
TNFα and microRNA-15b expression changes in experimental model of subarachnoid haemorrhage
Změny v expresi TNFα a microRNA-15b u experimentálního modelu subarachnoidálního krvácení
Cíl:
Cílem studie bylo prozkoumat změny v expresi pro-zánětlivého a pro-apoptotického cytokinu tumor nekrotizující faktor alfa (TNFα) a mikroRNA (miRNA), které se podílejí na jeho regulaci v časném období po subarachnoidálním krvácení (SAK).
Soubor a metodika:
Exprese miRNA (miR-125b, miR-146a, miR-346, miR-155, miR-15b) a mRNA (TNFα) byly stanoveny pomocí kvantitativní polymerázové řetězové reakce v reálném čase z mozkové tkáně experimentálních zvířat. Celkem 88 zvířat bylo rozděleno do skupin Sham (kontrolní operace bez indukce SAK), Lehké SAK, Těžké SAK, do časových intervalů 2, 4, 6 a 8 h (n = 7 ve skupině); 4 zvířata byla použita jako absolutní kontrola.
Výsledky:
Byly nalezeny statisticky významné rozdíly v expresi TNFα mezi skupinami Sham a Těžké SAK ve všech zkoumaných časových intervalech (p < 0,05), dále mezi skupinami Sham a Lehké SAK 4 h po indukci SAK (p < 0,05) a mezi skupinami Lehké SAK a Těžké SAK ve 2 a 6h časovém intervalu (p < 0,05). Dále byl pozorován významný rozdíl v expresi miRNA-15b mezi skupinami Sham a Těžké SAK 8 h po začátku SAK (p < 0,05). U dalších analyzovaných miRNA jsme v expresi nepozorovali žádné statisticky významné změny.
Závěr:
SAK bylo asociováno s časným nárůstem exprese TNFα a miR-15b, zejména u skupiny Těžké SAK. Navzdory komplexitě vzájemné regulace mezi cytokiny a mikroRNA, může informace o časné aktivaci zánětlivých/ apoptotických mechanismů několik hodin po SAK přispět k lepšímu poznání patofyziologie SAK. Pochopení mechanismů vzájemné regulace proapoptotických markerů TNFα a miR-15b může přispět ke zlepšení terapie této závažné patologie.
Klíčová slova:
subarachnoidální krvácení – časné poškození mozku – zánět – apoptóza – mikroRNA – perforační model
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Authors:
J. Lipkova 1; Z. Splichal 1; Michal Jurajda 1
![]() ; T. Madaraszova 2; A. Vasku 1; M. Smrcka 2; K. Duris 1,2
; T. Madaraszova 2; A. Vasku 1; M. Smrcka 2; K. Duris 1,2
Published in:
Cesk Slov Neurol N 2019; 82(1): 53-59
Category:
Original Paper
doi:
https://doi.org/10.14735/amcsnn201953
Overview
Aim:
The aim of the study was to investigate expression changes of pro-inflammatory and pro-apoptotic cytokine tumor necrosis factor alpha (TNFα) and microRNAs (miRNAs) involved in its regulation in early pathophysiological changes after subarachnoid haemorrhage (SAH).
Materials and methods:
MiRNAs (miR-125b, miR-146a, miR-346, miR-155, miR-15b) and mRNA (TNFα) expression were determined by quantitative real-time polymerase chain reaction in brain tissue samples. A total of 88 animals were divided to Sham (control surgery without induction of SAH), Mild SAH, Severe SAH groups in following time-points: 2, 4, 6 and 8 h (n = 7 per group); including 4 animals used as an absolute control.
Results:
We have found a statistically significant difference in TNFα expression between Sham and Severe SAH groups at all the time-points (p < 0.05), between Sham and Mild SAH groups 4 h after induction of SAH (p < 0.05) and between Mild and Severe SAH groups at 2 and 6 h time-points (p < 0.05). Furthermore, a significant difference in miR-15b expression between Sham and Severe SAH groups was observed 8 h after SAH (p < 0.05). All the other microRNAs have not been significantly changed.
Conclusions:
SAH was associated with an early increase in TNFα and miR-15b expression especially in Severe SAH group. Despite complex cross-regulation between cytokines and miRNA, any information about the activation of inflammation/ apoptotic mechanisms within a few hours after SAH may improve our knowledge of SAH pathophysiology. Furthermore, it can lead to therapeutic improvement using a combination of both pro-apoptotic markers TNFα and miR-15b.
Keywords:
subarachnoid haemorrhage – early brain injury – inflammation – apoptosis – microRNA – perforation model
TNFαmicroRNA-15b表达变化蛛网膜下腔出血的实验模型
目的:
这项研究的目的是探讨表达变化的促炎症细胞因子和pro-apoptotic肿瘤坏死因子α(TNFα)和小分子核糖核酸(microrna)参与监管在蛛网膜下腔出血(SAH)后早期病理生理变化。
材料与方法:
microrna (mir - 125 - b, mir - 146 a, mir - 346, mir - 155, miR-15b)和mRNA (TNFα)表达式定量实时聚合酶链反应测定脑组织中样本。将88只动物分为假手术组(无诱导SAH对照组)、轻度SAH组、重度SAH组,时间点分别为2、4、6、8 h(每组n = 7),其中4只动物作为绝对对照组。
结果:
我们发现了一个统计上的显著差异TNFα表达虚假的和严重的SAH组之间的跨度为虚假之间(p < 0.05)和轻度SAH组4 h感应SAH后(p < 0.05)和轻微和严重SAH组之间跨度为2和6 h (p < 0.05)。SAH后8 h,假手术组与重度SAH组miR-15b表达差异有统计学意义(p < 0.05)。其他所有的microrna都没有发生显著的改变。
结论:
长官与早期增加TNFα和miR-15b表达特别是严重SAH组。尽管细胞因子和miRNA之间存在复杂的交叉调控,但任何有关SAH后几小时内炎症/凋亡机制激活的信息都可能提高我们对SAH病理生理学的认识。此外,它会导致治疗改善使用pro-apoptotic标记TNFα和miR-15b的组合。
关键词:
蛛网膜下腔出血-早期脑损伤-炎症-凋亡-微rna -穿孔模型
Introduction
Aneurysmal subarachnoid haemorrhage (SAH), with an annual incidence of around 10 per 100,000 and case fatality rate of up to 50%, remains a severe condition which affects mainly patients of productive age [1]. Despite advances in surgical treatment and critical care in the last decades, the morbidity and mortality rate of patients suffering from SAH is still high. SAH is therefore a subject of intensive research in both clinical and experimental conditions.
Haemorrhage into the subarachnoid space initiates several pathological processes contributing to early brain injury (EBI), which occurs in the first 72 h following SAH. These include transient cerebral global ischaemia, oxidative stress or inflammation which may result in neuronal or endothelial cell death [2,3]. The mechanisms of the EBI and the brain‘s response to this injury are very complex and still not clear. Nevertheless, inflammation and apoptosis are among the most commonly proposed mechanisms which may contribute to EBI development [4–6]. One of the key mediators of inflammation is tumor necrosis factor alpha (TNFα) – a pleiotropic pro-inflammatory cytokine. Increased production of TNFα was measured in the brain after injury and its level rises in cerebrospinal fluid (CSF) and blood after stroke in humans [7]. TNFα has been proposed to play a crucial role in neuroinflammatory response after SAH, participating in SAH-related oxidative damage and is involved in cerebral vasospasm development [8]. Moreover, out of many pro-inflammatory cytokines, TNFα is the most potent inducer of apoptosis, representing a link between inflammation and cell death. TNFα mediates apoptotic cell death as well as cell proliferation and differentiation in a number of cell types [9–12].
On the molecular level, pathological changes induced by SAH are probably tightly orchestrated processes in which regulatory molecules named microRNA (miRNA), may play a pivotal role. MiRNAs are small non-coding RNAs able to mediate post-transcriptional regulation of genes. They are known to participate in fundamental cellular processes such as cellular metabolism [13], cell-cycle regulation, including apoptosis [14] or immune response [15]. Several studies have indicated that specific miRNAs are involved in the regulation of inflammation and apoptosis after SAH and are clinically associated with the severity of brain injury [16–18]. However, no details are known about the early dynamics of miRNAs after SAH and about its association with the early dynamics of pro-inflammatory cytokines. Temporal changes in the abundance of specific miRNAs after haemorrhage may provide a novel insight into their role in brain injury and the brain‘s response. Although several miRNAs were referred to be promising circulating biomarkers and potential diagnostic markers of acute stroke, the miRNAs are not yet used for diagnostic purposes in clinical practice [19].
The aim of the present study was to investigate the expression of pro-inflammatory and pro-apoptotic cytokine TNFα and several miRNAs involved in its regulation in brain tissue up to 8 h after induction of bleeding in an experimental model of SAH. We selected five miRNAs associated with TNFα (miR-15b, miR-125b, miR-146, miR-155, miR-346) which are involved in inflammation and/ or apoptosis. From those, miR-125b and miR-155 were evaluated as strong pro-inflammatory regulators of macrophage activation [20]. Moreover, miR-125b was described to inhibit glial cell proliferation, promotes cell apoptosis (via p53) and neuronal differentiation [21–23]. Similarly, miR-146 was described to be involved in cell proliferation, apoptosis and inflammatory response [24–26]. MiR-346 was determined as a negative regulator of inflammation with the ability to suppress cell proliferation [27,28], whereas miR-15b has been identified as a probably essential player in apoptosis by targeting anti-apoptotic genes in different cellular systems [29–32].
Material and methods
Animal experiments
All the experiments were performed in compliance with the Principles of Laboratory Animal Care (NIH Publication no. 86–23, revised 1985). The experimental protocol was approved by the Ethics Committee of the Masaryk University (Brno, Czech Republic). A total number of 88 adult male Sprague Dawley rats (260–300 g) was used for the purpose of this study. Animals were randomly assigned to Sham, SAH groups and divided into one of the following time-point groups: 2, 4, 6 or 8 h (n = 7 per group). Another four animals were used as an absolute control group. All animals were anesthetised using Isoflurane (AbbVie Ltd., Maidenhead, UK), intubated and kept on mechanical ventilation during surgery. SAH was induced by sharpened 4-0 nylon suture through the left internal carotid artery as described previously in Sham animals, the suture was inserted into the artery without perforation. Animals were sacrificed at scheduled time-points, a picture of the basal part of the brain was taken for SAH grade evaluation [33]. Then the samples of the brain, representing the area adjacent to haematoma, were collected (see below). According to SAH grade the animals were assigned to mild/ moderate SAH group (SAH grade 6–12; below labelled as Mild SAH) and Severe SAH group (SAH grade 13–18), while animals with SAH grade < 6 were excluded for insufficient SAH, as usual [33,34].
Samples
Brain samples were collected from the basal part of the left hemisphere (the area adjacent to haematoma, which is directly affected by haematoma) and frozen in RNA later at –80 °C. Tissue samples were collected at four different time-points after surgery 2, 4, 6 and 8 h). Isolation of total RNA from brain samples was performed with Tripure reagent (Qiagen GmbH, Hilden, Germany). Single-stranded cDNA was synthesized from 1,000 ng of total RNA using Transcriptor First Strand cDNA Synthesis Kit (Roche s.r.o., Praha, Czech Republic) MiRNA (from 300 ng of total RNA) was transcribed using TaqMan miRNA reverse transcription kit (Applied Biosystems Inc, Foster City, CA, USA). The probes for miRNA (rno-miR-125b-5p, assay ID: 000449; rno-miR-146a-5p, assay ID: 000468; rno-miR-346, assay ID: 001333; rno-miR-155-5p, assay ID: 002571; rno-miR-15b-5p, assay ID: 000390) and mRNA (TNFα, assay ID: Rn01525859_g1) were selected from the TaqMan gene expression assays (Life Technologies, Carlsbad, CA, USA). Expression was evaluated by quantitative real-time polymerase chain reaction at the LightCycler®480 II System (Roche s.r.o., Prague, Czech Republic). The amplified DNA was analysed by the comparative Ct method. Only genes where Ct were lower than 35 were considered significantly expressed. All samples were measured in triplicates. After normalisation with the reference miRNA (U6, assay ID: 001973) and mRNA (HPRT1, assay ID: Rn01527840_m1), the expression levels were presented as a relative fold change compared with the mean value of the absolute control group.
Statistical analyses
The data met criteria of log-normal distribution, the data were logarithmically transformed and the statistical analyses were performed on transformed data. Ordinary one-way ANOVA followed by Bonferroni‘s multiple comparison test was used to evaluate the data. A p value of <0.05 was considered as statistically significant. Statistical analyses were performed using GraphPad Prism version 7.03 for Windows (GraphPad Software, La Jolla, California, USA). Data are presented as a geometric mean and geometric standard deviation factor of the fold change normalised to the absolute control group.
Results
The expression of TNFα and miR-125b, miR-146a, miR-346, miR-155, miR-15b was quantitatively determined in a total number of 88 animals. The four animals were used as an absolute control and the rest of the 84 animals were divided in Sham, Mild SAH, Severe SAH groups and the following time-points: 2, 4, 6 and 8 h (n = 7 per group). Expression of the above-mentioned markers was evaluated in the area of the brain adjacent to haematoma, which is directly affected by bleeding.
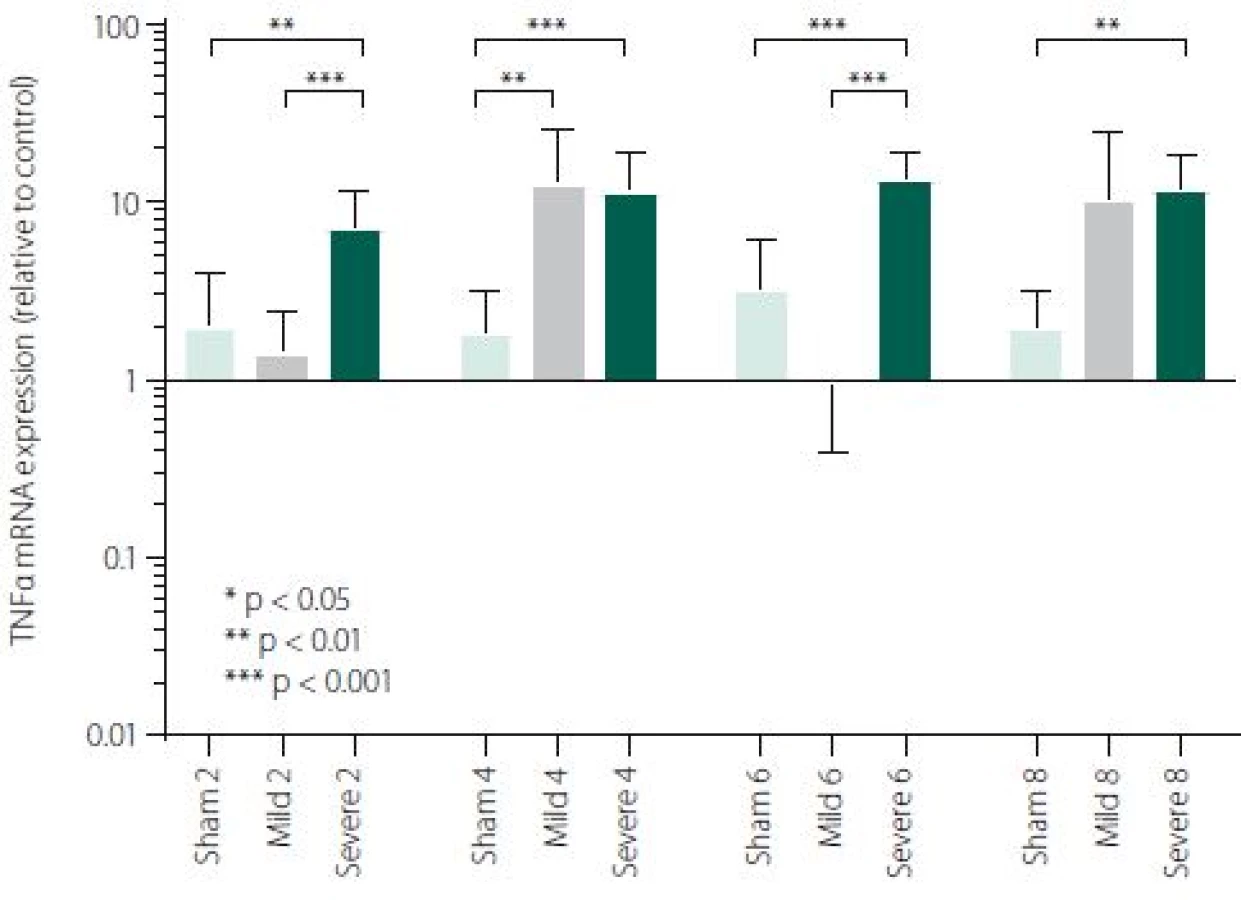
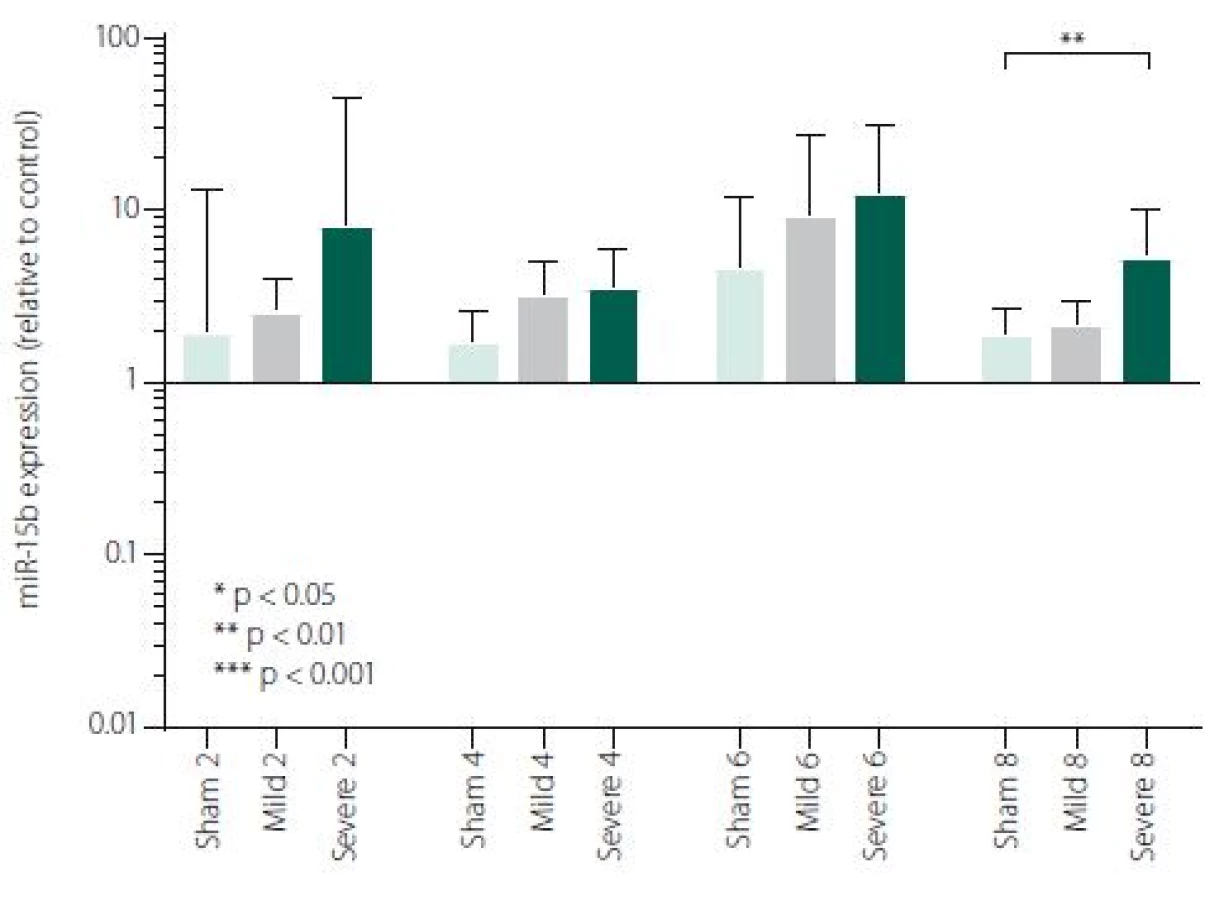
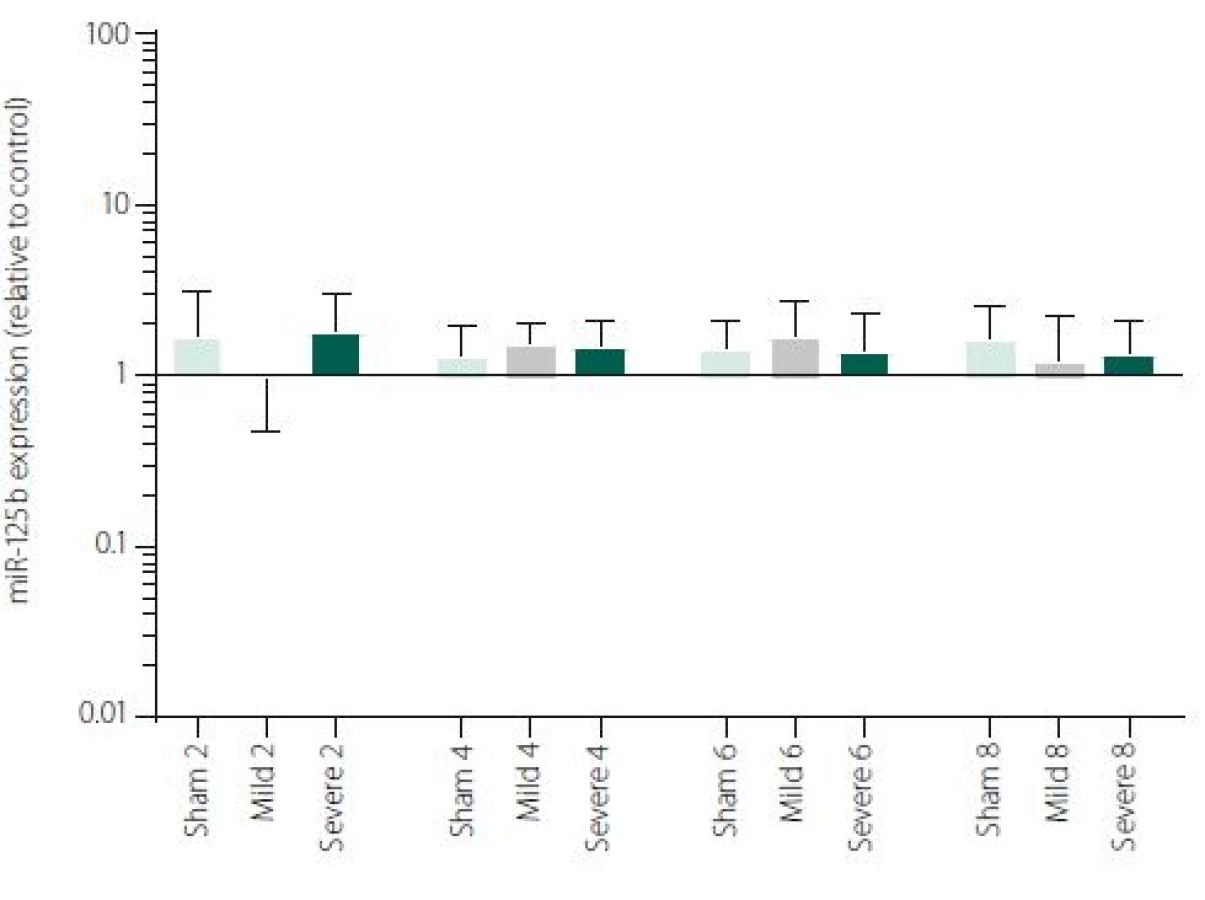
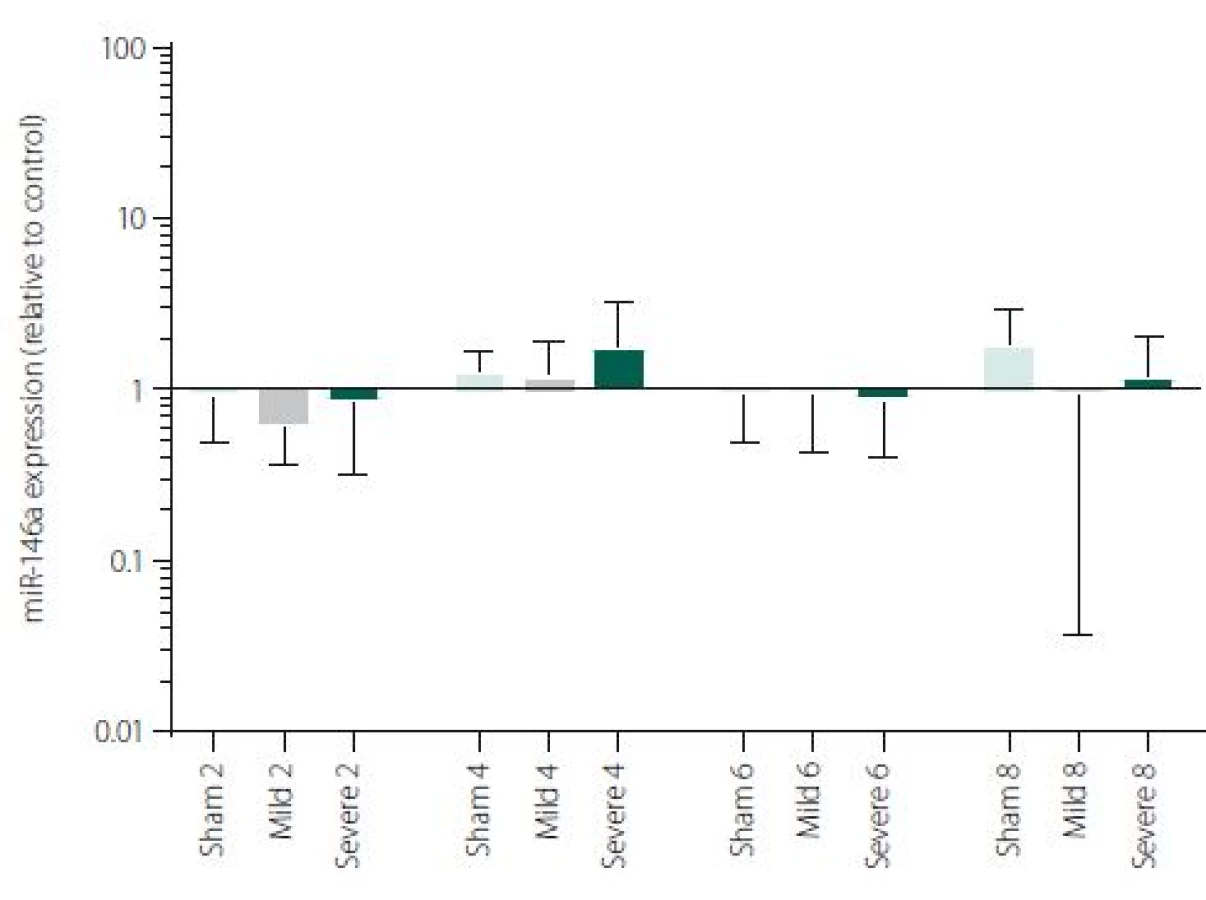
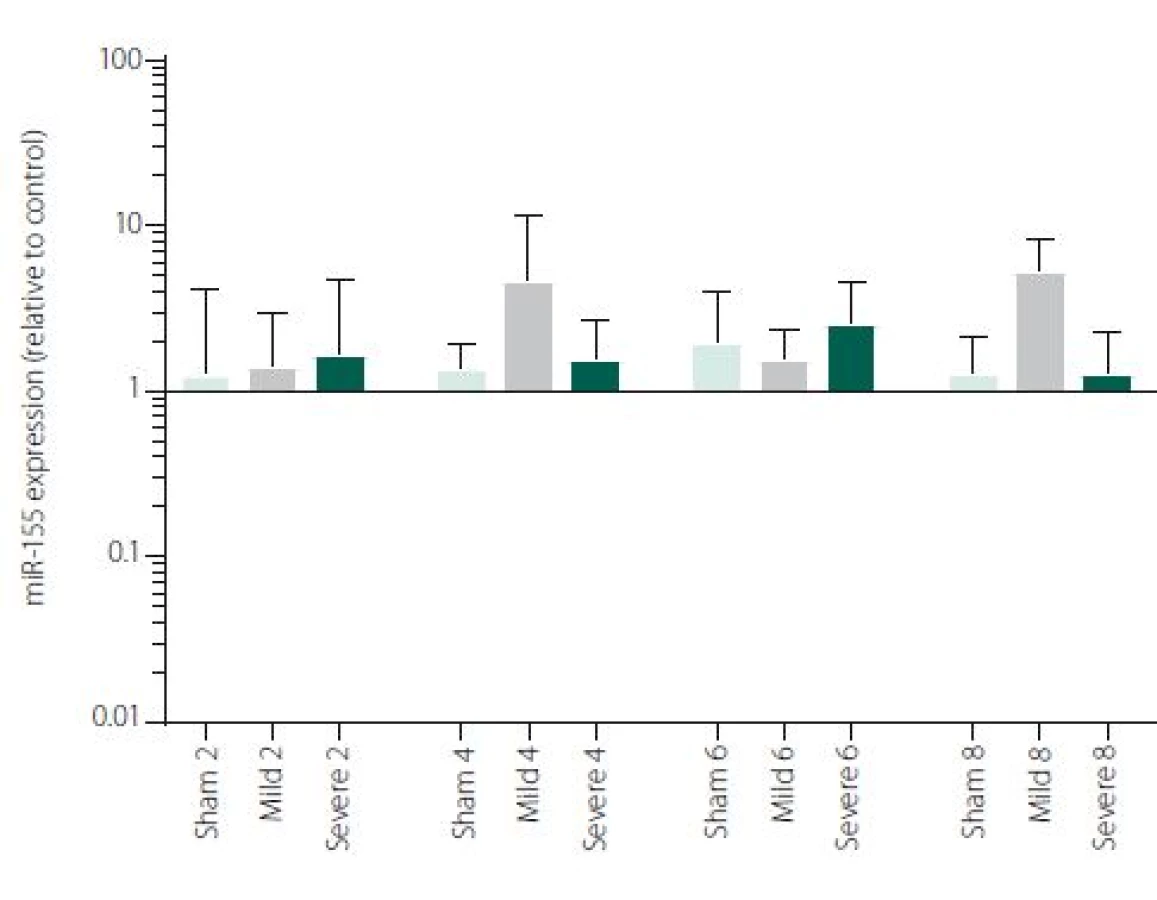
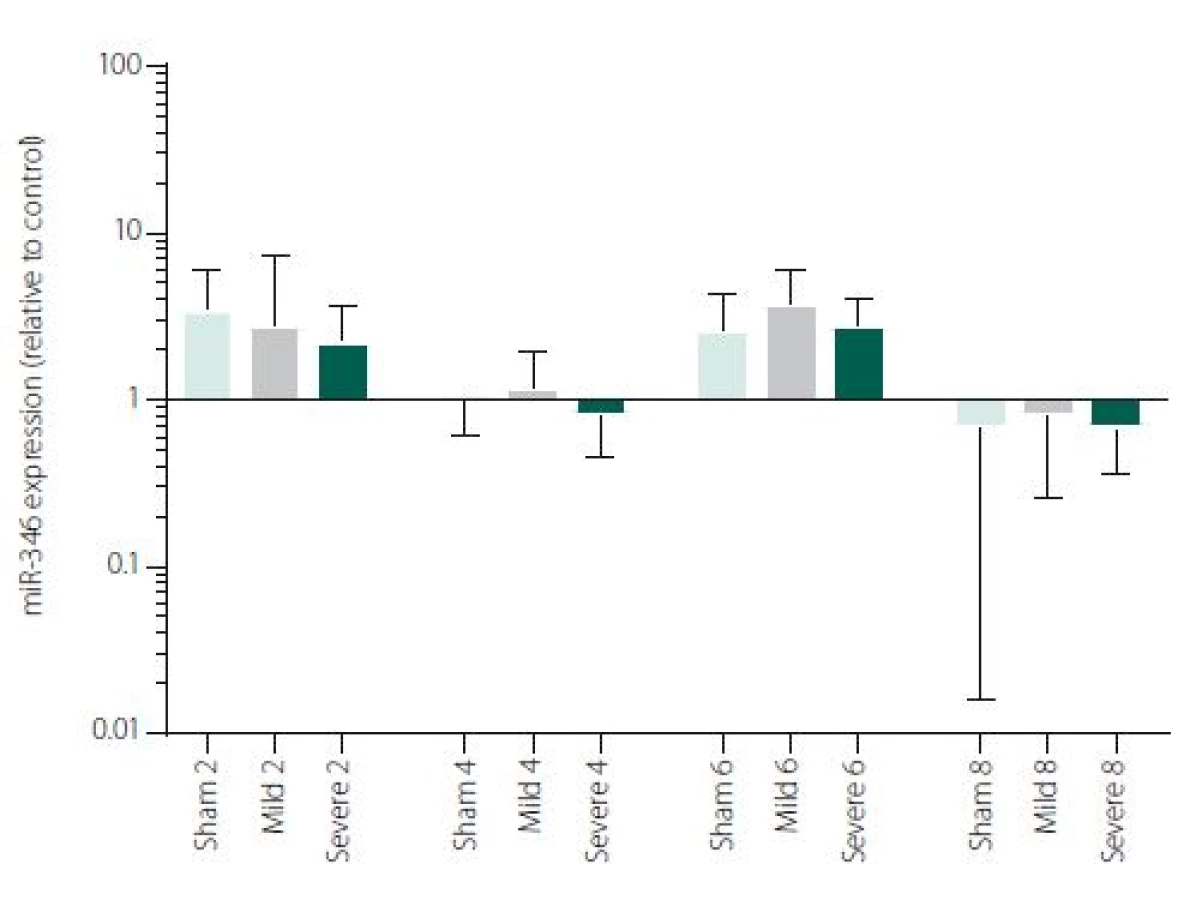
Figure 1 shows TNFα mRNA expression 2, 4, 6 and 8 h after induction of SAH. There is a statistically significant difference between Sham and Severe SAH groups at all the time-points (p < 0.05). The difference between Sham and Mild SAH groups is statistically significant 4 h after induction of SAH (p < 0.05). The difference between Mild and Severe SAH groups is statistically significant at 2 and 6 h time-points (p < 0.05). Fig. 2 shows miR-15b expression 2, 4, 6 and 8 h after induction of SAH. Significant difference between Sham and Severe SAH groups was observed 8 h after SAH (p < 0.05). Fig. 3 shows miR-125b expression 2, 4, 6 and 8 h after induction of SAH. No significant difference was observed between the groups at any time-point (p = NS). Fig. 4 shows miR-146a expression 2, 4, 6 and 8 h after induction of SAH. No significant difference was observed between the groups at any time-point (p = NS). Fig. 5 shows miR-155 expression 2, 4, 6 and 8 h after induction of SAH. No significant difference was observed between the groups at any time-point (p = NS). Fig. 6 shows miR-346 expression 2, 4, 6 and 8 h after induction of SAH. No significant difference was observed between the groups at any time-point (p = NS).
Discussion
Early inflammatory response, with the fundamental role of cytokine network, represents a hallmark in the pathology of SAH [35]. Within minutes of a haemor-rhage, a large number of proinflammatory cytokines (TNFα, IL-1b, IL-6, IL-8) are released into subarachnoid space. The levels of pro-inflammatory cytokines have been described to be globally elevated (brain tissue, circulation, CSF) after SAH in many studies [35,36]. Pro-inflammatory cytokines have a cytotoxic effect and significantly contribute to SAH associated EBI [12]. TNFα is considered as a major mediator of apoptosis [37] which is able to induce apoptosis directly in various cells, including endothelial cells [11] and neurons [38,39]. Apoptosis has been described in differ-ent intracranial pathologies such as Alzheimer‘s disease, Parkinson’s disease [40], cerebral ischaemia [41] or SAH [42].
In our study, an activation of early inflammatory response in the brain after experimentally induced SAH was evaluated on the level of crucial pro-inflammatory and pro-apoptotic cytokine TNFα. We have described a significant difference in TNFα mRNA expression between Severe SAH groups and Sham at all the time-points (2, 4, 6 and 8 h). However, the difference between Sham and Mild SAH groups was significant only 4 h after SAH induction. Finally, the difference between Mild and Severe SAH groups was statistically significant at 2 and 6-h time-points. These data suggest that activation of the immune system is strong in Severe SAH, while the immune response in Mild SAH may be variable. TNFα has a broad spectrum of biological activities and probably contributes to the exacerbation of neuronal damage following SAH. Previous studies have shown that TNFα is involved in BBB permeability disruption, in thrombogenic and vascular changes leading to neuronal damage and angiogenesis [43–46]. Furthermore, TNFα participates in the processes of cell death, both necrosis and apoptosis [37,47].
Apoptosis and inflammation are tightly regulated processes at the level of gene expression. Profiling studies in humans have shown that miRNAs participate in different post-stroke pathologies and their levels are changed globally after SAH in CSF and circulation [16,18]. MiRNAs can represent tissue-specific molecular profiles that further define significant pathological features and individual miRNA may have a specific protective or pathogenic role. As master regulators of gene expression, miRNAs participate in apoptosis via targeting both pro - and anti-apoptotic genes and death receptors. Those cell-surface receptors belong to the TNF superfamily and mediate TNF-induced apoptosis [48].
In the perspective of a more comprehensive understanding of the molecular mechanisms activated after SAH, we selected 5 miRNAs involved in TNFα regulation (miR-15b, miR-125b, miR-146a, miR-346, miR-155). We have found significant difference of miR-15b expression between Severe SAH and SHAM in 8-h time-point. Our data are in consensus with other stroke models. Shi et al. reported differential expression of miR-15b at both 24 and 72 h following middle cerebral artery occlusion (MCAO) compared to sham-operated controls [31]. Liu et al. detected increasing expression of miR-15b in embolic stroke model in rats through all time points (0–168 h after MCAO) [49]. The study focused on the effect of ischaemic preconditioning (PC) on cerebral “miRNAsome”, miR-15b showed increased expression at three time-points (6, 24 and 72 h) in the cerebral cortex of the rats subjected to PC by a 10-min transient MCAO [50]. MiR-15b exhibited robust up-regulation in ischaemic brain tissue representing thus a major contributor to neural damage following cerebral ischaemia. It was previously reported that miR-15b was involved in apoptosis via targeting the mRNA of the anti-apoptotic gene Bcl-2 in cancer cells [29,51]. Under ischaemic conditions, Bcl-2 protein level is suppressed and negatively correlated with miR-15b level [31]. Experimental suppression of miR-15b, exerting its anti--apoptotic effects due to Sevoflurane, has been described to contribute to its ischaemic neuroprotection [31]. Thus, targeting of miR-15b expression represents a novel neuroprotective strategy against ischaemic injury. It has also been reported that miR-15b acts as an upstream regulator of a mitochondrial signalling pathway and its inhibition can protect against cardiac ischaemic injury [30,32,52]. It has been described that miR-15b can regulate TNF--mediated apoptosis in hepatic cells via a Bcl-2 pathway [53]. Bcl-2 is a key anti-apoptotic protein, which protects cells from apoptosis in the TNF-dependent death receptors signalling pathway. An et al. suggested hypothesis, that miR-15bcan reduce TNFα at different levels (transcriptional and translational) via Bcl-2. Hence, decreased TNFα production following anti-miR15b treatment may contribute to reduced damage to cells [53].
Our results suggest that miR-15b together with TNFα may play important roles in the severity of brain damage in EBI after SAH. A combination of suppression of both pro-apoptotic TNFα and miR-15b may bring better results in therapeutic strategies for the treatment of SAH. While the EBI in terms of inflammatory response and cell death may be clearly characterised, regulatory mechanism of this response is very complex and hard to define. In our study, pro-apoptotic miR-15b was the only evaluated miRNA, which was influenced by SAH, suggesting that signalling pathways related to apoptosis could be triggered early after SAH [3,42].
Expression of other selected miRNAs was not overly affected by SAH, although these particular miRNAs were described to participate in the regulation of pro-inflammatory cytokine expression. MiRNAs and cytokine activities are strongly interconnected. Recent reports point to cross-regulation between cytokine and miRNA pathways [54,55]. It means, that not only miRNAs regulate cytokine expression, but also vice versa cytokines can modulate both miRNAs expression and secretion [56,57]. This most probably indicates the complexity of miRNAs-cytokine interactions and functions which are pleiotropic and influenced by many hardly defined factors. Moreover, several studies indicate that the selective manipulations with inflammatory response after stroke may be problematic due to the complexity of the signalling network [58,59]. We are, however, aware that we cannot make any conclusion to such a hypothesis based on our data and further study revealing the function of miR-15b in association with TNFα and description of their detailed dynamics is needed.
Limitations
In this study, we were focused on early dynamics of TNFα and associated miRNAs. We have evaluated selected parameters in a very short period of time following SAH and we have not studied the consequences of activation increased levels of TNFα and this has to be considered as a limitation. However, selected time-points represent the local immune response in brain mediated mainly by microglia and astrocytes, while later time-points (24 h or later) are associated with the second step of inflammatory response – inflammation mediated leukocytes from the periphery. Another limita-tion is the small number of evaluated miRNAs. Selected miRNAs are crucial for the regulation of TNFα expression, but we are aware that a larger panel of miRNAs could bring us much better information about TNFα regulations. Finally, we have not evaluated any inflammatory parameter other than TNFα. We were focused on this particular cytokine, because TNFα is one of the most important contributors to acute inflammatory response.
Conclusion
Subarachnoid haemorrhage is associated with an early increase in TNFα expression, which is sharp in severe and more variable in mild cases. MiRNAs involved in the regulation of TNFα expression has not been generally affected by SAH, except miR-15b which was increased in Severe SAH groups. Cytokine expression regulations are very complex and more comprehensive study is needed for better information about regulations/ deregulations of inflammatory response and apoptosis. Nevertheless, identification of miRNAs involved in the regulation of the mechanisms activated after a SAH and their potential targets may improve our knowledge of SAH pathophysiology and lead to identification of new therapeutic targets in order to minimise neurological impairment and enhance the functional recovery of the patients.
This work was supported by the Czech Science Foundation (GACR), project No. 14-23773P.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
MUDr. Michal Jurajda, Ph.D.
Institute of Pathological Physiology
Faculty of Medicine
Masaryk University
Kamenice 5
625 00 Brno
Czech Republic
e-mail: mjuraj@med.muni.cz
Accepted for review: 19. 7. 2018
Accepted for print: 21. 11. 2018
Sources
1. Al-Khindi T, Macdonald RL, Schweizer TA. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke 2010; 41(8): e519–e536. doi: 10.1161/ STROKEAHA.110.581975.
2. Chowdhury T, Dash HH, Cappellani RB et al. Early brain injury and subarachnoid hemorrhage: where are we at present? Saudi J Anaesth 2013; 7(2): 187–190. doi: 10.4103/ 1658-354X.114047.
3. Ostrowski RP, Colohan AR, Zhang JH. Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res 2006; 28(4): 399–414. doi: 10.1179/ 016164106X115008.
4. Sercombe R, Dinh YRT, Gomis P. Cerebrovascular inflammation following subarachnoid hemorrhage. Jpn J Pharmacol 2002; 88(3): 227–249.
5. Gallia GL, Tamargo RJ. Leukocyte-endothelial cell interactions in chronic vasospasm after subarachnoid hemorrhage. Neurol Res 2006; 28(7): 750–758. doi: 10.1179/ 016164106X152025.
6. Hasegawa Y, Suzuki H, Sozen T et al. Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochir Suppl 2011; 110(Pt1): 43–48. doi: 10.1007/ 978-3-7091-0353-1_8.
7. Clausen BH, Lambertsen KL, Babcock AA et al. Interleukin-1beta and tumor necrosis factor-alpha are expressed by different subsets of microglia and macrophages after ischemic stroke in mice. J Neuroinflammation 2008; 5 : 46. doi: 10.1186/ 1742-2094-5-46.
8. Vecchione C, Frati A, Di Pardo A et al. Tumor necrosis factor-alpha mediates hemolysis-induced vasoconstriction and the cerebral vasospasm evoked by subarachnoid hemorrhage. Hypertension 2009; 54(1): 150–156. doi: 10.1161/ HYPERTENSIONAHA.108.128124.
9. Polunovsky VA, Wendt CH, Ingbar DH et al. Induction of endothelial cell apoptosis by TNFα: modulation by inhibitors of protein synthesis. Exp Cell Res 1994; 214(2): 584–594.
10. Baxter GT, Kuo RC, Jupp OJ et al. Tumor necrosis factor-α mediates both apoptotic cell death and cell proliferation in a human hematopoietic cell line dependent on mitotic activity and receptor subtype expression. J Biol Chem 1999; 274(14): 9539–9547.
11. Messmer UK, Briner VA, Pfeilschifter J. Tumor necrosis factor-alpha and lipopolysaccharide induce apoptotic cell death in bovine glomerular endothelial cells. Kidney Int 1999; 55(6): 2322–2337.
12. Kimura H, Gules I, Meguro T et al. Cytotoxicity of cytokines in cerebral microvascular endothelial cell. Brain Res 2003; 990(1–2): 148–156.
13. Gauthier BR, Wollheim CB. MicroRNAs: “ribo-regulators” of glucose homeostasis. Nat Med 2006; 12(1): 36–38. doi: 10.1038/ nm0106-36.
14. Matsubara H, Takeuchi T, Nishikawa E et al. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17-92. Oncogene 2007; 26(41): 6099–6105.
15. Bi Y, Liu G, Yang R. MicroRNAs: novel regulators during the immune response. J Cell Physiol 2009; 218(3): 467–472. doi: 10.1002/ jcp.21639.
16. Bache S, Rasmussen R, Rossing M et al. MicroRNA changes in cerebrospinal fluid after subarachnoid hemorrhage. Stroke 2017; 48(9): 2391–2398.
17. Perry MM, Moschos SA, Williams AE et al. Rapid changes in microRNA-146a expression negatively regulate the IL-1beta-induced inflammatory response in human lung alveolar epithelial cells. J Immunol 2008; 180(8): 5689–5698.
18. Sepramaniam S, Tan JR, Tan KS et al. Circulating microRNAs as biomarkers of acute stroke. Int J Mol Sci 2014; 15(1): 1418–1432. doi: 10.3390/ ijms15011418.
19. Dewdney B, Trollope A, Moxon J et al. Circulating microRNAs as biomarkers for acute ischemicstroke: a systematic review. J Stroke Cerebrovasc Dis 2018; 27(3): 522–530. doi: 10.1016/ j.jstrokecerebrovasdis.2017. 09.058.
20. Chaudhuri AA, So AY, Sinha N et al. MicroRNA-125b potentiates macrophage activation. J Immunol 2011; 187(10): 5062–5068. doi: 10.4049/ jimmunol.1102001.
21. Le MTN, Xie H, Zhou B et al. MicroRNA-125b promotes neuronal differentiation in human cells by repressing multiple targets. Mol Cell Biol 2009; 29(19): 5290–5305. doi: 10.1128/ MCB.01694-08.
22. Pogue AI, Cui JG, Li YY et al. Micro RNA-125b (miRNA-125b) function in astrogliosis and glial cell proliferation. Neurosci Lett 2010; 476(1): 18–22. doi: 10.1016/ j.neulet.2010.03.054.
23. Gong J, Zhang JP, Li B et al. MicroRNA-125b promotes apoptosis by regulating the expression of Mcl-1, Bcl-w and IL-6R. Oncogene 2013; 32(25): 3071–3079. doi: 10.1038/ onc.2012.318.
24. Jude JA, Dileepan M, Subramanian S et al. miR-140-3p regulation of TNF-α-induced CD38 expression in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2012; 303(5): L460–L468. doi: 10.1152/ ajplung.00041.2012.
25. Wu P, Zuo X, Ji A. Stroke-induced microRNAs: The potential therapeutic role for stroke. Exp Ther Med 2012; 3(4): 571–576. doi: 10.3892/ etm.2012.452.
26. Liu G, Abraham E. MicroRNAs in immune response and macrophage polarization. Arterioscler Thromb VascBiol 2013; 33(2): 170–177. doi: 10.1161/ ATVBAHA.112.300068.
27. Semaan N, Frenzel L, Alsaleh G et al. miR-346 controls release of TNF-α protein and stability of its mRNA in rheumatoid arthritis via tristetraprolin stabilization. PLoS One 2011; 6(5): e19827. doi: 10.1371/ journal.pone.0019827.
28. Zhu W, Qian J, Ma L et al. MiR-346 suppresses cell proliferation through SMYD3 dependent approach in hepatocellular carcinoma. Oncotarget 2017; 8(39): 65218–65229. doi: 10.18632/ oncotarget.18060.
29. Guo CJ, Pan Q, Li DG et al. miR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: an essential role for apoptosis. J Hepatol 2009; 50(4): 766–778. doi: 10.1016/ j.jhep.2008.11.025.
30. Hullinger TG, Montgomery RL, Seto AG et al. Inhibition of miR-15 protects against cardiac ischemic injurynovelty and significance. Circ Res 2012; 110(1): 71–81. doi: 10.1161/ CIRCRESAHA.111.244442.
31. Shi H, Sun B, Zhang J et al. miR-15b suppression of Bcl-2 contributes to cerebral ischemic injury and is reversed by sevoflurane preconditioning. CNS Neurol Disord – Drug Targets 2013; 12(3): 381–391. doi: 10.2174/ 1871527311312030011.
32. Liu L, Zhang G, Liang Z et al. MicroRNA-15b enhances hypoxia/ reoxygenation-induced apoptosis of cardiomyocytes via a mitochondrial apoptotic pathway. Apoptosis 2014; 19(1): 19–29. doi: 10.1007/ s10495-013-0899-2.
33. Sugawara T, Ayer R, Jadhav V et al. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods 2008; 167(2): 327–334. doi: 10.1016/ j.jneumeth.2007.08.004.
34. Sugawara T, Ayer R, Jadhav V et al. Simvastatin attenuation of cerebral vasospasm after subarachnoid hemorrhage in rats via increased phosphorylation of akt and endothelial nitric oxide synthase. J Neurosci Res 2008; 86(16): 3635–3643. doi: 10.1002/ jnr.21807.
35. Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab 2012; 32(9): 1677–1698. doi: 10.1038/ jcbfm.2012.88.
36. Fassbender K, Hodapp B, Rossol S et al. Inflammatory cytokines in subarachnoid haemorrhage: association with abnormal blood flow velocities in basal cerebral arteries. J Neurol Neurosurg Psychiatry 2001; 70(4): 534–537.
37. Rath PC, Aggarwal BB. TNF-induced signaling in apoptosis. J Clin Immunol 1999; 19(6): 350–364.
38. Talley AK, Dewhurst S, Perry SW et al. Tumor necrosis factor alpha-induced apoptosis in human neuronal cells: protection by the antioxidant N-acetylcysteine and the genes bcl-2 and crmA. Mol Cell Biol 1995; 15(5): 2359–2366.
39. Sipe KJ, Dantzer R, Kelley KW et al. Expression of the 75 kDA TNF receptor and its role in contact-mediated neuronal cell death. Brain Res Mol Brain Res 1998; 62(2): 111–121.
40. Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 2000; 1(2): 120–129. doi: 10.1038/ 35040009.
41. Leker RR, Aharonowiz M, Greig NH et al. The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin alpha. Exp Neurol 2004; 187(2): 478–486. doi: 10.1016/ j.expneurol.2004.01.030.
42. Cahill WJ, Calvert JH, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 2006; 26(11): 1341–1353. doi: 10.1038/ sj.jcbfm.9600283.
43. Limb GA, Chignell AH, Green W et al. Distribution of TNF alpha and its reactive vascular adhesion molecules in fibrovascular membranes of proliferative diabetic retinopathy. Br J Ophthalmol 1996; 80(2): 168–173.
44. Barone FC, Arvin B, White RF et al. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 1997; 28(6): 1233–1244.
45. Mathiesen T, Edner G, Ulfarsson E et al. Cerebrospinal fluid interleukin-1 receptor antagonist and tumor necrosis factor – α following subarachnoid hemorrhage. J Neurosurg 1997; 87(2): 215–220. doi: 10.3171/ jns.1997.87.2. 0215.
46. Goukassian DA, Qin G, Dolan C et al. Tumor necrosis factor-alpha receptor p75 is required in ischemia-induced neovascularization. Circulation 2007; 115(6): 752–762. doi: 10.1161/ CIRCULATIONAHA.106.647255.
47. Zhang DW, Shao J, Lin J et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325(5938): 332–336. doi: 10.1126/ science.1172308.
48. Garoaflo M, Condorelli GL, Croce CM et al. MicroRNAs as regulators of death receptors signaling. Cell Death Differ 2009; 17(2): 200–208. doi: 10.1038/ cdd.2009.105.
49. Liu FJ, Lim KY, Kaur P et al. microRNAs involved in regulating spontaneous recovery in embolic stroke model. PLoS One 2013; 8(6): e66393. doi: 10.1371/ journal.pone.0066393.
50. Dharap A, Vemuganti R. Ischemic pre-conditioning alters cerebral microRNAs that are upstream to neuroprotective signaling pathways. J Neurochem 2010; 113(6): 1685–1691. doi: 10.1111/ j.1471-4159.2010.06735.x.
51. Xia L, Zhang D, Du R et al. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer 2008; 123(2): 372–379. doi: 10.1002/ ijc.23501.
52. Liu L, Johnson HL, Cousens S et al. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet 2012; 379(9832): 2151–2161. doi: 10.1016/ S0140-6736(12)60560-1.
53. An F, Gong B, Wang H et al. miR-15b and miR-16 regulate TNF mediated hepatocyte apoptosis via BCL2 in acute liver failure. Apoptosis 2012; 17(7): 702–716. doi: 10.1007/ s10495-012-0704-7.
54. Amado T, Schmolka N, Metwally H et al. Cross-regulation between cytokine and microRNA pathways in T cells. Eur J Immunol 2015; 45(6): 1584–1595. doi: 10.1002/ eji.201545487.
55. Benes V, Collier P, Kordes C et al. Identification of cytokine-induced modulation of microRNA expression and secretion as measured by a novel microRNA specific qPCR assay. Sci Rep 2015; 5 : 11590. doi: 10.1038/ srep11590.
56. Kutty RK, Nagineni CN, Samuel W et al. Inflammatory cytokines regulate microRNA-155 expression in human retinal pigment epithelial cells by activating JAK/ STAT pathway. Biochem Biophys Res Commun 2010; 402(2): 390–395. doi: 10.1016/ j.bbrc.2010.10.042.
57. Takahashi H, Kanno T, Nakayamada S et al. TGF- and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat Immunol 2012; 13(6): 587–595. doi: 10.1038/ ni.2286.
58. Enlimomab Acute Stroke Trial Investigators. Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology 2001; 57(8): 1428–1434.
59. Furuya K, Takeda H, Azhar S et al. Examination of several potential mechanisms for the negative outcome in a clinical stroke trial of enlimomab, a murine anti-human intercellular adhesion molecule-1 antibody: a bedside-to-bench study. Stroke 2001; 32(11): 2665–2674.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2019 Issue 1
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Hope Awakens with Early Diagnosis of Parkinson's Disease Based on Skin Odor
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
Most read in this issue
- Mild traumatic brain injury management – consensus statement of the Czech Neurological Society CMS JEP
- Chronic subdural haematoma
- Oligoclonal IgG and free light chains – comparison between agarose and polyacrylamide isoelectric focusing
- Ketogenic diet – effective treatment of childhood and adolescent epilepsies