebo Fas receptorů, poškozením DNA, popřípadě aktivací lysozomálních proteáz.
Cholinergním mechanizmem vyvolaná excitotoxicita hraje důležitou roli nejen v nastartování apoptického programu buněk, ale i v případě zániku neuronů, ztrátě nervových spojů a degeneraci axonů. Souhrnně jsou tyto patologické změny v CNS označovány jako sekundární neuronální poškození a mohou se vyskytovat 4 hod až 3 měsíce po intoxikaci v určitých částech mozku. Hipokampus a dorzolaterální jádra thalamu bývají zpravidla zasaženy 4 hod po intoxikaci, 24 hod po intoxikaci pak neuronální degenerace a edém postupuje do piriformní části mozkové kůry, 1 týden po intoxikaci jsou zaznamenávány významné ztráty neuronů v oblasti piriformního části mozkové kůry a zasažena jsou i jádra amygdaly. Přibližně 1 měsíc po intoxikaci bývá pozorována neuronální kalcifikace v oblasti thalamu a zároveň i piriformní části mozkové kůry. Hyalinní plaky, které jsou tvořeny atrofovanými zbytky buněk a jsou považovány za příznaky těžkého neuronálního poškození, jsou typicky distribuovány v oblasti hypothalamu [41,42].
Na základě rozvoje tohoto patologického stavu částí CNS lze charakterizovat následné, pozdní neurologické a neuropsychiatrické obtíže organofosfáty otrávených pacientů, které bývají často nevratné. Zdá se, že velká část těchto patologických změn může být vyvolána i subakutní intoxikací, a riziko je výrazně zvýšeno v případě opakovaných subakutních/ chronických otrav [43].
Pozdní centrální příznaky nejsou specifické a jsou popisovány jako závratě, úzkost, neklid, bolest hlavy, třes, zmatenost, ztráta koncentrace, kóma, křeče a útlum dýchání [7]. Pokud je překonána akutní fáze intoxikace, byly u některých jedinců zaznamenány změny povahy, střídání nálad, agresivní chování a psychotické epizody zahrnující schizoidní reakce, paranoiu, popřípadě se rozvíjejí dosud skryté psychiatrické problémy. Může se také zhoršit kvalita spánku, který je doprovázen nočními můrami a halucinacemi, popřípadě je rovněž zhoršena paměť a schopnost udržet pozornost [44,45].
Mezi další typické změny patří také zhoršení schopnosti propojovat informace, snížená schopnost orientace v prostoru a rozpoznávání, popřípadě snížení schopnosti dlouhodobého soustředění [46].
Celkový přehled progrese poškození CNS v případě intoxikace organofosforovými inhibitory je pro zlepšení přehlednosti shrnut ve schématu 2.

Potencionální necholinergní cílová místa účinku organofosforových inhibitorů AChE
Inhibitory AChE zasahují kromě výše popsaného do mnoha dalších biologických dějů, v současnosti se diskutuje o dalších změnách. Nejčastěji jde o ovlivnění dalších neurotransmiterových systémů, jako jsou např. serotonin, dopamin, gama aminomáselná kyselina [47]. Dochází také i k imunopatologickým změnám, anafylaktickým reakcím a změnám energetického metabolizmu mozku [46].
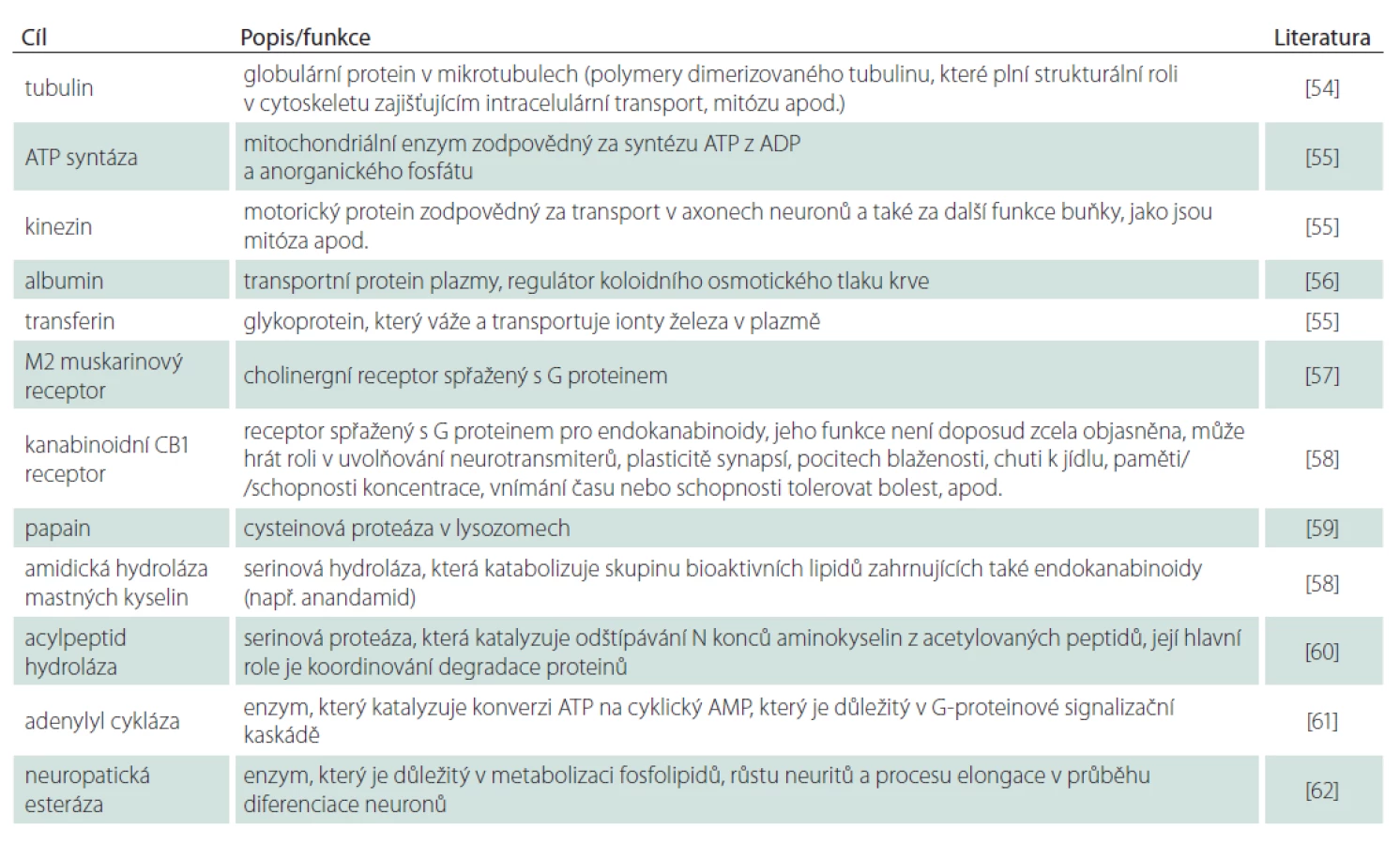
Některé z dalších relativně dobře dokumentovaných cílových míst, vč. jejich známých nebo teoretických funkcí, jsou shrnuty v tab. 1.
Závěr
Strategie léčby otrav způsobených inhibitory AChE, která je v současnosti doporučována, zahrnuje podání kombinace anticholinergně působící látky (atropin), antikonvulziva (diazepam) a oximu [48 – 52]. Tato kombinace léčiv/ antidot je doporučována na základě znalostí hlavního mechanizmu účinku inhibitorů AChE v CNS [53].
S rozšiřováním znalostí o následných patologických procesech v CNS je možné diskutovat o možném doplnění doporučené léčebné strategie za účelem zmírnění pozdních následků intoxikace.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. PharmDr. Jana Žďárová Karasová, Ph.D.
Katedra toxikologie a vojenské farmacie
Fakulta vojenského zdravotnictví
Univerzita obrany
Třebešská 1575
500 01 Hradec Kralové
e-mail: zdarova.jana@gmail.com
Přijato k recenzi: 10. 10. 2016
Přijato do tisku: 7. 11. 2016
Sources
1. Balali-Mood M, Balali-Mood K. Neurotoxic disorders of organophosphorus compounds and their managements. Arch Iran Med 2008;11(1):65 – 89.
2. Eddleston M, Buckley NA, Eyer P, et al. Management of acute organophosphorus pesticide poisoning. Lancet 2008;371(9612):597 – 607.
3. Vlček V, Pohanka M. Enviromentální aspekty užití organofosforových pesticidů schválených k užití v ČR. Chem Listy 2011;105 : 908 – 12.
4. Mzik M, Korabecny J, Nepovimova E, et al. An HPLC - MS method for the quantification of new acetylcholinesterase inhibitor PC 48 (7-MEOTA-donepezil like compound) in rat plasma: application to a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci 2016;1020 : 85 – 9. doi: 10.1016/ j.jchromb.2016.02.038.
5. Karasova JZ, Hroch M, Musilek K, et al. Small quaternary inhibitors K298 and K524: Cholinesterases inhibition, absorption, brain distribution, and toxicity. Neurotox Res 2016;29(2):267 – 74. doi: 10.1007/ s12640-015-9582-4.
6. Sepsova V, Karasova JZ, Tobin G, et al. Cholinergic properties of new 7-methoxytacrine-donepezil derivatives. Gen Physiol Biophys 2015;34(2):189 – 200. doi: 10.4149/ gpb_2014036.
7. Bajgar J. Organophosphates/ nerve agent poisoning: mechanism of action, diagnosis, prophylaxis, and treatment. Adv Clin Chem 2004;38 : 151 – 216.
8. Marrs TC. Organophosphate poisoning. Pharmacol Ther 1993;58(1):51 – 66.
9. Jokanovic M. Medical treatment of acute poisoning with organophosphorus and carbamate pesticides. Toxicol Lett 2009;190(2):107 – 15. doi: 10.1016/ j.toxlet.2009.07.025.
10. He YF, Zhu JH, Huang F, et al. Age-dependent loss of cholinergic neurons in learning and memory-related brain regions and impaired learning in SAMP8 mice with trigeminal nerve damage. Neural Regen Res 2014;9(22):1985 – 94. doi: 10.4103/ 1673-5374.145380.
11. Aigner TG. Pharmacology of memory: cholinergic.glutamatergic interaction. Curr Opin Neurobiol 1995;5(2):155 – 60.
12. Beierlein M. Synaptic mechanisms underlying cholinergic control of thalamic reticular nucleus neurons. J Physiol-London 2014;592(19):4137 – 45. doi: 10.1113/ jphysiol.2014.277376.
13. Santos MD, Pereira EF, Aracava Y, et al. Low concentrations of Pyridostigmine prevent soman-induced inhibition of GABA-ergic transmission in the central nervous system: involvement of muscarinic receptors. J Pharmacol Exp Ther 2003;304(1):254 – 65.
14. Jett DA. Neurological aspect of chemical terorism. Ann Neurol 2007;61(1):9 – 13.
15. Petras JM. Neurology and neuropathology of Soman-induced brain injury: an overiew. J Exp Anal Behav 1994;61(2):319 – 29.
16. Granacher RP. Traumatic brain injury: methods for clinical and forensic neuropsychiatric assessment. 2nd ed. Boca Raton: CRC Press 2007 : 26 – 32.
17. Job A, Baille V, Dorandeu F, et al. Distortion product otoacoustic emmission as non-invasive biomarkers as predictors of soman-induced central neurotoxicity: a preliminary study. Toxicology 2007;238(2 – 3):119 – 29.
18. Carpentier P, Delamanche IS, Le Bert M, et al. Seisure-related opening of the blood-brain barrier induced by soman: possible correlation with the acute neuropathology observed in poisoned rats. Neurotoxicology 1990;11(3):493 – 508.
19. Gokel Y. Subarachnoid hemorrhage and rhabdomyolysis induced acute renal failure complication organophosphate intoxication. Ren Fail 2002;24(6):867 – 71.
20. Hu CY, Hsu CH, Robinson CP. Effects of soman on calcium uptake in microsomes and mitochondria from rabit aorta. J Appl Toxicol 1991;11(4):293 – 6.
21. Pazdernik TL, Emerson MR, Cross R, et al. Soman-induces seisures: limbic activity, oxidative stress and neuroprotective proteins. J Appl Toxicol 2001;21(Suppl 1): S87 – 94.
22. Dhote F, Peinnequin A, Carpentier P, et al. Prolonged inflammatory gene response following soman-induced seisures in mice. Toxicology 2007;238(2 – 3):166 – 76.
23. Newey CR, Sarwal A, Hantus S. Continuous electroencephalography (cEEG) changes precede clinical changes in a case of progressive cerebral edema. Neurocrit Care 2013;18(2):261 – 5. doi: 10.1007/ s12028-011-9650-4.
24. Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 2009;118(1):103 – 13. doi: 10.1007/ s00401-009-0522-3.
25. Abbott NJ, Patabendige AAK, Dolman DEM, et al. Structure and function of the blood – brain barrier. Neurobiol Dis 2010;37(1):13 – 25. doi: 10.1016/ j.nbd.2009.07.030.
26. Abbott NJ, Hansson E. Astrocyte – endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 2006;7(1):41 – 53.
27. Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 2006;7(1):41 – 53.
28. Zolezzi JM, Inestrosa NC. Peroxisome proliferator-activated receptor and Alzheimer disease: Hitting the blood-brain barrier. Mol Neurobiol 2013;48(3):438 – 51. doi: 10.1007/ s12035-013-8435-5.
29. Damodaran TV, Bilska MA, Rahman AA, et al. Sarin cause early differential alteration and persistent overexpression in mRNAs coding for glialfibrillary acidic protein (GFAP) and vimentin genes in the central nervous system of rats. Neurochem Res 2002;27(5):407 – 15.
30. Angoa-Perez M, Kreipke CW, Thomas DM, et al. Soman increases neuronal COX-2 levels: possible link between seisures and protracted neuronal damage. Neurotoxicology 2010;31(6):738 – 46. doi: 10.1016/ j.neuro.2010.06.007.
31. Chapman S, Kadar T, Gilat E. Seisure duration following sarin exposure affects neuroinflamatory markers in the rat brain. Neurotoxicology 2006;27(2):277 – 83.
32. Rodgers KE, Ellefson DD. Mechanism of the modulation of murine peritoneal cell function and mast cell degranulation by low doses of malathion. Agents Actions 1992;35(1 – 2):57 – 63.
33. Flipo D, Bernier J, Girard D, et al. Combined effect of selected insecticides on humoral immune response in mice. Int J Immunopharmacol 1992;14(5):747 – 52.
34. Rodgers K, Xiong S. Effect of administration of malathion for 14 days on macrophage function and mast cell degranulation. Fundam Appl Toxicol 1997;37(1):95 – 9.
35. Banks ChN, Lein PJ. A review of experimental evidence linking neurotoxic organophosphorus compounds and inflammation. Neurotoxicology 2012;33(3):575 – 84. doi: 10.1016/ j.neuro.2012.02.002.
36. Ricciotti E, Fitzgerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 2011;31(5): 986 – 1000. doi: 10.1161/ ATVBAHA.110.207449.
37. Milatovic D, Gupta RC, Aschner M. Anticholinesterase toxicity and oxidative stress. Scientific World J 2006;6 : 295 – 310.
38. Hamilton MG, Posavad C. Alteration of calcium influx in rat cortical synaptosomes by soman. Neuroreport 1991;2(5):273 – 6.
39. Siesjo BK, Siesjo P. Mechanisms of secondary brain injury. Eur J Anesthesiol 1996;13(3):247 – 68.
40. Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 2000;1(2):120 – 9.
41. Kadar T, Cohen G, Sahar R, et al. Long-term study of brain lesions following soman, in comparison to DFP and metrazol poisoning. Hum Exp Toxicol 1992;11(6):517 – 23.
42. Kadar T, Shapira S, Cohen G, et al. Sarin-induced neuropathology in rats. Hum Exp Toxicol 1995;14(3):252 – 9.
43. Loh Y, Swanberg MM, Ingram MV, et al. Case report: long-term cognitive sequelae of sarin exposure. Neurotoxicology 2010;31(2):244 – 6. doi: 10.1016/ j.neuro.2009.12.004.
44. Karchmar AG. Anticholinesterases and war gases. In: Karchmar, ed. Exploring the vertebrate central cholinergic nervous system. New York: Springer 2007 : 237 – 310.
45. Marrs, TC, Vale JA. Management of organophosphorus pesticide poisoning. In: Gupta RC, ed. Toxicology of Organophosphorus and Carbamate Compounds. Amsterdam: Elsevier Academic Press 2006 : 715 – 33.
46. Terry AV jr. Functional consequence of repeated organophosphate exposure: potential non-cholinergic mechanisms. Pharmacol Therapeut 2012;134(3):355 – 65. doi: 10.1016/ j.pharmthera.2012.03.001.
47. Shih TM, Scremin OU, Roch M, et al. Cerebral acetylcholine and choline contents and turnover following low-dose acetylcholinesterase inhibitors treatment in rats. Arch Toxicol 2006;80(11):761 – 7.
48. Karasova JZ, Novotny L, Antos K, et al. Time-depending changes in concentration of two clinically used acetylcholinesterase reactivators (HI-6 and obidoxime) after administration in vivo by using HPLC techniques. Anal Sci 2010;26(1):63 – 7.
49. Karasova JZ, Kassa J, Jung YS, et al. Effect of several new and currently available oxime cholinesterase reactivators on tabun-intoxicated rats. Int J Mol Sci 2008;9(11):2243 – 52. doi: 10.3390/ ijms9112243.
50. Karasova JZ, Zemek F, Musilek K, et al. Time-dependent changes of oxime K027 concentrations in different parts of rat central nervous system. Neurotox Res 2013;23(1):63 – 8. doi: 10.1007/ s12640-012-9329-4.
51. Karasova JZ, Pavlik M, Chladek J, et al. Hyaluronidase: its effects on HI-6 dichloride and dimethanesulphonate pharmacokinetic profile in pigs. Toxicol Lett 2013;220(2):167 – 71. doi: 10.1016/ j.toxlet.2013.04.013.
52. Karasova JZ, Zemek F, Kassa J, et al. Entry of oxime K027 into the different parts of rat brain: comparison with obidoxime and oxime HI-6. J Appl Biomed 2014;12 : 25 – 9.
53. Karasova JZ, Bajgar J, Jun D, et al. Time-course changes of acetylcholinesterase activity in blood and some tissues in rats after intoxication by Russian VX. Neurotox Res 2009;16(4):356 – 60. doi: 10.1007/ s12640-009-9102-5.
54. Jiang W, Duysen EG, Hansen H, et al. 2010 Mice treated with chlorpyrifos or chlorpyrifos oxon have organophosphorylated tubulin in the brain and disrupted microtubule structures, suggesting a role for tubulin in neurotoxicity associated with exposure to organophosphorus agents. Toxicol Sci 2010;115(1):183 – 93. doi: 10.1093/ toxsci/ kfq032.
55. Grigoryan H, Lockridge O. Nanoimages show disruption of tubulin polymerization by chlorpyrifos oxon: implications for neurotoxicity. Toxicol Appl Pharmacol 2009;240(2):143 – 8. doi: 10.1016/ j.taap.2009.07.015.
56. Peeples ES, Schopfer LM, Duysen EG, et al. Albumin, a newbiomarker of organophosphorus toxicant exposure, identified by mass spectrometry. Toxicol Sci 2005;83(2):303 – 12.
57. Bomser JA, Casida JE. Diethylphosphorylation of rat cardiac M2 muscarinic receptor by chlorpyrifos oxon in vitro. Toxicol Lett 2001;119(1):21 – 6.
58. Quistad GB, Sparks SE, Casida JE. Fatty acid amide hydrolase inhibition by neurotoxic organophosphorus pesticides. Toxicol Appl Pharmacol 2003;173(1):48 – 55.
59. Chaiken IM, Smith EL. Reaction of a specific tyrosine residue of papain with diisopropylfluorophosphate. J Biol Chem 1969;244(15):4247 – 50.
60. Richards PG, Johnson MK, Ray DE. Identification of acylpeptide hydrolase as a sensitive site for reaction with organophosphorus compounds and a potential target for cognitive enhancing drugs. Mol Pharmacol 2000;58(3):577 – 83.
61. Auman JT, Seidler FJ, Slotkin TA. Neonatal chlorpyrifos exposure targets multiple proteins governing the hepatic adenylyl cyclase signaling cascade: implications for neurotoxicity. Brain Res Dev Brain Res 2000;121(1):19 – 27.
62. Lush MJ, Li Y, Read DJ, et al. Neuropathy target esterase and a homologous Drosophila neurodegeneration--associated mutant protein contain a novel domain conserved from bacteria to man. Biochem J 1998;332(1):1 – 4.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2017 Issue 2
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Hope Awakens with Early Diagnosis of Parkinson's Disease Based on Skin Odor
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Current Insights into the Antispasmodic and Analgesic Effects of Metamizole on the Gastrointestinal Tract
- Metamizole vs. Tramadol in Postoperative Analgesia
Most read in this issue
- Ulnar Nerve
- Stroke Incidence in Europe – a Systematic Review
- Anti-NMDAR Encephalitis in Children – a Case Report
- Febrile Seizures – Guidelines for Examination of a Child with Simple Febrile Seizures, Adapted from the Guidelines of the American Academy of Pediatrics