
Genetika neurodegenerativních demencí v deseti bodech – co může neurolog očekávat od molekulárního genetika?
Genetics of neurodegenerative dementias in ten points – what can a neurologist expect from molecular genetics?
Over the past three decades, significant advances have been made in understanding the molecular etiology of hereditary neurodegenerative dementias. Specific genes responsible for hereditary neurodegenerative diseases have been discovered, and studies on the development of disease-modifying therapies have been accelerated. Most neurodegenerative dementias are clinically different, although they share a common pathophysiological background. In neurodegeneration, neuronal atrophy due to apoptotic signaling pathway influenced by deposition of pathologically altered protein in the brain tissue are the leading mechanisms, thus, these diseases are called proteinopathies. In genetic (hereditary) neurodegenerations, conformational changes of proteins, gene aberrations or polymorphisms play crucial roles in pathophysiological mechanisms. Clinical manifestations and neuropathological findings of hereditary forms of neurodegenerative dementia are often inseparable from sporadic types, which increases an urgent need for molecular-genetic analysis of genes responsible for various neurodegenerations. The purpose of this work is to provide a brief overview of the most important genes related to the pathophysiology of neurodegenerative dementias in routine diagnostic practice and the possibilities of their detection.
Key words:
neurodegenerative disorder – dementia – pathogenic mutation
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
E. Parobková 1,2; R. Rusina 3,4; M. Matějčková 1; V. Gregor 1,5; R. Matěj 1,2,6
Authors‘ workplace:
Ústav patologie a molekulární medicíny, 3. LF UK a Thomayerova nemocnice, Praha
1; Ústav patologie, 3. LF UK a FN Královské Vinohrady, Praha
2; Neurologická klinika 3. LF UK a Thomayerova nemocnice, Praha
3; Neurologická klinika a Centrum klinických neurověd, 1. LF UK a VFN v Praze
4; Oddělení lékařské genetiky, Thomayerova nemocnice, Praha
5; Ústav patologie, 1. LF UK a VFN v Praze
6
Published in:
Cesk Slov Neurol N 2019; 82(1): 100-105
Category:
Neuropathological window
doi:
https://doi.org/10.14735/amcsnn2019100
Overview
Během posledních tří desetiletí byl zaznamenán významný pokrok v chápání molekulární etiologie dědičných neurodegenerativních demencí. Byly objeveny geny pro dědičné neurodegenerativní nemoci a množství studií pro vývoj terapií modifikujících onemocnění narůstá. Většina neurodegenerativních demencí se klinicky liší, ač mají společný patofyziologický dopad. U neurodegenerací dochází k zániku neuronů apoptotickou signální dráhou na podkladě ukládání depozit patologicky změněného proteinu v mozkové tkáni, tzv. proteinopatie. U genetických (dědičných) neurodegenerací hrají zásadní roli v patofyziologických mechanizmech konformační změny proteinů, genové aberace či polymorfizmy. Klinické projevy a neuropatologické nálezy dědičných forem neurodegenerativních demencí jsou často nerozlišitelné od sporadických typů, což zvyšuje potřebu molekulárně genetické analýzy genů zodpovědných za různé neurodegenerace. Účelem sdělení je provést stručný přehled genů, které jsou důležité v patofyziologii neurodegenerativních demencí v rutinní diagnostické praxi a možnosti jejich vyšetření.
Klíčová slova:
neurodegenerativní onemocnění – demence – patogenní mutace
Úvod
Neurodegenerativní onemocnění jsou podmíněna ukládáním patologických proteinových agregátů, specifických pro dané onemocnění, do mozkové tkáně. Tato depozita působí toxicky na neurony, spouštějí další děje (především apoptózu a uvolňování kyslíkových radikálů) a vedou k zániku neuronů nebo ztrátě funkce proteinů. Vedle sporadických forem neurodegenerací se uplatňuji i formy dědičné, kde významnou roli hrají odchylky nebo mutace v genech pro daný specifický protein.
Možnosti molekulárně genetických vyšetření neurodegenerativních demencí se v posledních letech zvyšují. Přehled nejčastějších patogenních mutací a polymorfizmů asociovaných s neurodegenerativními demencemi je možné najít na webových stránkách Alzforum [1] a AD/ FTD mutation database [2] (http:/ / www.molgen.ua.ac.be/ admutations, https:/ / www.alzforum.org). V následujícím textu přiblížíme některé častější mutace diagnostikovatelné v ČR.
1. Jaká je dostupnost genetického vyšetření u demencí v ČR a jak interpretovat výsledek genetického vyšetření?
Genetické poradenství a vyšetření je spojeno s fyzickou přítomností pacienta v ordinaci klinického genetika (odbornost 208), ovšem pro pacienty s pokročilou demencí nebo s prionovým onemocněním je přítomnost v ordinaci nepřínosná a reálně nedosažitelná. Proto je důležité, aby ošetřující neurolog, kromě podrobného neurologického vyšetření, zdokumentoval co nejširší rodinnou anamnézu s příbuznými pacienta.
V případě podezření na prionové onemocnění je ošetřující lékař povinen řídit se Metodickým listem TSE/ CJN, Praha 2000 (MZ ČR) [3] a WHO manual for surveillance of human transmissible spongiform encephalopathies [4].
Ke genetickému vyšetření se odebírají 1 – 2 zkumavky nesrážlivé krve, která by měla být uložena v chladu do doby předání do laboratoře. Krev se obvykle odebírá na pracovišti lékařské genetiky nebo také po domluvě u ošetřujícího neurologa. Zároveň je nutné, aby po náležitém poučení pacient (nebo jeho zákonný zástupce) podepsal informovaný souhlas s genetickým vyšetřením, který je pak odeslán s odebraným materiálem do laboratoře. Negativní nález je sdělen pacientovi (nebo jeho zákonnému zástupci, případně osobě blízké oprávněné získat informace o zdravotním stavu pacienta) indikujícím neurologem nebo genetikem, obvykle telefonicky. Pokud je identifikována patogenní mutace, je pacientovi nebo jeho rodinným příslušníkům zapotřebí nabídnout genetickou konzultaci a případně analýzu DNA pro danou mutaci. Presymptomatické testování je možné provést pouze u dospělých, kde je známa mutace u blízkého příbuzného.
Molekulárně genetické vyšetření v závislosti na jeho složitosti trvá obvykle 2 – 3 týdny, ale může trvat i několik měsíců. Interpretace nálezu je úkolem klinického genetika, který by měl podrobně seznámit probanda, případně rodinné příslušníky, s uvažovanou chorobou, možnostmi diagnostiky, léčby či prevence a zodpovědět veškeré dotazy ohledně genetické problematiky.
Testování před 18. rokem věku je možné pouze u specifické skupiny neurologických chorob se závažným průběhem a nástupem příznaků v dětském věku.
2. Liší se od sebe sporadické a genetické formy demencí v klinickém obraze?
Odlišit sporadickou a genetickou formu onemocnění klinickým vyšetřením jednoznačně nelze. Podezření na genetickou formu je odůvodněno při postižení více členů rodiny ve více generacích a rovněž i u časného začátku nemoci (většinou před 50. rokem života).
V klinickém obraze neurodegenerativních onemocnění dochází k překrývání symptomů. Tam, kde se podaří prokázat změnu sekvence DNA, u které je známa vazba ke konkrétní patologii, lze potvrdit genetický původ a upřesnit diagnózu daného onemocnění.
U prionových onemocnění nelze na základě pouze klinických projevů a výsledků pomocných vyšetření odlišit hereditární formu od sporadické. Dědičný původ potvrdí přítomnost patologické mutace v genu PRNP (jediný gen, u kterého je prokázáno, že jeho mutace způsobují geneticky přenosné lidské prionové onemocnění).
Také u mnoha dalších neurodegenerativních onemocnění může být klinicky neodlišitelná forma sporadická od geneticky podmíněné (např. u NMN s demencí). Na druhou stranu existují nemoci, u nichž jsou klinický obraz, doba nástupu i délka onemocnění u sporadické formy velmi odlišné od forem dědičných (typickým příkladem je Alzheimerova nemoc; AN).
Mutace v různých genech mohou mít podobné klinické manifestace (srovnatelný klinický obraz behaviorální varianty frontotemporální demence [bvFTD] při mutaci v genu pro progranulin nebo v genu pro tau protein). Naopak stejná mutace může vyvolávat rozmanité syndromy (např. mutace v genu MAPT – microtubule-associated protein tau [N279K] se může projevit někdy jako bvFTD a jindy jako progresivní supranukleární obrna).
3. Platí, že genetický podklad mají častěji formy demence s časným počátkem?
Genetický podklad je typický pro časné formy onemocnění, může se ale podílet i na vzniku pozdějších forem chorob. Platí tedy, že u většiny neurodegenerativních onemocnění je při rozvoji onemocnění v mladším věku větší pravděpodobnost záchytu specifické kauzální mutace; pokud se nemoc objeví ve vyšším věku, spíše se bude jednat o sporadickou formu. Rozhodnutí, zda genetické vyšetření indikovat, by mělo být individuální a vycházet z komplexního posouzení pacientova stavu (detailní rodinná anamnéza, průběh, klinický obraz i nález na MR).
Významnou roli v patofyziologii u některých neurodegenerativních onemocnění mají vedle vlastní patogenní mutace i polymorfizmy v příslušném genu. Genetický polymorfizmus je definován existencí dvou nebo více alel (variant genů) v jednom lokusu, převyšující svým výskytem 1% výskyt v populaci. Polymorfizmus samotný onemocnění nezpůsobí, ale může ovlivnit věk, kdy se objeví první příznaky, individuální rozvoj onemocnění nebo ovlivnit jeho průběh – pozoruhodným příkladem je mutace v kodonu 178 (N178D) genu prionového proteinu, která se projeví jednou jako hereditární Creutzfeldtova-Jakobova nemoc (CJN) (pokud je na mutované alele v kodonu 129 metionin) a podruhé jako fatální familiární insomnie (FFI) (pokud je na mutované alele v kodonu 129 valin).
4. Které geny vyšetřovat u Alzheimerovy nemoci?
Alzheimerova nemoc je nejčastějším neurodegenerativním onemocněním. U formy AN s časným začátkem (obvykle před 50. rokem života) bývají častým nálezem mutace v genu pro amyloidový prekurzorový protein (APP) a preseniliny (PS). Tyto hereditární formy s autozomálně dominantním typem dědičnosti nejsou početné a zahrnují 10 – 15 % případů AN [5].
Gen pro APP (chromozomální lokus 21q21) kóduje amyloid beta (Aß) prekurzorový protein, který je štěpen sekretázami buď na solubilní peptidy o 40 aminokyselinách (Aβ40), nebo na nerozpustný peptid o 42 nebo 43 aminokyselinách (Aβ42/ 43), který se pak patologicky ukládá do mozkové tkáně s formací amyloidních plak. Patogenní mutace APP jsou plně penetrantní, nacházejí se převážně v místech štěpení proteinu nebo v jejich blízkosti a vedou k rychlé progresi onemocnění, navíc často s doprovodnou amyloidovou angiopatií [6].
Sekretázy jsou enzymy, které zajištují proteolytické štěpení APP. Mutace v genech pro preseniliny PS1 a PS2 (chromozomální lokus 14q24.3 a 1q31 – 42) přímo ovlivňují proces štěpení APP a vedou ke zvýšené produkci Aβ42/ 43. Mutace PS1 jsou plně penetrantní a způsobují rychlou progresi nemoci s průměrným začátkem v 45 letech [7]. Mutace PS2 mají pozdější věk nástupu onemocnění (45–88 let), penetrance je neúplná, proto se u některých nositelů mutace onemocnění nemusí vůbec klinicky manifestovat [8].
5. Jaký je význam genetického vyšetření u podezření na prionový původ onemocnění a jaká je indikace k vyšetření?
Přibližně 15 – 20 % případů CJN je dědičných, přitom nemusí být přítomna přesvědčivá rodinná anamnéza [9].
Při podezření na prionové onemocnění se v ČR od roku 2001, kdy laboratoř vznikla, v souladu s vyhláškou Ministerstva zdravotnictví provádí pitva v Národní referenční laboratoři pro lidská prionová onemocnění v Thomayerově nemocnici v Praze. Ve své databázi má více než 650 případů podezřelých z CJN, diagnóza genetické formy prionového onemocnění byla potvrzena u více než 60 případů, sporadická forma byla potvrzena přibližně u 270 případů. Po průkazu patologických prionových depozit v mozkové tkáni se následně provádí sekvenování genu pro prionový protein (PRNP).
Mutace v genu PRNP jsou asociovány především s CJN, méně často s fatální familiární insomnií, Gerstmannovou-Sträusslerovou-Scheinkerovou nemocí (GSS) a kuru.
V ČR je nejčastější mutací PRNP genu E200K (většinou rodiny původem ze Slovenska, kde se tato mutace vyskytuje endemicky na Oravě, Spiši a u Rožňavy), zatímco mutace D178N, běžná v západní Evropě, je mnohem vzácnější (u nás byla zachycena naposledy v roce 2007). Klíčovou roli hrají i polymorfizmy v kodonu 129 a 219.
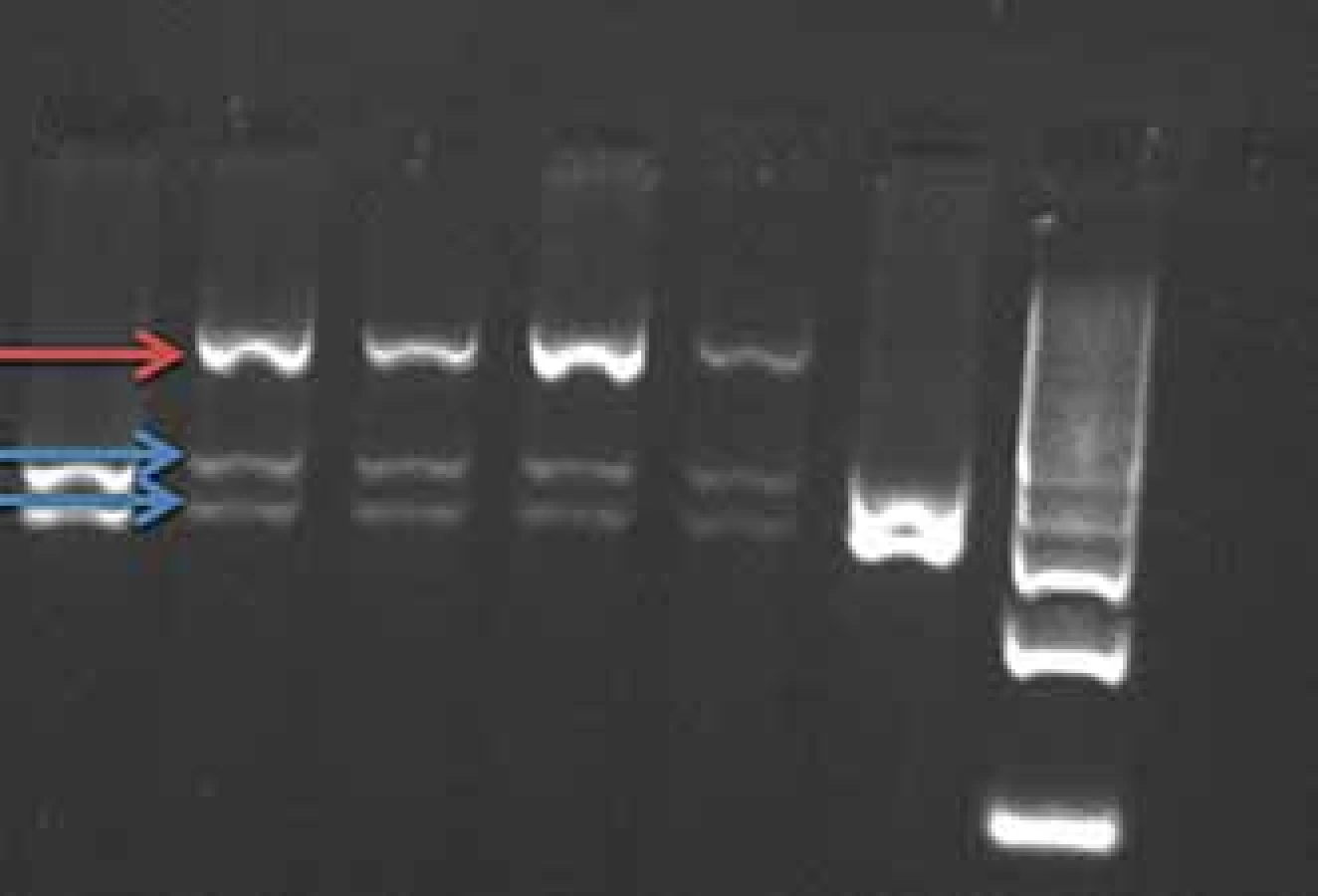
Mutace v genu PRNP se dědí autozomálně dominantně s různou penetrancí podle typu přítomné mutace 50 – 100 %, tedy část nosičů mutace nemá klinické příznaky onemocnění. Mutace E200K způsobuje hereditární CJN (obr. 1), mutace P102L hereditární formu prionového onemocnění GSS a mutace D178N může způsobit buď CJN nebo fatální familiární insomnii.
Fig. 1. BsmA1 restriction of PCR product
gene PRNP (mutation E200K). The E200K
mutation cancels the BsmA1 restriction
site.
Red arrow – uncut fragment of the mutated
allele, length 608 bp.
Blue arrows – split fragment of the wild-
-type allele, length 260–348 bp.
Atypický průběh onemocnění je v případě přítomnosti delecí nebo inzercí 120bp v nestabilní oblasti PRNP, která je bohatá na prolin, glycin a glutamin. Normální alely PRNP mají jeden nonapeptid (R1) a čtyři oktapeptidové repetice (R2_R2_R3_R4), které obsahují aminokyseliny: Pro-(His/ Gln)-Gly-Gly-Gly-(-/ Trp)-Gly-Gln. Delece dvou a více repetic jsou patogenní, stejně jako inzerce jedné nebo více repetic [10]. Fenotyp onemocnění s inzercemi je vysoce proměnlivý, často má rysy onemocnění CJN a GSS nebo naopak postrádá specifické histopatologické změny. Projevuje se neobvykle dlouhým trváním onemocnění s nástupem ve 3. – 6. dekádě života.
6. Mutace kterých genů nejčastěji vyvolají frontotemporální lobární degeneraci?
Frontotemporální lobární degenerace (FTLD) jsou poměrně heterogenní skupinou onemocnění, kterou dělíme na tauopatie (FTLD-tau) a tau negativní formy (FTLD-non tau) s přítomností patologických inkluzí obsahujících depozita různých bílkovin [11].
Mezi tauopatie řadíme Pickovu nemoc (FTLD s Pickovými tělísky), kortikobazální degeneraci, progresivní supranukleární obrnu nebo nemoc s argyrofilními zrny a některé formy primární progresivní afázie. Tauopatiím bude věnována samostatná pozornost (viz další otázka) [12].
K nejčastějším dědičným formám FTLD patří mutace v genu MAPT, genu GRN kódujícím protein progranulin, genu C9orf72 (kóduje protein, který hraje důležitou roli v regulaci endozomálního přenosu) a v genu TARDBP (kóduje přímo TDP-43). Vzácné jsou mutace lokalizované v genech CHMP2B, FUS, SOD1, VCP [13].
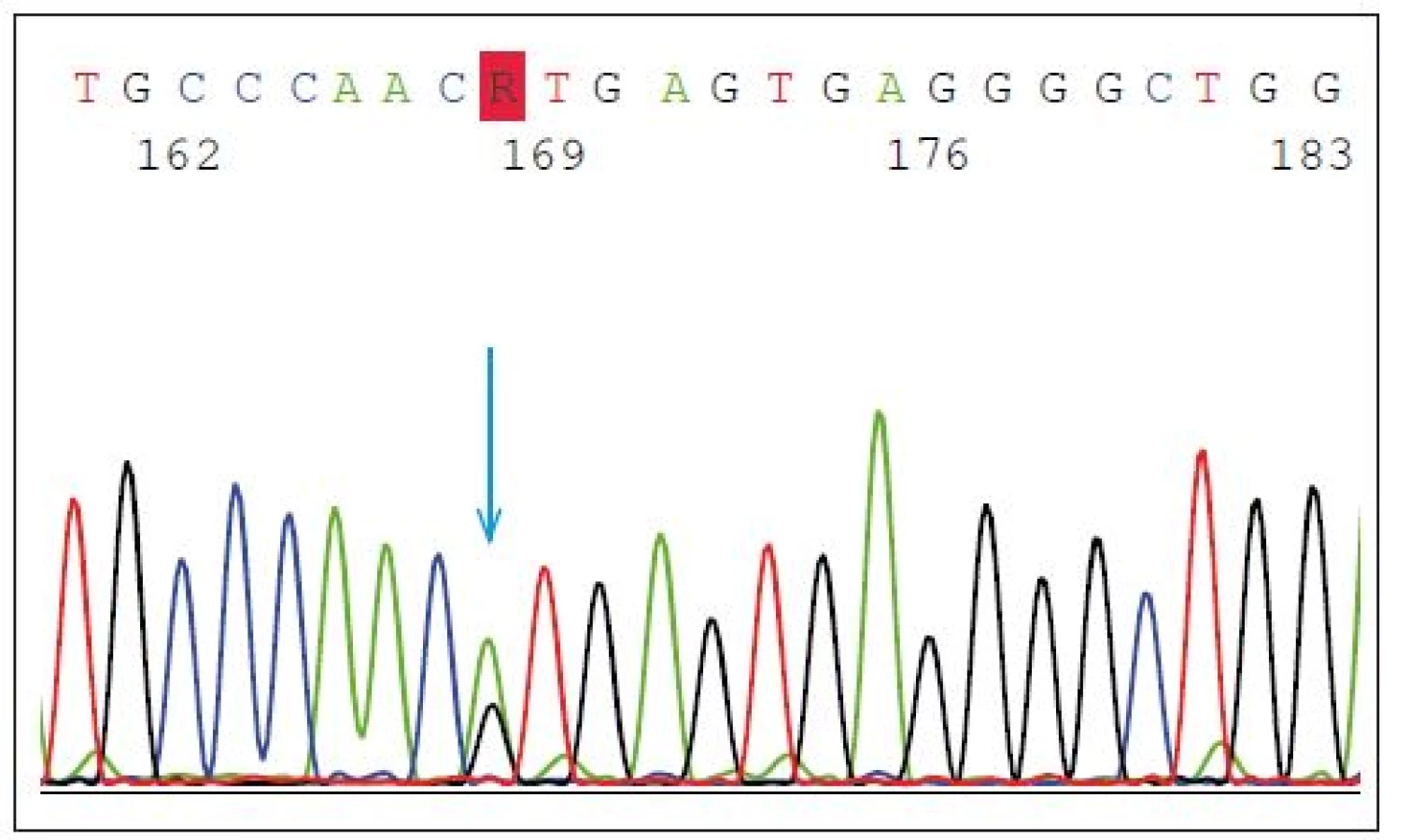
Mutace v GRN představují asi 5 % všech FTD a 20 % případů FTD s pozitivní rodinnou anamnézou. Dědičnost onemocnění je autozomálně dominantní, nicméně neexistuje jednoznačná korelace genotypu a fenotypu. Mezi častější mutace patří heterozygotní splice-site mutace p.Val200Glyfs*18(p.V200GfsX18) (mutace leží v místě, kde probíhá sestřih během zpracování prekurzorové mRNA do zralé mRNA) s fenotypem onemocnění FTD/kortikobazálního syndromu (FTD/CBS) (obr. 2) nebo inzerce IVS3 -46_-47insGTCA, která je spojována s vyšším rizikem amyotrofické laterální sklerózy [14].
Fig. 2. Heterozygous splice-site mutation p.Val200Glyfs*18 (p.V200GfsX18) in GRN gene.
The blue arrow shows the point mutation in intron 7 (IVS7 + 1G > A) splice donor site predicted
to cause exon 7 skipping, frameshift and premature translation termination, and is
demonstrated to result in transcript degradation.
Mutace v C9orf72 (obr. 3) je považována za druhou nejčastější mutaci asociovanou s FTLD s frekvencí 14 – 18 % u familiárních forem [15]. V oblasti mezi prvním a druhým exonem genu C9orf72 je přítomna repetitivní polymorfní oblast hexanukleotidové sekvence GGGGCC, která se může opakovat od 2 – 20 až do 700 – 1600 kopií. Patologická expanze GGGGCC v genu C9orf72 se vyskytuje u některých pacientů s amyotrofickou laterální sklerózou a demencí (FTLD-NMN). Pacienti s C9orf72 expanzí vykazují nižší věk nástupu příznaků, kratší přežití, nástup bulbárních symptomů a nápadnou náchylnost k psychózám a halucinacím [16].
Fig. 3. Determination of the copy number of C9ORF72 hexanucleotide expansion.
Reference values: normal allele < 20 repeats GGGGCC, premutation allele 20–29 repeats GGGGCC allele, full penetration > 29 repeats
GGGGCC. A blue arrow shows the position of the 20 GGGGCC repeats and the red arrow shows the position of 30 or more repetitions.
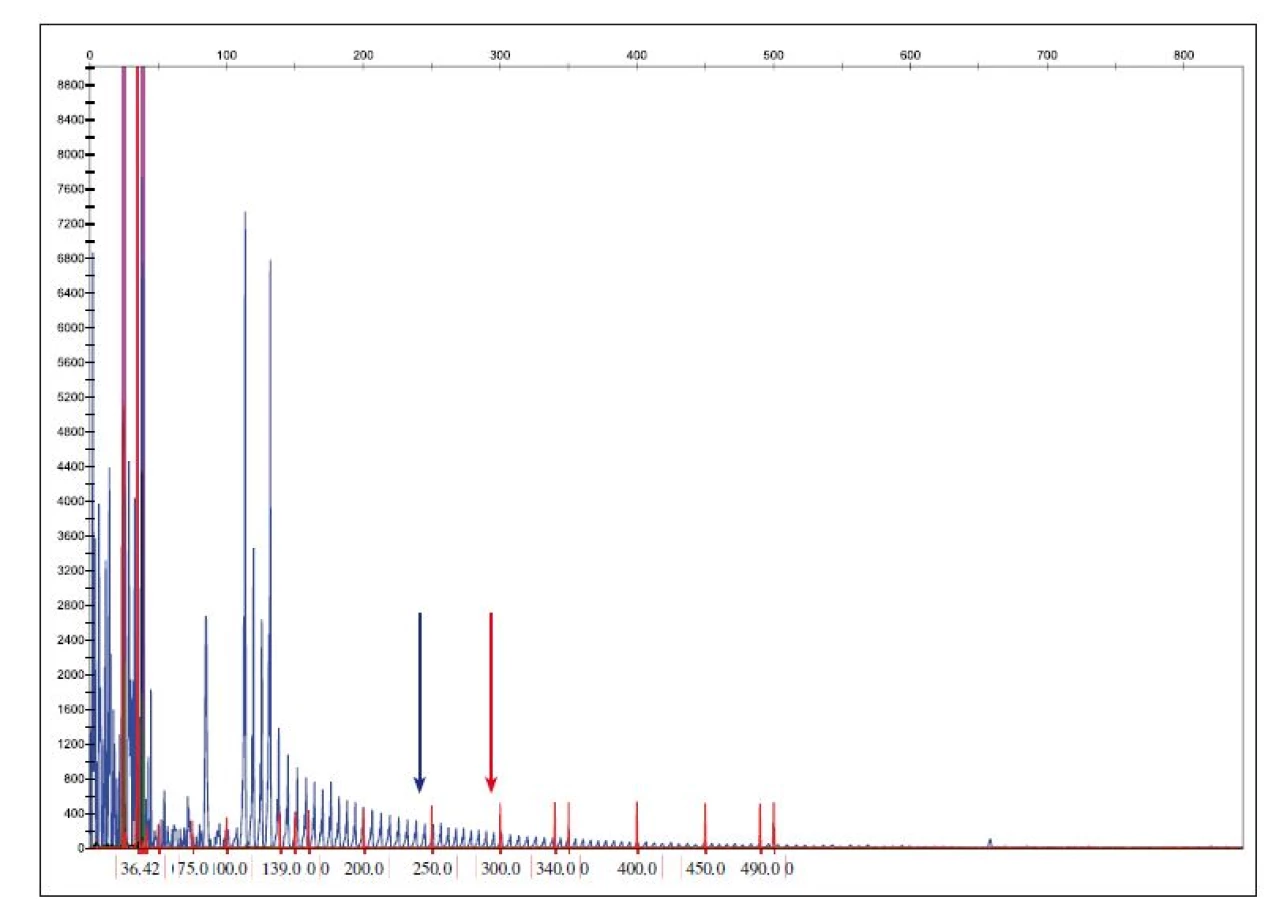
Rovněž mutace v genu TARDBP jsou asociovány především s FTLD-NMN. V ČR bylo zatím vyšetřeno 150 případů a všechny byly bez patogenní mutace (v několika případech byl nalezen polymorfizmus IVS5+14T/ C v heterozygotním stavu neznámé klinické signifikance).
7. Jaký je nejčastější gen, jehož mutace způsobuje tauopatie, a kdy ho vyšetřovat?
Nejběžnějším proteinem patologicky změněným u tauopatií je protein asociovaný s mikrotubuly tau, který je produktem alternativního sestřihu genu MAPT na chromozomálním lokusu 17q.21.32.
Mutace v genu MAPT vedoucí k akumulaci hyperfosforylovaného tau v neuronech nebo v gliových buňkách přímo způsobují některé typy AN, Pickovy nemoci, kortikobazální degenerace a progresivní supranukleární obrny a další vzácnější tauopatie [17].
Některé MAPT polymorfizmy se dědí jako dva haplotypy, H1 a H2. Převládající haplotyp H1 je asociován s progresivní supranukleární obrnou a kortikobazální degenerací, genotyp H1/ H1 s parkinsonismem při mutaci MAPT a H2 pak s hereditární FTD [18].
Frekvence MAPT mutací u tauopatií je celosvětově v některých pramenech odhadována až na 50 % [19]. Překvapivý je v tomto kontextu proto jejich téměř nulový záchyt v ČR (od roku 2007 bylo sekvenováno více než 150 případů s FTD či AN, přitom byla zachycena jediná patogenní mutace p.Ser305Asn s haplotypem H2/ H2).
8. Kdy vyšetřovat polymorfizmus apolipoproteinu E v klinické praxi?
Apolipoproteiny slouží jako informační molekuly, které zajišťují vazbu lipoproteinu na specifická místa. Apolipoprotein E (ApoE) v mozku má vliv na neuronální reparaci, růst dendritů, synaptickou plasticitu a je popsán i jeho protizánětlivý vliv. Gen na chromozomálním lokusu 19q13.2 je polymorfní a vyskytuje se ve třech kodominantních alelách: E2, E3 a E4.
Apolipoprotein E2 (četnost alely 7 %) způsobuje pomalejší odbourávání VLDL (lipoproteiny o velmi nízké hustotě) a chylomikronů a má nižší afinitu pro LDL (lipoproteiny o nízké hustotě) receptory a je spojován s hyperlipoproteinémií typu III i s rizikem Parkinsonovy nemoci. ApoE3 (četnost alely 79 %) je z hlediska rizikových faktorů neutrální. Četnost alely ApoE4 v české populaci je 15,7 %. Homozygotů E4/ E4 jsou v naší populaci asi 2 % a mají až 8× vyšší relativní riziko vzniku AN a nižší věk počátku příznaků ve srovnání s homozygoty E3/ E3 [20].
Vzhledem k tomu, že výskyt ApoE4 je sice spojen s několikanásobně vyšším rizikem rozvoje pozdní formy AN, přitom ale není přímou příčinou AN (tedy kauzální mutací), nedoporučuje se stanovování genotypu ApoE4 v rutinní klinické praxi [21]. Má však své významné místo v epidemiologických a výzkumných studiích, a provádí se proto ve specializovaných centrech.
9. Jaká je role nových metod sekvenování DNA u demencí?
S příchodem nové technologie tzv. masivního paralelního sekvenování či sekvenování nové generace došlo k výraznému zrychlení procesu identifikace známých, ale i nových genetických mutací.
Sekvenování nové generace nabízí možnost souběžně sekvenovat velký počet genomických oblastí a umožní v nich identifikovat všechny varianty genomu jednotlivce, zejména klinicky relevantní alely.
Velkým přínosem je možnost masivního sekvenování většího počtu kandidátních genů i jejich nekódujících oblastí, ve kterých se vyskytují regulační oblasti exprese. To umožní odhalit nové genetické příčiny u pacientů, kteří nenesou žádné známé kauzální mutace v dosud testovaných genech.
Užitím metody sekvenování nové generace je možné odhalit i méně frekventované mutace, na něž se v kontextu klinického obrazu a priori nemyslí. Běžnému užití sekvenování nové generace v rutinní diagnostice zatím brání velmi vysoká cena vyšetření a problematická interpretace získaných dat.
10. Má smysl indikovat genetické vyšetření u pacientů s atypickou demencí a negativní rodinnou anamnézou?
Odhadovaná četnost mutací u neurodegenerativních demencí se v různých pramenech významně liší, rozptyl je od jednotek po 20 % [22]. Jedná se především o časné formy.
Klinický průběh genetických forem může být zcela odlišný od sporadických typů onemocnění a bez znalosti patogenní mutace nelze mnohdy dojít ke správné diagnóze.
V mnoha případech u pacientů s prokázanou kauzální mutací nemusí být příznačná pozitivní rodinná anamnéza, neboť informace mohou být mnohdy zavádějící či neúplné. Kromě toho se patogenní mutace nemusí pouze dědit, ale může i vznikat de novo. Potomci takto postižených jedinců pak už dědí nově získanou mutaci standardním způsobem.
Proto v případě atypického klinického průběhu onemocnění může být přínosné provést molekulárně genetické vyšetření i přes negativitu rodinné anamnézy. K posouzení rentability genetického vyšetřování je vhodné konzultovat specializované centrum, které má zkušenosti s daným typem onemocnění a úzce spolupracuje s klinickým genetikem.
Závěr
Klinický obraz neurodegenerativních demencí bývá rozmanitý a symptomy se vzájemně překrývají. Vedle sporadických forem neurodegenerací se uplatňuji i formy dědičné, kde významnou roli hrají odchylky nebo mutace v genech pro daný specifický protein. Při rozvoji onemocnění v mladším věku je větší pravděpodobnost záchytu specifické kauzální mutace. Klinický průběh genetických forem může být zcela odlišný od sporadických typů onemocnění a bez znalosti patogenní mutace nelze mnohdy dojít ke správné diagnóze. I v případě atypických klinických průběhů tak může být přínosné provést molekulárně genetické vyšetření i v rodině se sporadickým výskytem onemocnění. Důležitá je úzká spolupráce neurologa s klinickým genetikem.
Práce byla podpořena MZ ČR projekty Thomayerova nemocnice-TN-0064190 a RVO-VFN 64165 a AZV NV18-04-00346, Univerzitou Karlovou (projekt Progres Q27 a Q28/ LF1) a OPPK projektem CZ.2.16/ 3.1.00/ 24509.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
prof. MUDr. Radoslav Matěj, Ph.D.
Ústav patologie a molekulární medicíny
3. LF UK
Thomayerova nemocnice
Vídeňská 800
140 59 Praha 4
e-mail: radoslav.matej@ftn.cz
Přijato k recenzi: 29. 6. 2018
Přijato do tisku: 13. 12. 2018
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2019 Issue 1
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Lehká mozková poranění – konsenzuální odborné stanovisko České neurologické společnosti ČLS JEP
- Chronický subdurální hematom
- Oligoklonální IgG a volné lehké řetězce – srovnání izoelektrické fokusace v agarózovém a polyakrylamidovém gelu
- Ketogenní dieta – účinná nefarmakologická léčba dětské a adolescentní epilepsie