
Hodnocení obsahové stejnoměrnosti tablet s nízkým obsahem léčivé látky s úzkým terapeutickým indexem
Evaluation of content uniformity of tablets with a low content of the active ingredient with a narrow therapeutic index
A short time ago there was a discussion about the use of NTI drugs such as warfarin. In the treatment by generic drugs some medical complications and deaths due to occult bleeding were retrospectively monitored. As a result of these observations, the generic substitutions with tablets containing warfarin were banned in the U.S. as there was a belief that generics have different pharmacokinetics. But a subsequent research found that different plasma levels are not due to generic drugs, but they are caused by different individual responses of the organism and especially by not uniform contents of warfarin in individual tablets. The different contents are caused by the method of direct compression where there is no granulation of the tablet blend before compressing. The method of direct compression, however, is very convenient in terms of time, technological feasibility and cost of cure. This report seeks to map the problems of direct compression in the production of tablets containing warfarin, concrete examples showing the relationships between selected parameters and the content uniformity of dosage form. Specifically, it is the influence of the density fillers and addition of the lubricant in various stages of mixing. On the basis of the application of statistical criteria according to the requirements of the FDA for the manufacture of tablets and with the use of the specific parameters of raw materials, the study has demonstrated that the technology of direct compression containing warfarin results not only in safe pharmacotherapy, but it also saves time, materials and cost of manufactured drugs.
Key words:
content uniformity • warfarin • GMP • validation • narrow therapeutic index
Received 19 September 2012 / Accepted 8 October 2012
Authors:
Jan Muselík; Aleš Franc
Authors‘ workplace:
Ústav technologie léků VFU, Brno
Published in:
Čes. slov. Farm., 2012; 61, 271-275
Category:
Original Articles
Overview
Nedávno proběhla diskuze ohledně používání léků s úzkým terapeutickým indexem (NTI), jako je warfarin. Při léčbě generiky docházelo ke zdravotním komplikacím a úmrtím na okultní krvácení. Proto bylo v USA zakázáno zahájit léčbu generickými náhradami s obsahem warfarinu v domnění, že léky mají rozdílnou farmakokinetiku. Výzkumy ale zjistily, že rozdílné plazmatické hladiny léčiva nejsou způsobeny generiky, ale rozdílnou odpovědí organismu a kolísáním obsahu warfarinu v tabletách. Rozdílný obsah byl způsoben použitou metodou přímého lisování výlisků, při které nedochází ke granulaci tabletoviny. Metoda přímého lisování je ovšem velice výhodná z hlediska času, technologické schůdnosti, a tím i ceny léku. Tato práce se snaží zmapovat úskalí výroby tablet s obsahem warfarinu metodou přímého lisování a na konkrétních příkladech ukazuje vztah mezi vybranými parametry a výslednou uniformitou lékové formy. Konkrétně se jedná o vliv hustoty plniva a vliv přísady kluziva v různém stupni mísení. Na základě uplatnění statistických kritérií požadovaných např. FDA pro výrobu tablet a dodržením kvality vstupních surovin se podařilo dokázat, že technologie přímého lisování s obsahem warfarinu vede nejen k bezpečné farmakoterapii, ale i k úspoře času, materiálů a snížení nákladů, resp. ceny vyráběných léků.
Klíčová slova:
obsahová stejnoměrnost • warfarin • SVP • validace • úzký terapeutický index
Úvod
Rozšiřující se trh s léky nutí výrobce stále více registrovat své přípravky na zahraničních teritoriích, což vede odborné pracovníky z výzkumu i výroby zvládat nejen odbornou, ale i ne vždy zcela přehlednou legislativní problematiku, spojenou s výrobou a registrací. Tato práce se zabývá problematikou validace obsahové stejnoměrnosti pevných lékových forem a na modelovém léčivu warfarinu ukazuje možnosti jak zvládnout přípravu lékové formy (tablet) za využití postupů SVP.
Léky musí splňovat jakostní kritéria, která země dovozců pro jejich registraci požadují. Dokumentem zabývajícím se validacemi v rámci Evropské unie je patnáctý dodatek Evropské komise týkající se validace a kvalifikace1). Kromě toho existují i odborná doporučení, která jsou vydávána od roku 1989 a která jsou publikována ve formě neustále aktualizovaných svazků. Sem patří i doporučení z roku 2012, zabývající se procesní validací2). V České republice se validací v oblasti technologie lékových forem zabývá pokyn SÚKLu, který je překladem již zmíněného pokynu patnáctého dodatku Evropské komise. Tyto pokyny jsou rovněž dostupné v podobě aktualizovaných vydání3). V USA je právní oblast farmaceutické výroby obsažena v zákonech jednotlivých zemí a ve federálních zákonech. Oblast léčiv a léků je zahrnuta v samostatných předpisech, které jsou jejich součástí a kam patří i část týkající se problematiky SVP4). Podrobné podmínky pro validaci obsahové stejnoměrnosti pevných lékových forem byly pak rozvedeny například v návrzích odborných doporučení FDA z roku 19995) a 20036).
Validace obsahové stejnoměrnosti pevných lékových forem
Přijatelná obsahová stejnoměrnost je nezbytnou podmínkou a jedním z nejdůležitějších kritických parametrů k tomu, aby mohl být léčivý přípravek uveden do výroby. Po validaci výroby a zavedení konkrétního výrobního postupu do praxe se provádí při každé výrobě rutinní, výstupní kontrola. Zkouška na obsahovou stejnoměrnost při rutinní kontrole, je-li požadována, musí vyhovovat požadavkům lékopisu. Zkouška je založená na stanovení jednotlivých obsahů léčivé látky v předepsaném počtu jednotek zkoušeného přípravku a určení, zda jednotlivé obsahy jsou v povolených mezích vzhledem k průměrné hodnotě obsahu. Pro vyhodnocení, zda tablety vyhovují nebo nevyhovují zkoušce na obsahovou stejnoměrnost, se použije zkouška A článku 2.9.6 ČL 2009, která se používá v případě tablet, prášků pro parenterální použití, očních inzertů a suspenzí pro injekce. Tato zkouška není zcela vypovídající, protože stanovuje pouze to, zda je množství léčivé látky v požadovaném rozmezí průměrného obsahu skutečného množství léčivé látky v tabletách, nikoliv jejího teoretického množství. Proto došlo ke změně požadavků ČL 2009, který byl harmonizován s Ph.Eur. (čl. 2.9.40). Namísto pevného procentuálního rozmezí od nalezeného průměru se zde používá výpočtu, který zohledňuje i teoretický obsah.
Před započetím řádné výroby se nejprve vyžaduje její validace. Při ní musí být splněny přísnější požadavky než při běžné rutinní kontrole již validovaného výrobního procesu. Nestačí splnit jen lékopisná kritéria. Cílem validace je zajistit, že na určité hladině statistické významnosti vyhoví následně vyrobené šarže daným lékopisným kritériím. Jako nejjednodušší se z hlediska informační výpovědi a proveditelnosti jeví tzv. indexy způsobilosti, jako jsou K-index správnosti procesu, Cp-index přesnosti procesu a nebo Cpk-index standardnosti procesu. Poslední z nich pak v sobě zahrnuje jak správnost, tak přesnost procesu a lze jej navíc snadno vypočítat na základě veličin, jako jsou průměr, maximum, minimum a směrodatná odchylka7). Z tohoto důvodu se obvykle pro ověření požadovaných kritérií pro testování obsahové stejnoměrnosti tablet používá Cpk-index pro limity dle ČL 2009 (čl. 2.9.6). Cpk-index je definován jako minimum z jeho horního (upper) a spodního (lower) indexu.

kde USL představuje horní specifikovaný limit, LSL představuje dolní specifikovaný limit, xi je aritmetický průměr z naměřených hodnot a s je směrodatná odchylka výběrového souboru. Pokud po dosazení patřičných proměnných obdržíme u prvních tří následně vyrobených šarží výsledek, který je větší nebo roven 1, znamená to, že nejméně 99,7 % šarží, které budou vyráběny tímto postupem, vyhoví použitému kritériu přijatelnosti. Tento limit je zároveň akceptovatelným pro validaci obsahové stejnoměrnosti tabletové směsi8). Pro ověření, že následně vyráběné šarže spolehlivě vyhoví článku 2.9.40, se používá tzv. Bergumovo rozdělení. Jedná se o tabelované hodnoty RSD, které na definované hladině spolehlivosti (obvykle α = 0,1) s 95% pravděpodobností zaručí, že následně vyrobené šarže vyhoví tomuto lékopisnému limitu9).
Farmakologické aspekty obsahové stejnoměrnosti léčivých přípravků s obsahem warfarinu
V roce 1997 došlo k soudnímu sporu mezi USA a firmou Barr Laboratories, která opakovaně vyráběla tablety, jejichž obsah přesahoval limit povoleným americkým lékopisem USP, což ohrožovalo bezpečnost farmakoterapie10). Po registraci přípravků dalších generických firem bylo potvrzeno, že terapeutická rizika spojená s podáváním warfarinu obvykle nebývají způsobena používáním warfarinu od různých výrobců11), nýbrž interindividuální variabilitou v odpovědi na podaný lék a kolísáním obsahu v jednotlivých tabletách12). Proto se parametry vztahující se k obsahové stejnoměrnosti jeví pro bezpečnost léčby jako zcela zásadní. Z tohoto důvodu požaduje monografie článku v USP i užší limit pro obsah léčiva v tabletě s obsahem warfarinu. Namísto běžného rozmezí pro obsah v tabletě 90–110 % je požadováno rozmezí 95–105 %. Odchylka v obsahu jednotlivých tablet by totiž mohla způsobit krvácení a následnou smrt pacienta, byť zavedeného na léčbu warfarinem od stejného výrobce. Dosažení vysoké obsahové stejnoměrnosti tablet se tak jeví jako velmi podstatný faktor bezpečnosti perorální lékové formy s obsahem warfarinu.
Technologické aspekty pro dosažení vyhovující obsahové stejnoměrnosti produktu
Při výrobě lékových forem s nízkým obsahem účinné látky je obtížné dosáhnout přísného limitu používaného pro validaci obsahové stejnoměrnosti tablet13).
Farmaceutická technologie proto nabízí několik možností výroby tablet s nízkým obsahem účinné složky, jako je vlhká granulace, suchá granulace, granulace tavením, fluidní granulace a v poslední době i impregnace14).
Některé z těchto metod však vždy nemusejí vést k očekávanému efektu a navíc se jedná o metody pracné a zdlouhavé, případně komplikované. Proto je zde stále, pokud je to možné, tendence preferovat metodu přímého lisování.
Metoda přímého lisování se začala ve farmaceutické technologii prosazovat zejména v sedmdesátých a osmdesátých letech 20. století. Její princip spočívá v tom, že se odvážené účinné a pomocné látky smísí v suchém stavu, a to buď v jednom kroku, nebo postupně. Vzniklá směs se bez dalších úprav ztabletuje. Mezi klasické pomocné látky patří plniva, která upravují objem výsledného výlisku a zlepšují lisovatelnost tabletoviny. Dále tzv. „suchá pojiva“, která zvyšují kohezivitu, resp. soudržnost výlisku, a často i lisovatelnost směsi, ze které vznikl. Pak jsou to rozvolňovadla, která vedou k rozpadu tablet v prostředí trávících šťáv. V neposlední řadě jde o kluziva usnadňující bezproblémové vysunutí výlisku z lisovnice tabletovačky. Aplikace kluziv, zejména stearanu hořečnatého, který se používá u drtivé většiny tabletových směsí, však při přímém lisování může způsobovat značné komplikace. Pokud se stearan hořečnatý mísí ve směsi po celou dobu již od počátku, mohou jeho jemné částice vytvořit na povrchu částic účinné látky, plniv a pojiv souvislý film. Ten pak díky omezení kohezivních vlastností směsi znesnadní proces lisování. Výsledkem jsou výlisky o nedostatečné pevnosti. Protože rovnoměrný hydrofobní film zároveň zabraňuje smáčivosti výlisku, resp. penetraci vody a aktivizaci rozvolňovadel, dosahují takovéto výlisky i pomalejší disoluce15). Proto se stearan hořečnatý přidává již k předmísené směsi a celková doba homogenizace obvykle nepřesahuje několik minut nebo dokonce sekund. Dojde přitom k „promazání“ směsi, ale již se nestačí vytvořit souvislý film. Jedná se o určitý kompromis mezi homogenitou stearanu hořečnatého ve směsi a účinností lubrikace. Na druhou stranu dlouhodobější doba mísení se stearanem hořečnatým může zlepšovat celkovou homogenitu výlisku nebo naopak jeho nehomogenitu vlivem akcelerace procesu segregace jednotlivých složek směsi právě v procesu mísení16).
Mezi hlavní nevýhody procesu přímého lisování patří nesnadné dosažení obsahové stejnoměrnosti. Ta je způsobena zejména rozdružováním při manipulaci se směsí a nedostatečným promísením v mísícím zařízení, kde zpravidla dochází k tzv. „hluchým místům“, ve kterých díky konstrukci mísících zařízení dochází často k nedostatečnému promísení17). Aby k tomuto jevu nedocházelo, bývají zde využívány účinnější 3D mísiče, kde se mísící kontejner otáčí kolem tří os. Případně se používají výkonné tzv. „rychloběžné mísiče“, které jsou však velice nákladné a nemají výhody 3D mísičů, kde stačí obvykle do mísiče upnout kontejner s ingrediencemi18).
Obsahovou stejnoměrnost směsi je rovněž nutné ošetřit vhodnou volbou fyzikálních vlastností použitých pomocných látek. Uplatňuje se zde řada teoretických i empirických pravidel, kam patří co nejpodobnější poměr podílu účinné látky a látek pomocných; účinná látka nemá být elektrostatická a zároveň hustota a velikost částic účinných a pomocných látek má být co nejpodobnější19).
U mísení sypkých směsí, kam přímé lisování z hlediska farmaceutické taxonomie patří, je rovněž nutné velmi opatrně volit dobu mísení. Neplatí zde pravidlo, že s délkou mísení vzrůstá i stupeň homogenity směsi. Naopak, po určité době mísení dojde k tzv. přemísení (overblending), kdy vlivem odlišných fyzikálně chemických vlastností jednotlivých komponent směsi dojde k jejich rozdružování a tyto se začnou oddělovat, resp. segregovat20). Proto musí být zvolena i optimální doba mísení, kterou nelze jednoznačně predikovat, ale je nutné ji stanovit experimentálně.
Souhrnně lze tedy konstatovat, že při výrobě tablet metodou přímého lisování je třeba věnovat zvýšené úsilí nejen složkám směsi, jejich fyzikálně chemickým vlastnostem (hustota, velikost a tvar částic, elektrostatický náboj) a jejich koncentraci ale i délce a rychlosti mísení, včetně velikosti násady v mísícím zařízení.
Pokusná část
Příprava tablet
Složení směsí a základní fyzikálně chemické vlastnosti jednotlivých složek jsou uvedeny v tabulce 1. Proměnnými byly hustota použitého plniva (laktosa DCL 11 nebo dihydrogen fosforečnan vápenatý Di-cafos 92-12) a technologie přípravy tabletových směsí.
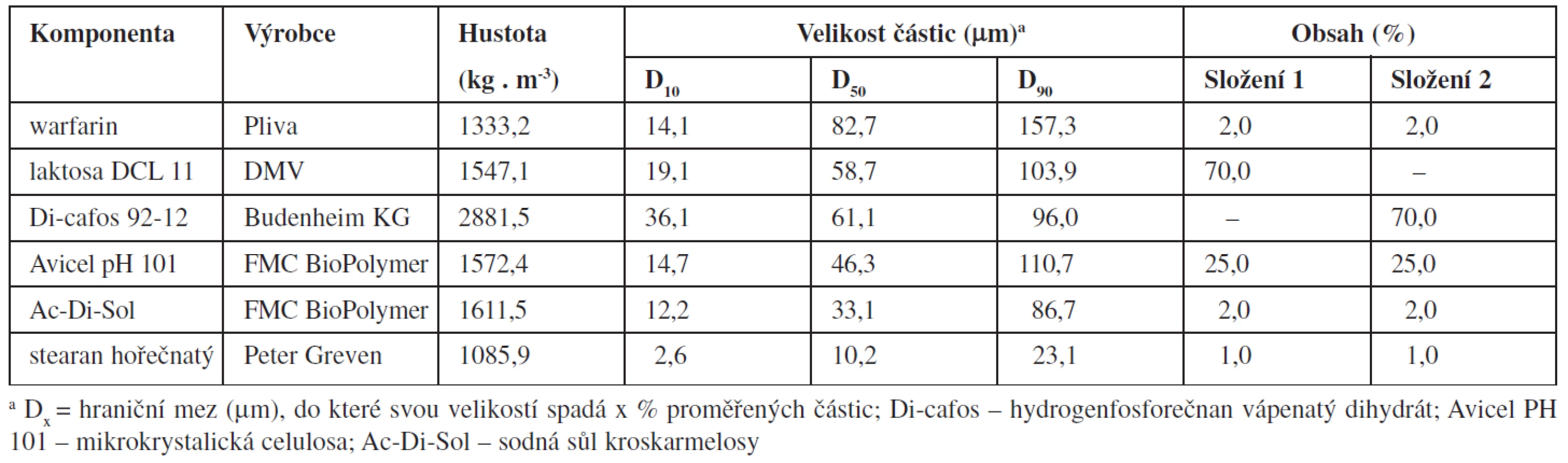
Jednotlivé látky (tab. 1.) byly přesítovány sítem o velikosti oka 250 ∝m a míseny 15 minut (technologie A) nebo byly jednotlivé látky míseny 10 minut bez přídavku stearanu hořečnatého, který byl přidán následně a mísení pokračovalo dalších 5 minut (technologie B). Mísení probíhalo v homogenizačním zařízení Turbula (T2C, Švýcarsko) rychlostí 40 otáček za minutu. Celkem byly připraveny čtyři různé tabletové směsi (dvě různá složení a dvě technologie mísení), z nichž od každé byly připraveny dvě šarže. Z jednotlivých směsí se nalisovaly tablety o hmotnosti asi 200 mg pomocí excentrického lisu (Korsch EK0, Německo). Tvrdost tablet byla přibližně 70 N.
Stanovení obsahu warfarinu v tabletách
Během lisování bylo v pravidelných časových intervalech odebráno z každé šarže 10 tablet. Odebrané tablety byly zváženy na analytických vahách, vloženy do odměrných baněk a ponechány po dobu 12 hodin v rozpouštědle (voda : methanol v poměru 9 : 1; v/v). Poté byla směs doplněna rozpouštědlem po rysku a nerozpuštěné pomocné látky byly odstraněny centrifugací (10 minut, 15 000 ot. min-1). Takto připravené vzorky byly analyzovány pomocí HPLC a obsah warfarinu byl stanoven na základě kalibrační křivky.
Pro stanovení obsahu warfarinu byl použit kapalinový chromatograf YL 9100 (Young Lin Instrument, Jižní Korea) s kvarternární pumpou, automatickým dávkovačem a detektorem s diodovým polem. Chromatografické separace byly provedeny na koloně BDS HYPERSIL C18 (150 ×⋅ 4,6 mm; velikost částic 5 ∝m). Složení mobilní fáze bylo 64 % methanolu a 36 % kyseliny mravenčí (0,04 M). Průtok mobilní fáze byl 1,4 ml min-1, teplota kolony byla 25 °C. Spektra byla snímána při 280 nm. Vždy bylo dávkováno 20 ∝l vzorku. Celková délka analýzy byla 7 minut.
Statistické zpracování výsledků
Výsledky stanovení jednotlivých tablet z každé šarže (počet vzorků n = 10) byly přepočteny relativně vůči teoretickému obsahu warfarinu. Z těchto hodnot byl pro každou připravenou šarži vypočítán průměrný obsah, směrodatná odchylka, relativní směrodatná odchylka, Cpk-indexy pro limity dle ČL 2009 čl. 2.9.6 (85–115 %) a získané hodnoty RSD byly porovnány s tabelovanými hodnotami Bergumova rozdělení.
Výsledky a diskuze
Připravené šarže tablet se odlišovaly jednak hustotou použitého plniva (laktosa nebo Di-cafos) a přísadou lubrikantu v různém stupni mísení (technologie A nebo B). Protože obsahová stejnoměrnost může být ovlivněna také velikostí částic pomocných látek, byla použita plniva o přibližně stejné distribuci velikosti částic (tab. 1). Celkový čas mísení a použité suroviny byly zvoleny na základě předchozích pokusů, jejichž výčet ani popis nejsou součástí tohoto článku.
Z výsledků vyplývá, že zvolené proměnné ovlivňovaly výslednou obsahovou stejnoměrnost tablet (tab. 2). Všechny připravené šarže vyhověly požadavkům ČL 2009 (čl. 2.9.6 a čl. 2.9.40) a statistickému hodnocení pomocí Cpk-indexu. Některé šarže však nevyhověly nejpřísnějšímu Bergumovu rozdělení. Pro ověření, zda připravené tablety vyhovují všem zvoleným kritériím, byly pro každé složení a obě technologie mísení připraveny a hodnoceny dvě šarže. Bergumovu rozdělení vyhověly pouze tablety s obsahem hydrogen fosforečnanu vápenatého (Di-cafos) jako plniva, připravené technologií mísení B, tedy s přídavkem stearanu hořečnatého v průběhu mísení. Během mísení technologií B na sebe částice hydrogen fosforečnanu vápenatého pravděpodobně absorbují warfarin, který je zde fixován díky svému elektrostatickému náboji, což dobře ukazují shodné hodnoty průměrného obsahu a RSD. U technologie A (šarže 1-2A_D61) tento efekt pozorován nebyl, pravděpodobně v důsledku narušení elektrostatických interakcí přídavkem stearanu hořečnatého hned v počátku mísení směsí.
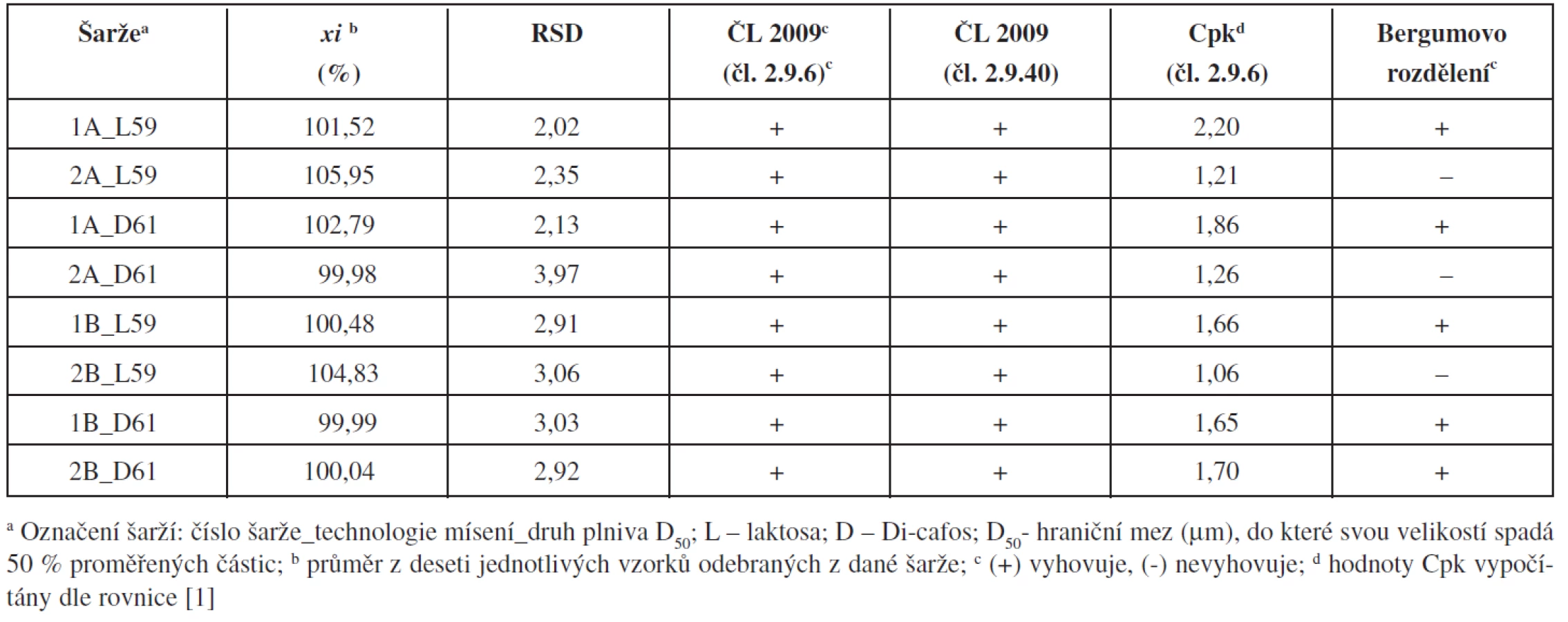
Vliv typu plniva použitého k přípravě tablet je pozorovatelný u zjištěných průměrných obsahů warfarinu v deseti analyzovaných tabletách z každé připravené šarže. Při analýze tablet připravených s použitím plniva s vyšší hustotou (Di-cafos) byly zjištěny menší odchylky průměrného obsahu warfarinu od teoretické hodnoty, než v případě použití plniva s menší hustotou (laktosa). Toto zjištění bylo potvrzeno u tablet připravených oběma technologiemi mísení.
Z provedených pokusů vyplývá, že přídavek lubrikantu (stearan hořečnatý) v průběhu mísícího procesu pozitivně ovlivňuje výslednou obsahovou stejnoměrnost tablet. Při jeho absenci na počátku mísení může zřejmě dojít k vytvoření souvislého filmu warfarinu na částicích plniva, který již dodatečná přísada lubrikantu neohrozí. Větší hustota plniva zlepšuje pohyblivost, resp. mísitelnost směsi během mísení, částice účinné látky lépe migrují v celém objemu kontejneru, čímž se zvýší i výsledná uniformita produktu.
Z nalezených hodnot lze konstatovat, že se podařilo nalézt vhodné složení pro přípravu tablet technologií přímého lisování s nízkým obsahem sodné soli warfainu klathrátu (šarže 1-2B_D61). Obsahová stejnoměrnost tablet vyhoví nejen lékopisným požadavkům, ale na potřebné statistické hladině významnosti vyhoví i kritériím používaným při procesní validaci, jako jsou indexy Cpk nebo Bergumovo rozdělení.
Seznam zkratek
ČL 2009 – Český lékopis 2009
FDA – Americký úřad pro potraviny a léčiva
NTI – úzký terapeutický index
Ph.Eur. – Evropský lékopis
SVP – Správná výrobní praxe
SÚKL – Státní ústav pro kontrolu léčiv
USP – Lékopis USA
Střet zájmů: žádný.
Došlo 19. září 2012 / Přijato 8. října 2012
Jan Muselík, Aleš Franc
Adresa pro korespondenci:
Mgr. Jan Muselík, Ph.D.
Ústav technologie léků VFU
Palackého tř. 1/3, 612 42 Brno
e-mail: muselikj@vfu.cz
Sources
1. European Commission: Annex 15 to the EU Guide to Good Manufacturing Practice - Qualification and Validation. Enterprise Directorate-General (EUDRALEX 2001).
2. E.U. European Medicines Agency, Sciences Medicines Health: Guideline on Process Validation. (EMA/CHMP/CVMP/QWP/ 70278/2012-Rev1 2012).
3. ČR. Státní ústav pro kontrolu léčiv: Pokyny pro správnou výrobní praxi - doplněk č. 15: Kvalifikace a validace (VYR 32 2003).
4. CFR – Code of Federal Regulations/Revised as of April 1, 2011. Current Good Manufacturing Practice for Finished Pharmaceutical (Title 21, Volume 4).
5. U.S. Department of Health and Human Services Food and Drug Administration: ANDAs: Blend Uniformity Analysis (CDER 1999).
6. U.S. Department of Health and Human Services Food and Drug Administration: Powder Blends and Finished Dosage Units — Stratified In-Process Dosage Unit Sampling and Assessment (CGMPs 2003).
7. Montgomery D. C. Introduction to Statistical Quality Control. 6th ed. New York: Wiley 2004; 734 s.
8. Pearn W. L., Shu M. H. Manufacturing capability control for multiple power-distribution switch processes based on modified C-pk MPPAC. Microelectron Reliab 2003; 43, 963–975.
9. Bergum J. S., Li H. Acceptance limits for the new ICH USP 29 content-uniformity test. Pharm. Tech. 2007; 31, 90–100.
10. The Federal Reporter: 386 F.3d 485 (2004). http://bulk. resource.org/courts.gov/c/F3/386/386.F3d.485.02-9346.02-9222.html (6. 8. 2012).
11. Henderson J. D., Esham R. H. Generic substitution: Issues for problematic drugs. South. Med. J. 2001; 94, 16–21.
12. Benson S. R., Vance-Bryan K. In favor of Coumadin over generic warfarin. Am. J. Health-Syst. Ph. 1998; 55, 727–729.
13. Carstensen J. T., Dali M. V. Blending validation and content uniformity of low-content, noncohesive powder blends. Drug Dev. Ind. Pharm. 1996; 22, 285–290.
14. Franc A., Rabišková M., Goněc R. Impregnation: a progressive method in the production of solid dosage forms with low content of poorly soluble drugs. Eur. J. Parent. Pharm. Sci. 2011; 16, 85–93.
15. Bolhuis G. K., Lerk F., Zijlstra H. T., de Boer A. H. Film formation by magnesium stearate during mixing and its effect on tableting. Pharmaceut. Weekbl. Sci. Ed. 1975; 10, 317–325.
16. Perrault M., Bertrand F., Chaouki J. An investigation of magnesium stearate mixing in a V-blender through gamma-ray detection. Powder. Technol. 2010; 200, 234–245.
17. Deveswaran R., Bharath S., Basavaraj B. V., Abraham S., Furtado S., Madhavan V. Concepts and techniques of pharmaceutical powder mixing process: A current update. Research J. Pharm. and Tech. 2009; 2, 245–249.
19. Siraj M. S., Radl S., Glasser B. J., Khinast J. G. Effect of blade angle and particle size on powder mixing performance in a rectangular box. Powder. Technol. 2011; 211, 100–113.
20. Chalabala M., Rabišková M., Pešák R., Masteiková R., Šolc J., Šrámek D. Základní operace a všeobecné postupy v technologii léků. In: Komárek P., Rabišková M. (eds). Technologie léků, 3. vyd. Praha: Galén 2006.
21. Moakher M., Shinbrot T., Muzzio F. J. Experimentally validated computations of flow, mixing and segregation of non-cohesive grains in 3D tumbling blenders. Powder. Technol. 2000; 109, 58–71.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2012 Issue 6
Most read in this issue
- Hodnocení obsahové stejnoměrnosti tablet s nízkým obsahem léčivé látky s úzkým terapeutickým indexem
- Analýza farmaceutické péče při dispenzaci léčiva orlistat v režimu OTC
- Halloysit – zajímavý nanotubulární nosič pro léčiva
- Porovnanie produkcie sanguinarínu suspenznými kultúrami rastlín čeľade Papaveraceae